





ABSTRACT
Posttraumatic osteoarthritis (PTOA) is associated with abnormal and increased subchondral bone remodeling. Inhibiting altered remodeling immediately following joint damage can slow PTOA progression. Clinically, however, inhibiting remodeling when significant joint damage is already present has minimal effects in slowing further disease progression. We sought to determine the treatment window following PTOA initiation in which inhibiting remodeling can attenuate progression of joint damage. We hypothesized that the most effective treatment would be to inhibit remodeling immediately after PTOA initiation. We used an animal model in which a single bout of mechanical loading was applied to the left tibia of 26‐week‐old male C57Bl/6 mice at a peak load of 9 N to initiate load‐induced PTOA development. Following loading, we inhibited bone remodeling using daily alendronate (ALN) treatment administered either immediately or with 1 or 2 weeks' delay up to 3 or 6 weeks post‐loading. A vehicle (VEH) treatment group controlled for daily injections. Cartilage and subchondral bone morphology and osteophyte development were analyzed and compared among treatment groups. Inhibiting remodeling using ALN immediately after load‐induced PTOA initiation reduced cartilage degeneration, slowed osteophyte formation, and preserved subchondral bone volume compared to VEH treatment. Delaying the inhibition of bone remodeling at 1 or 2 weeks similarly attenuated cartilage degeneration at 6 weeks, but did not slow the development of osteoarthritis (OA)‐related changes in the subchondral bone, including osteophyte formation and subchondral bone erosions. Immediate inhibition of subchondral bone remodeling was most effective in slowing PTOA progression across the entire joint, indicating that abnormal bone remodeling within the first week following PTOA initiation played a critical role in subsequent cartilage damage, subchondral bone changes, and overall joint degeneration. These results highlight the potential of anti‐resorptive drugs as preemptive therapies for limiting PTOA development after joint injury, rather than as disease‐modifying therapies after joint damage is established. © 2021 American Society for Bone and Mineral Research (ASBMR).
INTRODUCTION
Osteoarthritis (OA) is a degenerative joint disease that causes joint pain, stiffness, and lack of mobility.(1‐3) End‐stage OA is treated through total joint replacement surgery, but currently few nonsurgical disease‐modifying treatments exist that effectively slow OA progression.(4‐6) Simultaneous progressive changes in both articular cartilage and the underlying subchondral bone are key factors in OA pathogenesis and suggest that bone‐cartilage interactions play an important role in OA development.(7‐9) Due to the presence of subchondral bone sclerosis in osteoarthritic joints, high subchondral bone mass and stiffness have long been theorized to increase stress on cartilage leading to degeneration and OA development.(10) However, recent studies demonstrated that subchondral plate thickening alone is insufficient to increase OA development without an associated increase in subchondral bone remodeling.(11‐13) Additionally, in a subset of patients with both OA and osteoporosis, OA severity was more strongly correlated to increased subchondral bone resorption than to measures of bone mass.(14) These results suggest that increased subchondral bone remodeling rather than subchondral bone mass and stiffness may be a driving factor in OA development and increased OA risk.
Many studies investigating the role of subchondral bone remodeling in OA progression examine posttraumatic OA (PTOA). PTOA occurs secondary to a traumatic joint injury or overload, and therefore has a defined time point at which disease initiation occurs.(15) In preclinical models, PTOA development following joint injury or overload is used to track early‐stage changes in the joint and the progression of joint degeneration.(16‐19) PTOA development is associated with increased subchondral bone remodeling and an uncoupling of normal remodeling activity, but the role of subchondral bone remodeling in PTOA progression remains unclear.(20,21) Early‐stage PTOA is associated with reduced subchondral bone mass resulting from increased bone resorption and remodeling.(20,22‐25) Late‐stage PTOA is associated with increased subchondral bone mass and sclerosis occurring secondary to increased bone formation.(20,22,24,25) Given that abnormal and increased subchondral bone remodeling are one of the earliest markers of PTOA development(21) and may play a role in OA development,(13) preventing subchondral bone remodeling following joint injury may inhibit PTOA development and provide a treatment target for slowing disease progression.
Bisphosphonates, including alendronate (ALN), are clinically‐approved osteoporosis treatments that suppress bone resorption and turnover(26) and can inhibit subchondral bone remodeling following OA initiation. In preclinical studies, inhibiting remodeling with ALN treatment immediately following PTOA initiation slows the progression of cartilage degeneration and osteophyte formation and inhibits subchondral bone changes.(27‐31) However, results were mixed in clinical studies when bisphosphonate treatments were applied to patients with more advanced radiographically‐diagnosed OA, and inhibiting bone remodeling generally failed to prevent further cartilage degeneration or the progression of OA symptoms.(32‐35) Together, these results suggest a treatment window following OA initiation during which inhibiting the initial increase in subchondral bone remodeling can slow future OA development. Previously, Mohan et al.(36) showed that immediate ALN treatment following monosodium iodoacetate injection to induce OA pathology inhibited the progression of cartilage damage and subchondral bone changes; however, delayed ALN treatment beginning either 2 or 6 weeks following OA initiation was far less effective in altering disease progression,(36) again suggesting that an early window following OA initiation likely exists for effective treatment. Based on these findings, we were interested in determining how soon after load‐induced PTOA initiation subchondral bone resorption should be inhibited to effectively attenuate joint damage progression.
In the present study, we investigated inhibiting subchondral bone remodeling with ALN treatment starting within the 2‐week window immediately following load‐induced PTOA initiation to determine the role of early changes in subchondral bone remodeling on development of joint damage. We subjected mice to a single bout of high‐magnitude cyclic compressive tibial loading, a previously established model for PTOA development,(37) to noninvasively initiate PTOA progression. Following the single bout of tibial loading, we began daily ALN treatment either immediately after PTOA initiation, at a 1‐week delay, or at a 2‐week delay. PTOA development was then examined at 3 weeks and 6 weeks following loading through assessment of cartilage damage and thickness, osteophyte formation, and subchondral bone morphology. We hypothesized that inhibiting bone remodeling with ALN treatment immediately after PTOA initiation will most effectively inhibit load‐induced PTOA progression.
MATERIALS AND METHODS
Cyclic mechanical loading
A single bout of cyclic mechanical loading was applied to the lower left limb of 26‐week‐old male C57B1/6 mice (n = 7 to 8/group, 52 animals total, Figure 1; Jackson Laboratory, Bar Harbor, ME, USA) at a peak load magnitude of 9 N.(37) Right limbs served as contralateral controls in loaded groups. Mice were placed under general anesthesia during loading (2% isoflurane, 2.0 L/min, Webster, Devens, MA, USA). Loading was applied to left tibiae at 4 Hz for 1200 cycles (5 min). Mice were housed one to four per cage with ad libitum access to food and water (fed a standard diet; Harlan Teklad LM‐485; Harlan Laboratories, Indianapolis, IN, USA). All experimental procedures were approved by the Institutional Animal Care and Use Committee at Cornell University and the United States Army Medical Research and Development Command (USAMRMC) Animal Care and Use Review Office (Fort Detrick, MD, USA).
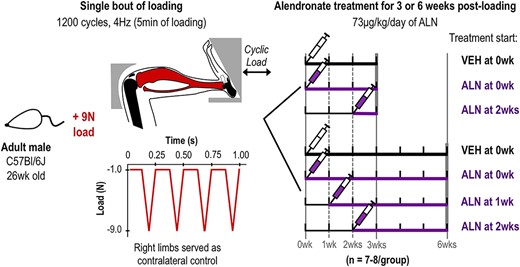
Experimental design and loading model. 26‐week C57Bl/6 male mice underwent a single bout of cyclic tibial compression. In the 3‐week group, mice then received ALN treatment immediately (0 week) or 2 weeks after loading until euthanasia. In the 6‐week group, mice began ALN treatment immediately (0 week), at 1 week, or 2 weeks after loading until euthanasia. In the control groups, VEH was administered immediately following the single bout of loading until mice were euthanized at either 3 or 6 weeks post‐loading. Contralateral limbs served as loading controls at each time point and treatment group. Abbreviations: ALN, alendronate; VEH, vehicle.
ALN treatment
Following the single bout of loading, mice were randomly separated into seven different treatment groups (Figure 1). Two groups received daily ALN treatment and were examined at 3 weeks post‐loading (73 μg/kg/day ip, 5 days/week, Sigma‐Aldrich, St. Louis, MO, USA); one group beginning immediately after the single bout of loading (0 week ALN, n = 7) and another beginning 2 weeks after the single bout of loading (2 week ALN, n = 7). Three groups received daily ALN treatment and were examined at 6 weeks post‐loading (73 μg/kg/day, ip, 5 days/week); one group beginning immediately after the single bout of loading (0 week ALN, n = 7), another beginning 1 week after the single bout of loading (1 week ALN, n = 8), and the third beginning 2 weeks after the single bout of loading (2 week ALN, n = 8). Two control groups received daily vehicle (VEH) treatment (saline, same volume, ip, 5 days/week, n = 7–8) beginning immediately after the single bout of loading for either 3 or 6 weeks post‐loading (Figure 1). Mice in each group resumed normal cage activity for 3 or 6 weeks following the single loading session. In the 3‐week experiment six of 21 were housed singly due to fighting; one of seven in the VEH‐treated group and five of seven in the 0 week ALN group, and none of seven in the 2‐week ALN group. In the 6‐week experiment 13 of 31 were housed singly due to fighting; five of eight in the VEH group, three of seven in the 0 week ALN group, five of eight in the 1 week ALN group, and none of eight in the 2 week ALN group. At 3 or 6 weeks following initial loading, mice in all treatment groups were euthanized. Knee joints were harvested and fixed in 4% paraformaldehyde overnight at 4°C.
Micro–computed tomography
Bone morphological differences in the tibia were assessed using micro–computed tomography (μCT). After overnight fixation in 4% paraformaldehyde, tissues were washed and transferred to 70% ethanol. Intact knee joints were scanned with an isotropic voxel resolution of 10 μm (μCT35; Scanco, Brüttisellen, Switzerland; 55 kVp, 145 μA, 600 ms integration time). Medial and lateral cortical subchondral plate thickness, tissue mineral density (TMD), and bone volume were measured. Subchondral plate thickness and TMD are presented as an average of the medial and lateral tibial plateaus, and bone volume is presented for the medial tibial plateau. Bone volume fraction (BV/TV) and TMD were assessed in the cancellous epiphysis. In all regions, bone was isolated by manually contouring the desired volume of interest (VOI). The VOI for the subchondral bone plate included the region of cortical bone beginning at the proximal end of the tibia and extended distally to the start of the cancellous bone in the epiphysis. The medial and lateral subchondral plates were visually segmented, and the full medial‐lateral width of the subchondral plate was included, excluding the tibial notch and osteophytes. The VOI for the epiphysis included cancellous bone distal to the subchondral plate and proximal to the growth plate.
Histological analysis
After completion of μCT scanning, knee joints were decalcified in 10% EDTA for 2 weeks, dehydrated in an ethanol gradient, and embedded in paraffin for histological analyses. Coronal sections were obtained from posterior to anterior at a 6 μm thickness using a rotary microtome (Leica RM2255; Leica, Wetzlar, Germany). Cartilage damage was assessed at 90‐μm intervals throughout the joint with Safranin O/Fast Green stained slides of the medial and lateral tibial plateaus using histological scoring performed by a blinded researcher. Structural cartilage damage in both loaded and control limbs was assessed by the Osteoarthritis Research Society International (OARSI) scoring system.(38) Histological scores are reported as averages for the whole joint, medial plateau, and lateral plateau. Cartilage thickness was measured on three representative Safranin O/Fast Green stained sections (posterior, middle, and anterior) on both medial and lateral tibial plateaus as described(39,40) and are reported as averages for the whole joint, medial plateau, and lateral plateau.
Medial tibial osteophyte formation was also assessed from Safranin‐O/Fast Green–stained sections in loaded limbs, because osteophyte formation only occurred in loaded limbs. The slide from each loaded knee containing the largest portion of the medial tibial osteophyte was analyzed. Osteophyte size was defined as the medial‐lateral width of the osteophyte at its widest point, measured from the medial edge of the epiphysis to the edge of the ectopic bone.(39) Osteophyte maturity was scored on a scale of 0 to 3 (0 = no osteophyte, 1 = primarily cartilaginous, 2 = mixture of cartilage and bone, or 3 = primarily bony structure) as described.(41)
Safranin‐O/Fast Green–stained sections also were used to examine loss of subchondral bone and calcification of the meniscus and medial collateral ligament (MCL). Gross loss of bone from the subchondral plate, epiphysis and, in several cases, metaphysis was observed in a portion of loaded limbs. The fraction of loaded limbs from each treatment group with a gross loss of subchondral bone was determined. In addition, both the meniscus and MCL were significantly calcified in a portion of loaded limbs at 6 weeks; the fraction of loaded limbs from each treatment group with these calcifications was determined.
Tartrate resistant acid phosphatase staining and anti–pro‐collagen I immunohistochemistry
Bone remodeling in the epiphysis and metaphysis were assessed by immunohistochemistry. Sections were stained for tartrate‐resistant acid phosphatase (TRAP) or using anti pro‐collagen I–specific antibodies (undiluted, SP1.D8; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) as described.(42) The numbers of positively stained osteoclasts (TRAP) and osteoblasts (pro‐collagen I) in the cancellous metaphysis were quantified and normalized to measured bone surface (OsteomeasureXP, v3.2.1.7; OsteoMetrics, Decatur, GA, USA). Osteoclast number was quantified for two representative slides per limb (n = 7 to 8/group), and osteoblast number was quantified for one representative slide per limb (n = 4/group).
Statistical analysis
The effects of loading and treatment group were determined using a linear mixed‐effects model for each duration post‐loading with fixed effects of loading (contralateral control or loaded) and treatment group (0 week ALN, 2 week ALN, or VEH for 3 weeks post‐load and 0 week ALN, 1 week ALN, 2 week ALN, or VEH for 6 weeks post‐load), and a random mouse effect (JMP Pro, Version 15, SAS Institute Inc., Cary, NC, USA). Post hoc analysis was performed using Tukey's test for significant interaction effects and t test for individual effects. The effect of treatment on the presence of MCL calcifications in loaded limbs was evaluated with Fischer's exact test and the Bonferroni correction for post hoc pairwise comparisons between difference treatment groups. Normality was confirmed visually using histograms and QQ plots of the residuals. Significance was set at p < 0.05 for linear mixed‐effects model and p < 0.008 when using the Bonferroni correction for post hoc pairwise comparisons (six hypotheses) following Fischer's exact test.
RESULTS
Inhibiting bone remodeling after PTOA initiation attenuated cartilage damage
Cartilage damage occurred in all loaded limbs at both 3 weeks (p < 0.001) and 6 weeks post‐loading (p < 0.001, Figure 2A,B). More severe damage occurred on the medial tibial plateau compared to the lateral (Figure 2A, Figure S1A). Full thickness cartilage erosions occurred on the medial plateau, whereas vertical clefts into cartilage and smaller erosions were visible on the lateral plateau. Following PTOA initiation, immediately inhibiting bone remodeling attenuated cartilage damage at 3 weeks (p = 0.043), while inhibiting bone remodeling regardless of when treatment was started attenuated cartilage damage at 6 weeks (p = 0.007, Figure 2A,B). At 3 weeks cartilage damage was only attenuated with ALN treatment on the lateral tibial plateau (p = 0.017, Figure S1B). At 6 weeks, the attenuation of cartilage damage by ALN treatment was similar on both the medial (p = 0.037, Figure S1A) and lateral tibial plateaus (p = 0.016, Figure S1B).
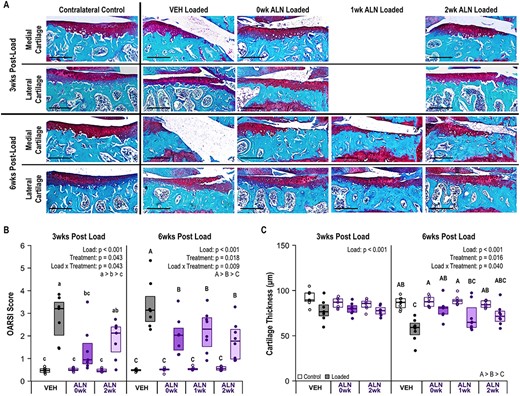
ALN treatment attenuated load‐induced cartilage damage. (A) Representative histology images of medial and lateral tibial plateau cartilage damage at 3 and 6 weeks following a single bout of loading. The representative medial cartilage image in the 6‐week VEH‐treated group also shows severe erosion of the medial tibial plateau. Scale bar = 250 μm. (B) Cartilage damage occurred in all loaded limbs (p < 0.001), but severity of damage was attenuated by ALN treatment at both 3 weeks (p = 0.043) and 6 weeks (p = 0.009). (C) Cartilage thickness decreased in loaded limbs (p < 0.001) and, at 6 weeks, loss of cartilage thickness was attenuated by ALN treatment (p = 0.040). Statistical test: linear mixed‐effects model. Box plot indicates median and interquartile range. Individual datum points (n = 7–8/group) are overlaid. Abbreviations: ALN, alendronate; VEH, vehicle.
Average cartilage thickness also decreased in loaded limbs at both 3 weeks (p < 0.001) and 6 weeks (p < 0.001) after the single bout of loading (Figure 2A,C). At 3 weeks, ALN treatment regardless of start time did not inhibit the loss of cartilage thickness. At 6 weeks, immediate ALN treatment most effectively attenuated loss of cartilage thickness when averaged across the whole joint (p = 0.043, Figure 2C), but did not affect either the medial (p = 0.246) or lateral (p = 0.130) tibial plateaus individually (Figure S1C,D). Overall, inhibiting bone remodeling with ALN attenuated load‐induced cartilage damage. Immediate ALN treatment most effectively slowed the development of cartilage damage at the earlier time point, whereas cartilage damage was attenuated similarly regardless of ALN‐treatment start time at the later time point.
Inhibiting bone remodeling immediately after PTOA initiation slowed osteophyte development
All loaded limbs developed osteophytes on the medial aspect of the tibia at 6 weeks post‐loading. The majority of loaded limbs developed osteophytes at 3 weeks post‐loading; two limbs in the immediate ALN treatment group and one limb in the delayed ALN‐treated group did not have osteophytes at 3 weeks post‐loading (Figure 3A). Osteophytes were absent in all non‐loaded contralateral control limbs. Immediate ALN treatment inhibited osteophyte growth after loading, resulting in smaller osteophytes at 3 weeks (p = 0.031) and smaller, less mature osteophytes at 6 weeks (p = 0.039, p = 0.004, Figure 3A–C). Delayed ALN treatment did not alter osteophyte size relative to VEH‐treated mice at either time point post‐load. At 3 weeks post‐load all osteophytes were at least partially cartilaginous. When ALN treatment began immediately after loading, the majority of osteophytes remained cartilaginous at 6 weeks following PTOA initiation. In contrast, delayed ALN or VEH treatment resulted in osteophytes that were partially to fully calcified at 6 weeks. Inhibiting bone remodeling immediately after PTOA initiation most effectively slowed periarticular bone formation.
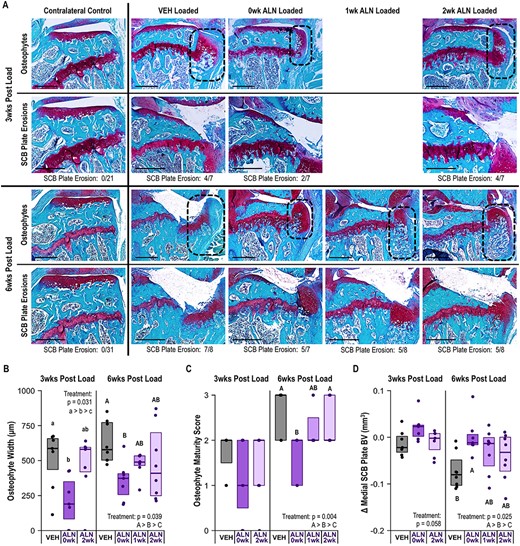
ALN treatment beginning immediately after loading slowed load‐induced osteophyte formation and attenuated medial subchondral plate erosions. (A) Representative histology images of osteophyte formation (delineated by dotted line) and medial subchondral plate erosions at 3 weeks and 6 weeks following a single bout of loading. The proportion of limbs in each group with subchondral plate erosions are listed below the images. Scale bar = 500 μm. (B) ALN treatment beginning immediately after loading decreased the osteophyte width in loaded limbs at both 3 weeks (p = 0.031) and 6 weeks (p = 0.039). (C) ALN treatment beginning immediately after loading slowed osteophyte maturity and reduced osteophyte calcification at 6 weeks (p = 0.004). (D) Immediate ALN treatment prevented loss of bone volume in the medial subchondral plate at 6 weeks following loading (p = 0.025). Statistical test: linear mixed effects model. Box plot indicates median and interquartile range. Individual datum points (n = 7–8/group) are overlaid. Δ indicates difference between loaded and control limb measures within a single animal. Abbreviations: ALN, alendronate; VEH, vehicle.
Inhibiting bone remodeling immediately after PTOA initiation reduced subchondral bone loss
A distinct loss of subchondral bone tissue from the medial tibial plateau was observed histologically in many loaded limbs at both time points post‐loading. Inhibiting bone remodeling immediately post‐loading decreased the relative number of limbs with medial subchondral plate erosions compared to VEH (Figure 3A), based on the total bone volume in the medial subchondral plate. Despite the presence of subchondral plate erosions in a portion of limbs at 3 weeks post‐loading, medial subchondral plate bone volume was not significantly reduced regardless of treatment. At 6 weeks post‐loading, inhibiting bone remodeling immediately prevented loss of medial subchondral plate bone volume (p = 0.025, Figure 3D). In contrast, subchondral plate bone volume decreased at 6 weeks following loading in the VEH and delayed ALN groups.
When examining subchondral bone morphology across the medial and lateral tibia, subchondral plate thickness decreased following the single bout of loading in all treatment groups at both 3 weeks (p < 0.001) and 6 weeks post‐loading (p = 0.005, Figure 4A). At the 6‐week time point, ALN treatment, regardless of start time, systemically increased subchondral plate thickness across both the loaded and contralateral control limbs (p = 0.012, Figure S2A), but did not alter the loss of subchondral tissue following loading (p = 0.187, Figure 4A). Similarly, subchondral plate TMD decreased following loading at both 3 weeks (p < 0.001) and 6 weeks (p < 0.001, Figure 4B). Subchondral TMD also was systemically increased by ALN treatment at 6 weeks (p = 0.002, Figure 2SB), but the loss of TMD following loading was not attenuated by ALN treatment (p = 0.259, Figure 4B).
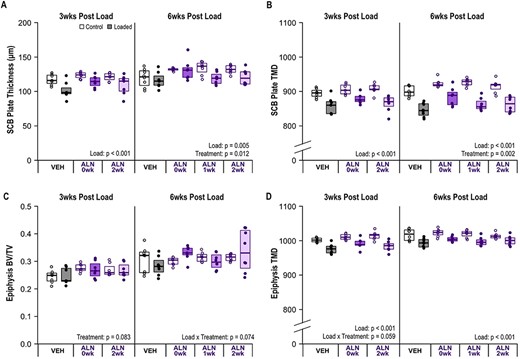
ALN treatment caused a systemic increase in subchondral plate thickness (p = 0.012) and TMD at 6 weeks (p = 0.002). (A) Plate thickness decreased in loaded limbs at 3 weeks (p < 0.001) and 6 weeks (p = 0.005) following a single bout of loading. (B) Subchondral plate TMD decreased in loaded limbs at 3 weeks (p < 0.001) and 6 weeks (p < 0.001). (C) Epiphysis BV/TV was not altered with loading at either time point. (D) Epiphysis TMD decreased with loading (p < 0.001). Statistical test: linear mixed‐effects model. Box plot indicate median and interquartile range. Individual datum points (n = 7–8/group) are overlaid. Abbreviations: ALN, alendronate; BV/TV, bone volume fraction; TMD, tissue mineral density; VEH, vehicle.
In the cancellous epiphysis, at the 3‐week time point BV/TV was not altered by loading (p = 0.694, Figure 4C) but was systemically increased by immediate ALN treatment (p = 0.029, Figure S2C). At 6 weeks post‐loading, BV/TV was not altered by loading (p = 0.657, Figure 4C) or ALN treatment (p = 0.223, Figure S2C). TMD in the epiphysis was decreased with loading at both 3 weeks (p < 0.001) and 6 weeks post‐loading (p < 0.001, Figure 4D), but was not altered by ALN treatment at either time point (p = 0.066 at 3 weeks, p = 0.382 at 6 weeks, Figure S2D).
Overall, inhibiting bone remodeling with ALN treatment immediately following loading most effectively attenuated erosions of the subchondral bone at 6 weeks following a single bout of mechanical loading. Both loading and ALN treatment, regardless of start time, had a limited effect on cancellous subchondral bone morphology.
Inhibiting bone remodeling immediately after PTOA initiation decreased MCL and meniscus calcification
Calcifications of the MCL and meniscus were observed histologically in 21 of 31 loaded limbs at 6 weeks after a single bout of loading (Figure 5). Inhibiting bone remodeling altered the relative number of limbs in which soft tissue calcifications were present (p = 0.010. Figure 5), but no paired treatment comparisons were significant. At 3 weeks calcifications of the MCL and meniscus were only observed in two limbs following loading, both in the VEH group (data not shown). Eighteen of 21 limbs had increased proteoglycan staining and signs of chondrogenesis in the meniscus and MCL at 3 weeks.
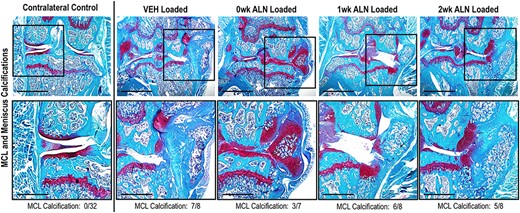
ALN treatment immediately after loading reduced the calcification of the meniscus and MCL at 6 weeks following loading. Representative images (4×, scale bar = 1 mm) of Safranin‐O–stained limbs of different treatment groups at 6 weeks after loading. Squares indicate areas selected for higher magnification photomicrographs in the lower panels (10×, scale bar = 500 μm). The number of calcified MCLs is indicated below each treatment group. The relative number of limbs with calcifications present was altered with inhibition of bone remodeling after loading (p = 0.010, Fischer's exact test), but no paired treatment comparisons were significant. Abbreviations: ALN, alendronate; MCL, medial collateral ligament; VEH, vehicle.
ALN treatment effectively inhibited bone remodeling
The presence of osteoclasts, as examined through TRAP staining, was decreased by ALN treatment at both 3 weeks (p = 0.004) and 6 weeks (p < 0.001, Figure 6A,B). In the 3‐week group, ALN decreased osteoclasts similarly regardless of start time. After 6 weeks, the immediate ALN‐treatment group had the largest decrease, the 1‐week‐ALN‐delay group the next largest, and the 2‐week‐ALN‐delay group had the smallest decrease relative to the VEH‐treated group. Osteoclast presence was decreased with loading at 6 weeks (p = 0.020), but ALN treatment had no further effect on load‐induced changes (p = 0.167). Presence of active osteoblasts, examined through procollagen‐I staining, was decreased with ALN treatment regardless of treatment start time at both 3 weeks (p < 0.001) and 6 weeks (p < 0.001, Figure 6A,C). Loading did not alter the presence of active osteoblasts at 3 weeks (p = 0.595) or 6 weeks (p = 0.099).
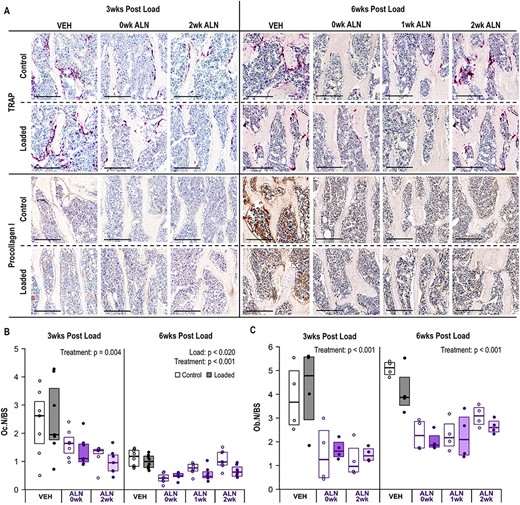
ALN treatment reduced bone remodeling. (A) Representative photomicrographs of TRAP (osteoclasts) and anti‐procollagen I (active osteoblasts) stained bone tissues in the metaphysis at 3 and 6 weeks following a single bout of loading. Scale bar for TRAP and Procollagen I = 200 μm. (B) ALN treatment decreased presence of osteoclasts at both 3 weeks (p = 0.004) and 6 weeks (p < 0.001). Loading also decreased osteoclasts at 6 weeks (p = 0.020). (C) ALN treatment decreased presence of active osteoblasts. Statistical test: linear mixed‐effects model. Box plot indicates median and interquartile range. Individual datum points (n = 7–8/group for TRAP, n = 4/group for Procollagen I) are overlaid. Abbreviations: ALN, alendronate; TRAP, tartrate‐resistant acid phosphatase; VEH, vehicle.
DISCUSSION
PTOA is associated with abnormal subchondral bone remodeling, and early‐stage increases in bone resorption may be a driving factor in PTOA development.(20) In the present study, we investigated the 2 weeks immediately following PTOA initiation as a treatment window for inhibiting subchondral bone remodeling to prevent joint damage progression. Inhibiting subchondral bone remodeling resulted in a similar attenuation of cartilage damage at 6 weeks following PTOA initiation regardless of when ALN treatment was started, but immediate inhibition of bone remodeling most effectively inhibited the development of OA‐associated features in bone and other tissues.
Inhibiting bone remodeling after PTOA initiation attenuated the severity of load‐induced cartilage damage. At 3 weeks post‐loading immediate inhibition of bone remodeling was more effective than a 2‐week delay, but inhibiting bone remodeling regardless of start time was similarly effective at 6 weeks post‐loading. Based on the relative degree of cartilage damage at 3 weeks compared to 6 weeks, the majority of structural cartilage damage occurred within the first 3 weeks following loading. Both the VEH and delayed ALN groups had similar severity of cartilage damage at both times examined; only the severity of the immediate ALN group increased from 3 to 6 weeks. Similar results are reported with other preclinical traumatic joint injury models in which ALN treatment immediately after injury also attenuated the progression of cartilage damage.(27,29‐31) However, these prior studies only examined treatment applied immediately after joint injury, whereas we demonstrated similar results when starting treatment up to 2 weeks after PTOA initiation, suggesting a wider treatment window for attenuating cartilage damage through inhibiting bone remodeling.
In contrast, inhibiting bone remodeling immediately after PTOA initiation most effectively inhibited osteophyte formation, subchondral bone loss, and soft tissue calcifications. Previous work has similarly showed that ALN treatment immediately after traumatic joint injury can alter periarticular bone formation.(27,28,40,43) Osteophyte formation occurs through the development of a cartilaginous outgrowth at the periphery of the joint that calcifies over time through endochondral ossification and grow through localized bone formation.(44) In the present study, both osteoclast and osteoblast presence were decreased by ALN treatment indicating an inhibition of bone remodeling, which would alter endochondral ossification and the initial calcification of osteophytes, in addition to slowing calcified osteophyte growth. Previously immediate ALN treatment directly inhibited secondary remodeling and the calcification of osteophytes, resulting in smaller, less mature osteophytes.(28) Osteophyte formation has been observed as early 2 to 3 days following PTOA initiation,(45,46) and we have previously demonstrated osteophyte formation at both 1 and 2 weeks following a single bout of loading.(37) Given osteophyte formation begins so quickly after disease initiation, immediate inhibition of remodeling is most effective in slowing osteophyte development.
The relative presence and maturity of calcifications in the meniscus and MCL were also decreased by immediate ALN treatment. Historically, meniscal and ligament pathology have not been well described in preclinical OA models. More recently, however, other studies have reported similar calcifications and chondrogenesis in these soft tissues.(47,48) Thus, pathological changes in the meniscus and ligaments may be important hallmarks of PTOA development and should be better characterized in models moving forward. In our study, inhibiting bone remodeling immediately after PTOA initiation attenuated degenerative changes in the meniscus and MCL, suggesting that these degenerative changes initiate early after PTOA development and that the remodeling process is implicated in the ensuing calcification. Heterotopic calcification of the meniscus and ligaments likely occurs through endochondral ossification similar to osteophyte formation.(48) ALN treatment likely inhibits this process through a similar mechanism to inhibiting osteophyte formation, as discussed previously. In addition to decreasing the relative number of joints with calcifications of the meniscus and MCL, mice receiving immediate ALN treatment also exhibited less complete calcifications and the presence of more cartilaginous tissue remaining within the meniscus or MCL of loaded limbs (Figure 5).
Immediate ALN treatment also reduced subchondral plate erosions and prevented loss of medial subchondral plate volume, but did not attenuate subchondral plate thinning occurring with PTOA development. In previous studies, PTOA development secondary to traumatic joint injury was associated with subchondral bone loss,(22) and immediately inhibiting bone remodeling prevented the loss of subchondral bone volume and subchondral plate thinning.(31) In addition to directly inhibiting subchondral bone resorption in the medial subchondral plate, immediate ALN treatment may also have helped reduced the presence of subchondral plate erosions by slowing the development of ligament calcification, thereby helping maintain a greater joint range of motion and prevent a localized stress concentration from forming on the medial articular surface. In contrast, in our study inhibiting subchondral bone remodeling did not attenuate the relative reduction in subchondral plate thickness in loaded limbs relative to control limbs regardless of when ALN treatment started. Inhibiting remodeling did cause a systemic increase in both subchondral plate thickness and volume of both control limbs over time. The balance between increased resorption during early stage OA development and inhibited resorption with ALN treatment may have kept subchondral plate thickness relatively consistent in ALN‐treated limbs following loading. On the other hand, in ALN‐treated control limbs, in which normal levels of remodeling were present, inhibiting resorption led to increased subchondral plate thickness over time. Overall, inhibiting bone remodeling immediately after PTOA initiation slowed the progression of OA‐like changes in subchondral bone.
Both osteoclast and osteoblast presence were decreased by ALN treatment at 3 and 6 weeks following loading, as expected. In addition, osteoclast presence was lower in OA limbs compared to control limbs at 6 weeks. Abnormal bone remodeling follows a distinct pattern following OA initiation and throughout OA development. Initiation and early‐stage OA development are associated with increased bone resorption resulting from increased osteoclast activity, whereas later‐stage OA is associated with increased bone formation resulting from a relative increase in osteoblast activity and decreased osteoclast activity.(20) Given the severe damage present across the joint at both end time points, our results in loaded limbs likely represent osteoclast presence during late‐stage OA development. Therefore, osteoclast presence would be expected to be relatively lower compared to the high levels of resorption present in early stages of OA development or even normal levels of bone resorption observed in control limbs, consistent with our 6‐week findings.
At both 3 and 6 weeks following the single bout of loading, a loss of joint structure and collapse of the medial tibial plateau were present in a subset of limbs, similar to joint damage observed in anterior cruciate ligament (ACL) rupture models.(49,50) Previously we showed that the ACL was not directly damaged by a single bout of cyclic loading and no structural damage to cartilage, bone, ligaments, or other joint tissues was present at 1 h following loading.(37) Given the repeatable nature of the cyclic loading model and work examining ACL damage in various loading configurations,(51) we are confident that the ACL was not ruptured by loading in the present study. However, a single bout of loading could have induced a subcritical level of damage to the ACL and other tissues that initiated further joint degradation. In addition, no major disruptions of ACL integrity were visible at 3 weeks following loading when significant OA‐like pathology was already present in the joint (Figure S3), suggesting that structural ACL damage is not the primary driver of OA development. However, we did observe the presence of chondrocyte‐like cells in the ACL developing at 3 and 6 weeks post‐loading that was attenuated by ALN treatment, suggesting chondrogenesis developing within the ligament. Comparable ACL pathology has been reported in the destabilization of the medial meniscus (DMM) model(48) and could indicate a progression toward ligament calcification similar to what we observed at 6 weeks in the MCL. For these reasons, we do not believe that the joint damage following the single bout of loading was driven by ACL rupture and associated knee instability; rather, we hypothesize that the ACL changes present post‐loading are part of the whole‐joint pathology and degeneration.
Counterintuitively, joint damage developed at 6 weeks following a single load was more severe than joint damage observed following 6 weeks of daily repetitive loading at the same load magnitude, a model we have used previously to replicate OA development.(39,40,52) Neither subchondral plate erosions nor MCL and meniscus calcifications were observed in our daily loading model. Cartilage damage on the medial condyle was also more severe at 6 weeks following a single load, although lateral cartilage damage was similar between loading regimens. Several factors may contribute to increased damage severity following a single load compared to daily loading, in particular reduced joint range of motion over time in the single‐load model with the development of ligament calcifications that do not form with daily loading, perhaps due to daily loading maintaining range of motion within the joint. The roles of daily cage activity, repeated exposure to anesthesia, daily injections, and potential protective effects of daily loading(53) in relation to OA development and progression need to be investigated further to better understand the difference between these two load‐induced preclinical OA models.
The present study has several limitations. We did not assess the direct effects of bisphosphonate treatment on cartilage metabolism, although previous studies indicated ALN treatment may alter chondrocyte function directly.(54,55) We only studied time points within 6 weeks of PTOA initiation. Considering later time points could provide more information regarding how effectively inhibiting bone remodeling inhibits longer‐term PTOA progression. In addition, we were unable to examine protein or gene expression in cartilage due to the severe loss of cartilage at both 3 and 6 weeks, particularly within the VEH‐treated group. Examining earlier time points within hours and days of loading could identify cellular and molecular changes associated with PTOA development and ALN treatment. Future work should investigate the early changes in biomolecular signaling to better understand the mechanisms through which inhibiting subchondral bone remodeling immediately after PTOA initiation slows the development of both cartilage and bone pathology. In addition, ALN treatment may alter the inflammatory environment of the joint, which could also directly influence PTOA development.(56) Localized inflammation is also hypothesized to play a role in the initiation of osteophyte formation and heterotopic bone formation,(57) and may be altered by ALN treatment. In the present study, we observed a visual decrease in synovial thickening and joint capsule fibrosis in limbs receiving ALN treatment (Figure S4). The interaction between joint inflammation resulting from PTOA development and ALN should be further investigated. Overall, determining the direct effects of ALN on different joint tissues would allow for a better understanding of the role of inhibiting bone remodeling on PTOA progression.
In conclusion, our findings suggest a potential role of ALN treatment as a prophylactic treatment following traumatic joint injury to slow future PTOA development. The window in which treatment would need to be started to effectively inhibit early‐stage bone changes associated with PTOA must be determined prior to clinical application. Future work should examine an earlier treatment window on the scale of hours to days to better understand how early‐stage changes in bone remodeling lead to the development of OA‐related pathology in subchondral bone. The duration of treatment required to attenuate OA development should also be investigated. In terms of clinical application, long‐term daily treatment may not be an option for preventative treatment following joint injury, especially within younger patients, due to negative effects of long‐duration bisphosphonate usage.(58) A short‐term course of treatment following injury to attenuate or prevent long‐term PTOA progression may be feasible, and should be explored.
ACKNOWLEDGMENTS
Funding for this study was provided by DOD PRMRP W81XWH‐17‐1‐0540, NIH T32‐AR007281, and the Clark Foundation. The sponsors had no role in the writing of the manuscript or in the decision to submit the manuscript for publication. We thank Drs. Steven and Mary Goldring, Dr. Tania Pannellini, Anna Ashford, Emma Briggs, Allison Brown, Ali Corkran, Erin Hudson, Ariana Otto, Lyudamila Lukashova, Cornell Statistical Consulting Unit, and the Cornell CARE staff.
DISCLOSURES
Marjolein van der Meulen has grants/grants pending from NIH, NSF, and DOD, is an associate editor for the Journal of Bone and Mineral Research, and is the Vice President of the Orthopaedic Research Society. All other authors declare that they have no conflicts of interest and nothing to disclose.
AUTHOR CONTRIBUTIONS
Conception and design of the study: Sophia N. Ziemian, Ana Witkowski, Timothy M. Wright, Miguel Otero, Marjolein C. H. van der Meulen. Acquisition of data: Sophia N. Ziemian, Ana Witkowski. Analysis and interpretation of data: Sophia N. Ziemian, Ana Witkowski, Timothy M. Wright, Miguel Otero, Marjolein C. H. van der Meulen. Drafting of the article: Sophia N. Ziemian, Ana Witkowski, Marjolein C. H. van der Meulen. Critical revision of the article for important intellectual content: Sophia N. Ziemian, Ana Witkowski, Timothy M. Wright, Miguel Otero, Marjolein C. H. van der Meulen. Final approval of the article: Sophia N. Ziemian, Ana Witkowski, Timothy M. Wright, Miguel Otero, Marjolein C. H. van der Meulen. Sophia N. Ziemian and Marjolein C. H. van der Meulen take responsibility for the integrity of the work as a whole, from inception to finished article.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4397.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}