





ABSTRACT
Osteosclerotic metaphyseal dysplasia (OSMD) is a rare autosomal recessive sclerosing skeletal dysplasia. We report on a 34‐year‐old patient with sandwich vertebrae, platyspondyly, osteosclerosis of the tubular bones, pathologic fractures, and anemia. In the third decade, he developed osteonecrosis of the jaws, which was progressive in spite of repeated surgical treatment over a period of 11 years. An iliac crest bone biopsy revealed the presence of hypermineralized cartilage remnants, large multinucleated osteoclasts with abnormal morphology, and inadequate bone resorption typical for osteoclast‐rich osteopetrosis. After exclusion of mutations in TCIRG1 and CLCN7 we performed trio‐based exome sequencing. The novel homozygous splice‐site mutation c.261G>A in the gene LRRK1 was found and co‐segregated with the phenotype in the family. cDNA sequencing showed nearly complete skipping of exon 3 leading to a frameshift (p.Ala34Profs*33). Osteoclasts differentiated from the patient's peripheral blood monocytes were extremely large. Instead of resorption pits these cells were only capable of superficial erosion. Phosphorylation of L‐plastin at position Ser5 was strongly reduced in patient‐derived osteoclasts showing a loss of function of the mutated LRRK1 kinase protein. Our analysis indicates a strong overlap of LRRK1‐related OSMD with other forms of intermediate osteopetrosis, but an exceptional abnormality of osteoclast resorption. Like in other osteoclast pathologies an increased risk for progressive osteonecrosis of the jaws should be considered in OSMD, an intermediate form of osteopetrosis. © 2020 The Authors. Journal of Bone and Mineral Research published by American Society for Bone and Mineral Research.
Introduction
Osteosclerotic metaphyseal dysplasia (OSMD; OMIM #615198) is a rare sclerosing skeletal dysplasia first described by Nishimura and Kozlowski in 1993.1 It is characterized by osteosclerosis of the long bones (predominantly at the metaphyses) and vertebrae, ribs, clavicles, and iliac crest. Developmental delay, hypodontia, and seizures were also reported to be part of the clinical picture.1 It is a very rare autosomal‐recessive disease with a prevalence of <1:1,000,000, caused by mutations in the leucine‐rich repeat kinase 1 (LRRK1) gene. To date, four families with five affected individuals carrying biallelic LRRK1 mutations have been reported.2, 3, 4 LRRK1 is a protein of the ROCO family composed of four ankyrin repeat (ANK) domains, seven leucine‐rich (LRR) domains, a Ras of complex proteins (Roc) guanosine triphosphatase (GTPase) domain, a C‐terminal of Roc (COR) domain, a serine/threonine kinase‐domain, and WD40 domain.5, 6 In murine osteoclasts it was shown to play a role in bone resorption by regulating the formation of the sealing zone.7 Lrrk1 associates with C‐terminal Src‐kinase (Csk) and phosphorylates the cellular Rous sarcoma oncogene c‐Src as well as the actin regulating proteins L‐plastin and Cdc42.8, 9
Osteonecrosis of the jaw (ONJ) was described by Edwards and colleagues in 2002 as a side effect of treatment with anti‐resorptive drugs such as intravenous bisphosphonates.10 Risk factors for medication‐related osteonecrosis of the jaws (MRONJ) include diabetes, malignancies, oral surgical procedures, and poor oral hygiene,11, 12 ONJ has also be observed in patients with osteopetrosis.13, 14
Here, we report on a patient with severe ONJ and clinical, radiological, and histological signs of osteoclast‐rich osteopetrosis caused by a LRRK1 splice site mutation. We compare this case with other patients of LRRK1‐related OSMD and describe the effect of the loss of the LRRK1 kinase on the function of human osteoclasts.
Patients and Methods
Biopsy studies
An iliac crest bone biopsy was obtained to further characterize the detailed skeletal characteristics. The specimen was fixed in 3.7% formaldehyde, dehydrated, embedded in methyl methacrylate, and cut on a microTec rotation microtome (CVT 4060E; microTec, Walldorf, Germany). Five‐micrometer (5‐μm) sections were stained by toluidine blue, trichrome Masson‐Goldner, and von Kossa. Histomorphometric analysis was performed according to the ASBMR standards15 and compared to reference values.16
Additionally, the bone mineralization density distribution (BMDD) in the trabecular bone was analyzed by quantitative backscattered electron imaging (qBEI; LEO 435 VP; LEO Electron Microscopy Ltd., Cambridge, UK) as described.17 Six images per specimen were taken (magnification ×100) and evaluated using a custom MATLAB‐based program (TheMathWorks, Inc., Natick, MA, USA). Histomorphometry and BMDD values were furthermore compared to previously published patients with CLCN7‐related autosomal dominant osteopetrosis type II16 as well as to reference values from the literature.18
Next generation sequencing
Before next generation sequencing was initiated, mutations in TCIRG1 and CLCN7 were excluded by Sanger sequencing. For the patient, the two siblings and his parents, libraries for trio‐based exome sequencing (WES) were prepared using NEBNext DNA Library Prep Master Mix Set for Illumina (New England BioLabs, Ipswich, MA, USA). Enrichment of the target regions was done using the SureSelect Human Exome Kit V6 (Agilent Technologies, Santa Clara, CA, USA) and run on a HiSeq 2500 Sequencer (Illumina, San Diego, CA, USA) with a 150‐bp paired‐end sequence length. The sequences were aligned to the human genome (GRCh37) with Burrows‐Wheeler Alignment tool (BWA)19 and the variants were detected with the Genome Analysis Toolkit (GATK), version 2.6 (https://gatk.broadinstitute.org/hc/en-us).20 The exome genotyping accuracy was estimated to be above 0.9999 based on the variant calls.21 Variants were detected with SAMtools (http://www.htslib.org/), annotated with ANNOVAR (http://annovar.openbioinformatics.org/en/latest/).22 All exons with pathogenic variants in ClinVar (NIH, NCBI, Bethesda, MD, USA; https://www.ncbi.nlm.nih.gov/clinvar/) were covered at a minimum of 20 reads. Ninety‐five percent (95%) of the remnant exome target region was also covered by more than 20 reads.
Bioinformatic filtering of variants
The variants were analyzed by the Gene‐Talk platform23 and the pathogenic potential of individual candidate variants was evaluated by MutationTaster (http://www.mutationtaster.org/).24 For further analysis, we only analyzed homozygous variants that were not represented by more than three individuals in the 1000 Genomes Project (https://www.internationalgenome.org/) cohort, Genome Aggregation Database (gnomAd; https://gnomad.broadinstitute.org/) or Exome Aggregation Consortium (ExAC25) and which were not annotated as benign polymorphisms by MutationTaster.
PCR and Sanger sequencing
The respective exons and exon–intron boundaries of LRRK1 (NM_024652) were amplified using oligonucleotide primers (sequences available upon request) and sequenced on an ABI3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
Culturing of patient mesenchymal stem cells and osteoblasts
Primary human osteoblasts and mesenchymal stem cells (MSCs) were received from the Core‐Facility “Tissue Harvesting” of the Berlin Institute of Health (BIH) Center for Regenerative Therapies (BCRT). Written informed consent was given, and ethics approval was obtained from the local ethics committee/institutional review board (IRB) of the Charité University Hospital. Cells were isolated from metaphyseal bone marrow (BM) or cancellous bone biopsies, respectively, from patients undergoing hip replacement at Charité University Hospital as described.26, 27 Briefly, MSCs were isolated by density gradient centrifugation using Histopaque‐1077 (Sigma‐Aldrich, St. Louis, MO, USA) and subsequent plastic adherence. Cells were cultured and expanded in Dulbecco's modified Eagle's medium (DMEM Low Glucose; Gibco, Grand Island, NY, USA) with 10% fetal calf serum (Biochrom, Berlin, Germany), 5mM L‐alanyl‐l‐glutamine (Gibco, Grand Island, NY, USA), 100 U/mL penicillin plus 100 μg/mL streptomycin (Biochrom, Berlin, Germany). MSC phenotype and in vitro differentiation potential was confirmed according the minimal criteria for defining multipotent mesenchymal stem cells of the International Society for Cellular Therapy.28 To isolate human osteoblasts, cancellous bone specimens from the same patients were extensively washed in PBS and subsequently cut into small pieces with a scalpel. The pieces were transferred into a culture flask and expanded in the same medium used for MSCs for 3 to 4 weeks until reaching approximately 80% confluency. Osteogenic potential of the isolated cells were validated using Alizarin staining and alkaline phosphatase (ALP) activity.29
In vitro osteoclastogenesis
To generate human osteoclasts, peripheral blood was collected and peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation and later differentiated into active osteoclasts by growth factors. The experiment was done with two independent 30‐mL heparinized blood samples from the patient and a healthy donor. The blood sample was split into two parts and each half was diluted by PBS in a 1:2 solution (15 mL peripheral blood + 15 mL PBS). In each 50‐mL Falcon tube, 15 mL Biocoll Separating Solution (1077 g/mL; Biochrom, Berlin, Germany) were transferred as the bottom layer and carefully topped with 30 mL blood solution (1:2 dilution with PBS) without mixing the layers. The tubes were centrifuged for 30 min at 400g without break at room temperature (RT). The supernatant was removed and the interphase (PBMCs) was harvested and collected in a new Falcon tube. A maximum of 10 mL PBMCs were topped with 40 mL PBS and centrifuged for another 10 min at 300g. The supernatant was removed and the PBMC pellet resuspended in 5 mL PBS. The PBMC suspension of each Falcon tube was pooled and filled up to 50 mL. The cell number was determined and the PBMCs were seeded out at density of 3 × 105 cells per 24‐well plate. The cells were resuspended in medium composed of Alpha‐MEM (Gibco, Grand Island, NY, USA) + 10% FCS (Lonza, Basel, Switzerland) + 1% Ultraglutamine (Gibco, Grand Island, NY, USA), 1% Penicillin/Streptomycin (Gibco, Grand Island, NY, USA) + 15 ng/mL recombinant human macrophage colony‐stimulating factor (rhM‐CSF) (R&D Systems, Minneapolis, MN, USA; #216‐MCC). The medium was changed every 2 to 3 days. In a 24‐well plate, 700 μL was removed and replaced with fresh medium. At day 3, recombinant human RANKL (rhRANKL) (PeproTech, Rocky Hill, NJ, USA; #310‐01) was first added to the medium at a concentration of 50 ng/mL. At day 5, the first osteoclasts were detected. At day 14, osteoclast staining was performed.
Osteoclast staining
In vitro differentiated osteoclasts were fixed at day 14 with 4% paraformaldehyde (PFA) and permeabilized with 0.1% saponin. Osteoclasts were stained for 15 min at RT for tartrate resistant acid phosphatase (TRAP) activity using 0.1 mg/mL Napthol AS‐MX phosphate disodium salt (Sigma‐Aldrich, Taufkirchen, Germany) and 0.6 mg/mL Fast Red Violet LB Salt (Sigma‐Aldrich, Taufkirchen, Germany) in 40mM sodium acetate and 10mM sodium tartrate. Actin in osteoclasts was stained overnight at 4°C with fluorescently‐labeled phalloidin (Alexa Fluor 488; Life Technologies, Inc., Grand Island, NY, USA; 1:400) and 4′,6‐diamidino‐2‐phenylindole (DAPI) (Life Technologies, Darmstadt, Germany) was used to stain nuclei (15 min RT, 1 μg/mL). Fluorescence microscopy of stained osteoclasts was performed with an Olympus BX60 microscope (Olympus, Waltham, MA, USA). Two independent experiments were performed, each with three coverslips.
Resorption assay
At day 6 of osteoclast differentiation, osteoclasts were lifted by incubating the culture with StemPro Accutase (Thermo Fisher Scientific, Waltham, MA, USA) for 10 min at 37°C and then detached with a cell scraper. Osteoclasts were seeded in Alpha‐MEM with 10% FBS Superior, 1% Penicillin/Streptomycin, 1% Ultraglutamine, 50 ng/mL rhM‐CSF and rhRANKL at a density of 6.7 × 104/dentine slice in 96‐well plates and osteoclast culture on dentine was continued for 5 days. Half of the culture medium was replaced by fresh medium 2 days after osteoclast transfer. At culture termination, the cells were removed from the dentine slices and resorption events were visualized with 0.25% toluidine blue (Sigma‐Aldrich). Three independent experiments were performed, each including three dentine slices. For 3D visualization of resorption traces cleaned dentine discs were labeled by incubation with 1% WGA‐Texas Red (Life Technologies) in 1% BSA/PBS and imaged using a LSM 700 confocal microscope (Zeiss, Inc., Thornwood, NY, USA) as described.30
RNA isolation
Trizol‐based (Life Technologies) RNA isolation of human MSCs (hMSCs), human osteoblasts (hOBs), and human osteoclasts (hOCs) was performed with the Direct‐zol RNA kit (Zymo Research, Irvine, CA, USA). Two 25‐cm2 wells were lysed for each cell identity. RNA concentration and quality was measured by the NanoDrop system (Thermo Fisher Scientific).
cDNA sequencing
RNA was reverse transcribed into cDNA with the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Hennigsdorf, Germany) and LRRK1 was sequenced with oligonucleotide primers (sequences available upon request) and sequenced on an ABI3730xl DNA Analyzer (Applied Biosystems, Dreieich, Germany).
Immunoblot analyses
Cultured osteoblasts and MSCs were lysed. Cell lysates (20 μg) were transferred to a NuPAGE 3‐8% Tris‐Acetate protein gel (Invitrogen, Carlsbad, CA, USA). The Phospho‐Ser5‐L‐plastin antibody (Signalway Antibody, College Park, MD, USA; #12455), the L‐plastin Antibody (Abcam, Cambridge, MA, USA; #ab206322), the β‐Actin Antibody (13E5) (Cell Signaling Technology, Beverly, MA, USA; #4970) and Cathepsin K antibodies (Abcam, Cambridge, MA, USA ;#ab49893) were used for protein detection. Lysates from two independent osteoclast cultures were analyzed. Although differences in L‐plastin phosphorylation are evident, no significance levels were calculated because no blood sample for a third independent experiment was obtained.
Ethics
This study has been approved by the local ethics committee (EA2/010/19).
Results
Case report
The male patient from a Bulgarian family without known consanguinity first presented in hospital at the age of 23 years with circumscribed necrosis in the premolar region of the right mandible and molar region of the left maxilla (Fig. 1A–D; Supporting Information Fig. S1), causing infraorbital fistulas (Fig. 1E). Two years prior to presentation, the molars in the left mandible had been removed due to chronic inflammation and pain. Otherwise, his childhood and adolescence were uneventful. Also, the parents and his two siblings have no history of any bone disease or fractures.
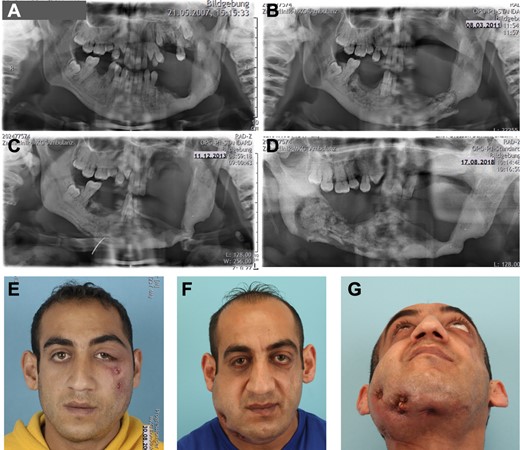
Craniofacial phenotype. (A–D) Panoramic X‐ray of the jaws. (A) Status post teeth removal in left mandible and sclerosing bone transformation. (B) After 4 years, loss of teeth in left upper jaw and mandible, pathologic fracture of the left mandible. (C) Spontaneous bone healing within two years, after intermittent sequester removal. (D). Five years later, progressive bone necrosis and sequester formation of right mandible resulting in loss of all teeth in lower mandible. (E) Initial presentation at 23 years of age with infraorbital fistulas due to necrotic maxillary bone. (F,G) Presentation at 34 years of age with perimandibular and submandibular fistulas caused by multiple sequestra of the right mandible.
The patient's clinical management was performed as follows: the necrotic jaw bone was conservatively removed without jeopardizing the integrity and stability of the mandible, as it is frequently practiced in MRONJ. Despite this conservative treatment regimen, an atraumatic fracture of the left mandible occurred (Fig. 1B), which healed up within 2 years after intermittent sequester removal (Fig. 1C). Osteosynthesis of the mandibular fracture was deferred due to poor bone quality at the fracture site and limited soft tissue coverage. Between ages 32 and 34 years, the patient developed disintegration and sequester formation on the right side of his mandible. In the context of conservative sequestrectomy, also the remnant teeth of his mandible were removed (Fig. 1D). ONJ led to soft tissue infection and fistula formation, initially in the zygomatic prominence (Fig. 1E) and years later in the submandibular region (Fig. 1F,G). Surgical drainage and antibiotics enabled the local control of the necrotic and infected bone.
Although the course of the osteonecrosis was not unusual for MRONJ there was no history of any medication explaining the pathology. Instead, the patient presented with additional skeletal findings. Kyphoscoliosis (Fig. 2A,I) and short stature were noted and radiographs of the spine revealed osteosclerotic sandwich vertebrae with endplate deformities and mild flattening (Fig. 2A–D). The diaphyseal cortical bone of the right femur appeared thinned, whereas (Fig. 2E) the cortical bone of the left femur was thickened (Fig. 2F). The differences in cortical thickness could be due to unloading because there was a history of several pathologic fractures (Fig. 2E,F). At the age of 25 years, he suffered from a right‐sided proximal femur shaft fracture and a pelvic fracture without adequate trauma (Fig. 2E), and sustained another right‐sided femur shaft fracture at the age of 31 years. These fractures were surgically treated (Fig. 2E,G) and healing was uneventful. The femurs showed modeling defects with metaphyseal widening (Fig. 2H). The radiological picture was compatible with the diagnoses of autosomal dominant osteopetrosis type 2 (ADOII) or dysosteosclerosis. His intellectual development is in the normal range. The phenotype is summarized in Table 1 and compared to published patients with LRRK1 mutations for which sufficient clinical details were available.
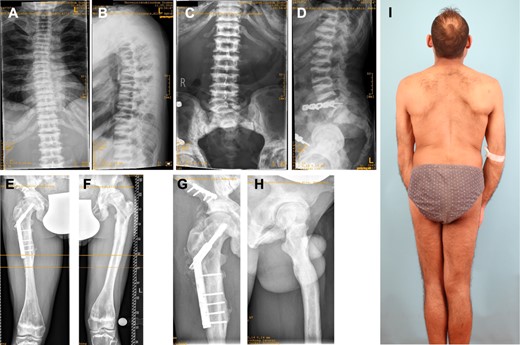
Osteopetrotic changes in the axial and appendicular skeleton. (A) Scoliosis of cervical spine with irregular corticospongious appearance of the ribs. (B) Kyphosis, sandwich appearance of vertebrae and platyspondyly. (C,D) Lumbar spine and sacroiliac joint revealing sclerotic sandwich vertebrae. (E) Osteosynthetic material after right‐sided pathologic femur shaft fracture. (F) Diaphyseal thickening and metaphyseal modeling defect of femur. (G) Osteosynthesis after pathologic pelvis fracture and (H) a modeling defect of proximal femur. (I) Full‐body picture of patient's back shows kyphoscoliosis and leg asymmetry.
Clinical Presentation and LRRK1 Mutations Found in OSMD
| Characteristic | This report | Iida and colleagues3 | Guo and colleagues2 | |
| Gender | Male | Male | Male | Female |
| Age today | 34 years | 2 years | 12–14 years | 25 years |
| Ethnicity | Bulgarian | Moroccan | Indian | Indian |
| Consanguinity | No | Yes | No | No |
| Affected siblings | No | ? | Yes | Yes |
| Gene | LRRK1 | LRRK1 | LRRK1 | |
| Mutation | c.261G>A; p.(Ala34Profs*33) | c.5938_5944delGAGTGGT (prolonged protein +29 AA) | c.5917_5972insG; p.(Glu1980Alafs*66) | |
| Zygosity | Homozygous | Homozygous | Homozygous | |
| Clinical manifestations (HPO terms) | ||||
| Pathologic fractures (HP:0002756) | + | + | + | + |
| Coxa vara (HP:0002812) | + | n.d. | + | n.d. |
| Short stature (HP:0004322) | + | + | − | − |
| Failure to thrive (HP:0001508) | − | + | − | − |
| Mild global developmental delay (HP:0011342) | − | + | − | − |
| Anemia (HP:0001903) | + | n.d. | − | n.d. |
| Hepatomegaly (HP:0002240) | n.d. | − | + | n.d. |
| Facial dysmorphism | + | n.d. | + | − |
| Abnormality of the teeth (HP:0000164) | + | +, hypodontia | +, crowding of teeth | − |
| Recurrent infections (HP:0002719) | + | n.d. | − | − |
| Radiographic findings | ||||
| Sandwich appearance of vertebral bodies (HP:0004618) | + | + | + | + |
| Flattened vertebrae (HP:0000926) | + | − | − | − |
| Erlenmeyer flask deformity of the femurs (HP:0004975) | + | n.d. | + | n.d. |
| Dense metaphyseal bands (HP:0100959) | + | + | + | + |
| Thickened ribs (HP:0000900) | + | n.d. | + | n.d. |
| Osteosclerosis of the calvaria and base of the skull (HP:0005746) | n.d. | − | + | n.d. |
| Characteristic | This report | Iida and colleagues3 | Guo and colleagues2 | |
| Gender | Male | Male | Male | Female |
| Age today | 34 years | 2 years | 12–14 years | 25 years |
| Ethnicity | Bulgarian | Moroccan | Indian | Indian |
| Consanguinity | No | Yes | No | No |
| Affected siblings | No | ? | Yes | Yes |
| Gene | LRRK1 | LRRK1 | LRRK1 | |
| Mutation | c.261G>A; p.(Ala34Profs*33) | c.5938_5944delGAGTGGT (prolonged protein +29 AA) | c.5917_5972insG; p.(Glu1980Alafs*66) | |
| Zygosity | Homozygous | Homozygous | Homozygous | |
| Clinical manifestations (HPO terms) | ||||
| Pathologic fractures (HP:0002756) | + | + | + | + |
| Coxa vara (HP:0002812) | + | n.d. | + | n.d. |
| Short stature (HP:0004322) | + | + | − | − |
| Failure to thrive (HP:0001508) | − | + | − | − |
| Mild global developmental delay (HP:0011342) | − | + | − | − |
| Anemia (HP:0001903) | + | n.d. | − | n.d. |
| Hepatomegaly (HP:0002240) | n.d. | − | + | n.d. |
| Facial dysmorphism | + | n.d. | + | − |
| Abnormality of the teeth (HP:0000164) | + | +, hypodontia | +, crowding of teeth | − |
| Recurrent infections (HP:0002719) | + | n.d. | − | − |
| Radiographic findings | ||||
| Sandwich appearance of vertebral bodies (HP:0004618) | + | + | + | + |
| Flattened vertebrae (HP:0000926) | + | − | − | − |
| Erlenmeyer flask deformity of the femurs (HP:0004975) | + | n.d. | + | n.d. |
| Dense metaphyseal bands (HP:0100959) | + | + | + | + |
| Thickened ribs (HP:0000900) | + | n.d. | + | n.d. |
| Osteosclerosis of the calvaria and base of the skull (HP:0005746) | n.d. | − | + | n.d. |
HPO = Human Phenotype Ontology; n.d. = not determined.
Clinical Presentation and LRRK1 Mutations Found in OSMD
| Characteristic | This report | Iida and colleagues3 | Guo and colleagues2 | |
| Gender | Male | Male | Male | Female |
| Age today | 34 years | 2 years | 12–14 years | 25 years |
| Ethnicity | Bulgarian | Moroccan | Indian | Indian |
| Consanguinity | No | Yes | No | No |
| Affected siblings | No | ? | Yes | Yes |
| Gene | LRRK1 | LRRK1 | LRRK1 | |
| Mutation | c.261G>A; p.(Ala34Profs*33) | c.5938_5944delGAGTGGT (prolonged protein +29 AA) | c.5917_5972insG; p.(Glu1980Alafs*66) | |
| Zygosity | Homozygous | Homozygous | Homozygous | |
| Clinical manifestations (HPO terms) | ||||
| Pathologic fractures (HP:0002756) | + | + | + | + |
| Coxa vara (HP:0002812) | + | n.d. | + | n.d. |
| Short stature (HP:0004322) | + | + | − | − |
| Failure to thrive (HP:0001508) | − | + | − | − |
| Mild global developmental delay (HP:0011342) | − | + | − | − |
| Anemia (HP:0001903) | + | n.d. | − | n.d. |
| Hepatomegaly (HP:0002240) | n.d. | − | + | n.d. |
| Facial dysmorphism | + | n.d. | + | − |
| Abnormality of the teeth (HP:0000164) | + | +, hypodontia | +, crowding of teeth | − |
| Recurrent infections (HP:0002719) | + | n.d. | − | − |
| Radiographic findings | ||||
| Sandwich appearance of vertebral bodies (HP:0004618) | + | + | + | + |
| Flattened vertebrae (HP:0000926) | + | − | − | − |
| Erlenmeyer flask deformity of the femurs (HP:0004975) | + | n.d. | + | n.d. |
| Dense metaphyseal bands (HP:0100959) | + | + | + | + |
| Thickened ribs (HP:0000900) | + | n.d. | + | n.d. |
| Osteosclerosis of the calvaria and base of the skull (HP:0005746) | n.d. | − | + | n.d. |
| Characteristic | This report | Iida and colleagues3 | Guo and colleagues2 | |
| Gender | Male | Male | Male | Female |
| Age today | 34 years | 2 years | 12–14 years | 25 years |
| Ethnicity | Bulgarian | Moroccan | Indian | Indian |
| Consanguinity | No | Yes | No | No |
| Affected siblings | No | ? | Yes | Yes |
| Gene | LRRK1 | LRRK1 | LRRK1 | |
| Mutation | c.261G>A; p.(Ala34Profs*33) | c.5938_5944delGAGTGGT (prolonged protein +29 AA) | c.5917_5972insG; p.(Glu1980Alafs*66) | |
| Zygosity | Homozygous | Homozygous | Homozygous | |
| Clinical manifestations (HPO terms) | ||||
| Pathologic fractures (HP:0002756) | + | + | + | + |
| Coxa vara (HP:0002812) | + | n.d. | + | n.d. |
| Short stature (HP:0004322) | + | + | − | − |
| Failure to thrive (HP:0001508) | − | + | − | − |
| Mild global developmental delay (HP:0011342) | − | + | − | − |
| Anemia (HP:0001903) | + | n.d. | − | n.d. |
| Hepatomegaly (HP:0002240) | n.d. | − | + | n.d. |
| Facial dysmorphism | + | n.d. | + | − |
| Abnormality of the teeth (HP:0000164) | + | +, hypodontia | +, crowding of teeth | − |
| Recurrent infections (HP:0002719) | + | n.d. | − | − |
| Radiographic findings | ||||
| Sandwich appearance of vertebral bodies (HP:0004618) | + | + | + | + |
| Flattened vertebrae (HP:0000926) | + | − | − | − |
| Erlenmeyer flask deformity of the femurs (HP:0004975) | + | n.d. | + | n.d. |
| Dense metaphyseal bands (HP:0100959) | + | + | + | + |
| Thickened ribs (HP:0000900) | + | n.d. | + | n.d. |
| Osteosclerosis of the calvaria and base of the skull (HP:0005746) | n.d. | − | + | n.d. |
HPO = Human Phenotype Ontology; n.d. = not determined.
Bone biopsy findings
The histological and histomorphometric analysis of the patient's transiliac bone biopsy was consistent with osteoclast‐rich osteopetrosis (Fig. 3). The trabecular bone displayed an osteopetrosis‐like increase in trabecular bone mass (Fig. 3A). Furthermore, mineralized cartilage remnants were detected throughout the biopsy (Fig. 3B). Although few eroded surfaces were seen, there were several multinucleated osteoclasts with a flat shape as well as no visible resorption lacunae both indicating impaired resorption activity (Fig. 3C). Bone histomorphometry indicated higher microstructural indices (primarily higher trabecular thickness), normal osteoid levels, and a higher osteoclast surface compared to previously published age‐matched reference values (Fig. 3D).
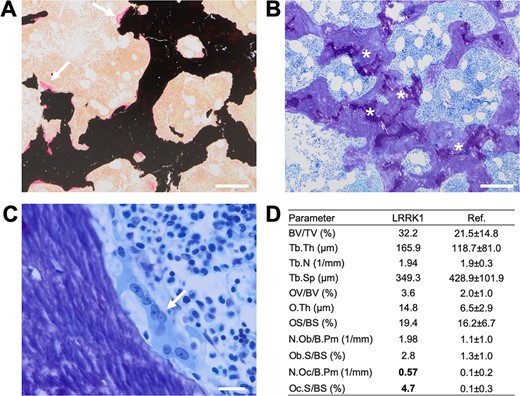
Histological characterization of an iliac crest biopsy. (A) Von Kossa/van Gieson staining showing a general increase in trabecular bone mass (primarily trabecular thickening) and no increased osteoid indices (white arrows). (B) Toluidine staining (overview) with cartilage remnants (asterisks). (C) Flat, multinucleated osteoclast (white arrow) and no visible resorption lacuna (ie, eroded surface) indicative of defective bone resorption. (D) Bone histomorphometry pointing to increased bone mass, slightly elevated osteoid levels, and a markedly higher osteoclast surface per bone surface compared to previously published age‐matched reference values.16
Backscattered electron microscopy revealed high matrix mineralization including the presence of hypermineralized cartilage remnants (Fig. 4A,B), which are similarly found in ADOII. BMDD histograms showed an overall shift toward higher calcium content with a second peak attributable to hypermineralized cartilage remnants (Fig. 4C), while overall calcium distribution values such as the mean calcium content (CaMean) and the heterogeneity of the calcium distribution (CaWidth) were comparable to those found in ADOII (Fig. 4D,E).
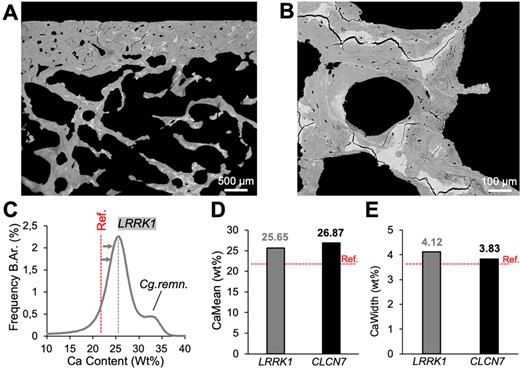
Backscattered electron imaging. (A) Biopsy overview indicating hypermineralized areas corresponding to cartilage remnants. (B) Confirmation of highly mineralized bone areas at higher magnification. (C) BMDD histogram demonstrates overall high matrix mineralization, partly due to cartilage remnants (second peak). (D,E) CaMean and CaWidth are overall higher compared to reference values18 and similar to CLCN7‐related autosomal dominant osteopetrosis type II.16
Identification of a LRRK1 splice site mutation leading to skipping of exon 3
Sanger sequencing of the main osteopetrosis genes CLCN7 and TCIRG1 revealed no mutations. In the following, we performed trio exome sequencing, which identified the homozygous variant c.261G>A in the gene LRRK1 in the patient. Segregation analysis proved the parental transmission; the parents and one sibling carry the mutation in a heterozygous state (Fig. 5A). The mutation is not listed in ExAC, gnomAD, or the 1000 Genomes project. The sequence change does not introduce a novel amino acid, but it affects the last nucleotide of exon 3. Accordingly, Human Splicing Finder (http://www.umd.be/HSF/) predicted an alteration of splicing due to an alteration of an exonic ESE site.31
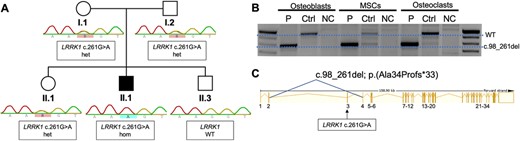
Genetic analysis. (A) Segregation of the mutation LRRK1 c.261G>A by Sanger sequencing. The index (II.1/P2) is affected and carries the mutation in a homozygous state. The clinically unaffected parents carry the mutation in a heterozygous state. (B) RT‐PCR of exons 2–6 of LRRK1 in patient‐derived osteoblasts, MSCs, and osteoclasts (P) compared to a healthy control (Ctrl) and a negative control (NC). The PCR product of the primer pairs (exon 2–6) yielded a 957‐bp product (WT). A smaller band of approximately 800 bp was detected in all cell lines carrying the LRRK1 splice site mutation indicating abnormal splicing. Sequencing of the lower band showed skipping of exon 3. (C) Schematic representation of the LRRK1 gene showing skipping of exon 3 leading to a frameshift mutation c.98_261del; p.Ala34Profs*33. MSC = mesenchymal stem cell.
We therefore performed RT‐PCR using mRNAs isolated from MSCs, osteoblasts, and osteoclasts from the patient an from healthy controls. Amplification of exons 2 to 6 resulted in a band of 900 bp in controls, but a shortened band of approximately 800 bp in all cells from the patient (Fig. 5B). Sanger sequencing of the shortened band revealed a skipping of exon 3 leading to deletion of 164 bases and a frameshift c.98_261del; p.(Ala34Profs*33)(Fig. 5C).
Increased size of LRRK1‐deficient osteoclasts and superficial bone erosion
In order to better understand the consequences of the identified LRRK1 mutation, we investigated osteoclasts differentiated from circulating monocytes from the patient and a healthy control individual. Osteoclasts carrying the mutation showed a faster differentiation compared to wild‐type osteoclasts resulting in giant osteoclasts, which is in line with the findings in the bone biopsy (Fig. 6A; Supporting Fig. 2). When the osteoclasts were seeded on dentine discs, control cells were able to form high numbers of resorption pits (Fig. 6A). In contrast, mutated osteoclasts formed broad areas with only superficial bone erosion that can best be visualized by confocal microscopy (Fig. 6A). This effect, sometimes called “pseudo‐resorption,” occurs much more rarely in healthy osteoclasts. This abnormal resorption behavior also becomes obvious when comparing the much lower number and area of resorption pits excavated by LRRK1‐deficient cells (Fig. 6B,C) with the total toluidine‐stained area indicating any surface roughness (Fig. 6D), which is comparable to control osteoclasts.
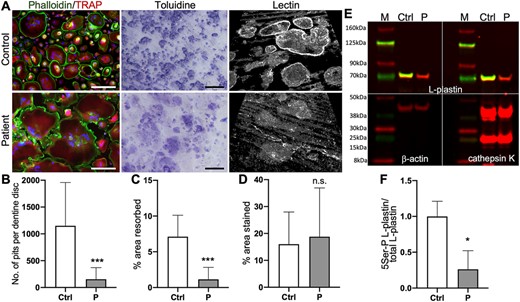
Osteoclast differentiation and functional characterization. (A) Fluorescence imaging of osteoclasts, resorption pits stained by toluidine and WGA lectin. Fluorescence images reveal increased size of patient‐derived osteoclasts compared to cells from healthy control. Staining: DAPI (blue) for nuclei detection, phalloidin (green), TRAP (red). Scale bar = 200 μm. Toluidine‐stained dentine disc from resorption assay reveals almost complete absence of resorption pits, but abundant superficial pseudo‐resorption by mutant osteoclasts. Scale bar = 200 μm. WGA lectin–stained resorption pits imaged by confocal microscopy Z‐stack. Control cells form deep resorption pits and some superficial erosions, while LRRK1‐deficient cells mostly produce superficial erosions. (B–D) Quantification of resorption assay. (B) Number of pits per dentine disc, (C) total pit area/dentine area, (D) total toluidine‐stained area/dentine area. (E) Immunoblot of osteoclast lysates from control (Ctrl) and patient (P). Reduced L‐plastin Ser5 phosphorylation (green) in patient‐derived osteoclasts, but comparable levels of unphosphorylated L‐plastin (red). β‐Actin was used as an endogenous control. Cathepsin K detection confirmed solid upregulation in patient and control typical for otherwise normally differentiated osteoclasts. (F) Quantification of 5Ser‐P L‐plastin/L‐plastin fold‐change. All osteoclast experiments were performed three times, representative images are shown. WGA = wheat germ agglutinin.
Because it was reported that LRRK1 phosphorylates L‐plastin, we compared levels of total L‐plastin levels to L‐plastin with Ser5 phosphorylation (Fig. 6E). We used this as a readout for LRRK1 function, because the determination of LRRK1 protein levels was impossible due to the lack of antibodies suitable for immunoblotting. LRRK1 mutated osteoclasts showed normal levels of unphosphorylated L‐plastin, but a strong reduction of the phosphorylated form to around 20% (Fig. 6F). High cathepsin K levels confirmed otherwise normal osteoclast differentiation.
Discussion
We describe the fourth known human LRRK1 mutation in an adult individual with OSMD. Besides pathologic long bone fractures the patient suffered from a severe progressive ONJ starting in the third decade and finally leading to loss of most teeth. We confirmed a LRRK1 loss of function and found striking morphological changes in cultured osteoclasts as well as an altered resorption behavior.
To date, four individuals from four different families have been described to carry biallelic mutations in LRRK1 leading to osteosclerosis. As shown in Table 1, the OSMD patients described so far closely resemble our patient.2, 3, 4 All presented with pathologic and recurrent fractures and osteosclerosis at multiple skeletal sites, predominantly at the metaphyses and vertebral bodies. Different degrees of osteosclerosis of ribs and skull and of Erlenmeyer flask deformity of the femurs were present. The clinical presentation of OSMD is thus highly overlapping with intermediate osteopetrosis due to CLCN7 mutations or TCIRG1 splice mutations, ADOII due to CLCN7 mutations, and dysosteosclerosis due to CSF1R, SLC29A3, and TNFRSF11A mutations.32, 33, 34, 35, 36 It thus seems justified to subsume all these disorders under the term osteopetrosis because they all share impaired osteoclast function as the mechanistic basis.
Our patient is the only one who presented with progressive ONJ with sequester formation causing local infections as frequently seen in MRONJ. The resection of the right‐sided mandible was considered but deferred on. In the setting of a slowly progressing destruction of the mandible, no clear resection border could be determined. Commonly practiced microvascular fibula graft reconstruction was deferred on due to the high perceived risk and increased donor site morbidity in a patient suffering from repeated pathologic fractures. Of the different factors triggering ONJ the patient had poor oral hygiene and a history of tooth extraction due to impacted teeth. Dental crowding and persistent primary teeth have been described in OSMD.2 Another risk factor for ONJ is impaired osteoclast function, which is often secondary to antiresorptive treatment in MRONJ or to hereditary osteopetrosis in ONJ.13, 14 Given its rarity it is difficult to judge whether ONJ is more frequent in OSMD than in other forms of osteopetrosis.
We report the first LRRK1 mutation affecting splicing. The mutation is placed at the last base pair of exon 3 and causes skipping of the entire exon 3. Although the resulting frameshift is predicted to lead to a premature stop codon we found no evidence for a significant nonsense‐mediated mRNA decay (NMD). The other described mutations described are more C‐terminal: the homozygous frameshift deletion LRRK1 c.5938_5944delGAGTGGT leading to a stop‐loss and a protein prolonged by 29 amino acids in a 2‐year‐old Moroccan male, the homozygous insertion LRRK1 c.5917_5972insG; p.(Glu1980Alafs*66) in Indian siblings, and the p.Glu929* nonsense mutation found in an Iranian patient.2, 3, 4 The phenotypic similarities do not point toward a milder effect of these C‐terminal mutations that could lead to a stable truncated protein. Because the large LRRK1 protein is difficult to detect by immunoblot no information is available on the exact consequences of these mutations. Therefore, we opted for a functional readout to test for the function of the protein. As shown by Si and colleagues8 in murine osteoclasts, our patient shows a reduction of L‐plastin Ser5 phosphorylation in LRRK1 mutated osteoclasts by around 80% compared to a healthy control.
Iliac crest histology revealed a dense trabecular bone microstructure associated with multinucleated osteoclasts with signs of inadequate bone resorption (ie. flat shape, few eroded surfaces). qBEI pointed to high matrix mineralization, which was partly due to hypermineralized cartilage remnants. Taken together, the micro‐morphological pattern is compatible with osteoclast‐rich osteopetrosis such as found in ADOII.16 Interestingly, hypermineralized cartilage remnants indicative of resorption defects were previously also found in gnathodiaphyseal dysplasia.18 The high abundance of large osteoclasts clearly distinguishes LRRK1‐related osteopetrosis from the differential diagnosis dysosteosclerosis, which rather is osteoclast‐poor.32, 33, 37
Murine osteoclasts lacking Lrrk1 revealed a reduced resorption pit area, and pits were smaller and shallower compared to knockout (KO) cultures, and only few osteoclasts formed a typical sealing zone.3, 7 Although osteoclasts from our patient formed broad toluidine‐stained eroded areas instead of pits or trenches, we think that this defect is also due to an abnormal sealing zone, which may lead to a leaking of protons and proteases out of the resorption lacuna. However, the increased size of LRRK1‐mutated osteoclasts cannot readily be explained by a cytoskeletal problem. This could be due to an enhanced migration facilitating fusion of a polykaryon with mononuclear precursors.38 Interestingly, also osteoclasts from Clcn7‐deficient mice and a patient with CLCN7‐related autosomal recessive osteopetrosis have a larger diameter for an unknown reason.30, 39, 40
Lrrk1−/− mice were resistant to ovariectomy (OVX)‐induced bone loss at the spine, femur, and tibia and showed elevated total bone mass density (BMD) at all three skeletal sites at time of OVX.7 LRRK1 has hence been contemplated as a potential target for osteoanabolic therapy.41 However, only few cases of LRRK1 mutations in patients are described to date and only little is known about osteoclast pathology in the LRRK1‐mutated setting. A deeper understanding of the effect LRRK1 mutations in humans have on osteoclasts is inevitable in the process of considering LRRK1 as a feasible drug target.
Conclusion
In conclusion, we describe the first patient with OSMD and ONJ caused by a LRRK1 splice‐site mutation. Although the micro‐morphological analyses conducted pointed to the presence of osteoclast‐rich osteopetrosis, our findings may lead to a better understanding of OSMD, the advancement of osteopetrosis, and potentially also osteoporosis treatment. When discussing LRRK1 as a potential drug target for treating low BMD/osteoporosis the possibility of MRONJ as a side effect should be kept in mind.
Disclosures
The authors report no conflict of interest.
Acknowledgments
This study was supported by a stipend from the Berlin Institute of Health (BIH) to AH. This project has received funding from the European Community's Seventh Framework Programme under grant agreement no. 602300 (SYBIL), and the German Federal Ministry of Education and Research (BMBF) within the project “Detection and Individualized Management of Early Onset Osteoporosis (DIMEOS)”. We are grateful to the patient and his family members whose cooperation made this study possible.
Authors' roles: Study design: AH, HPH, UK. Study conduct: AH, RO, UK; Data collection: AFH, AH, NS, SB, SG, TR, UR; Data analysis: AFH, AH, AK, JZ, MA, RO, TR, UK, UR; Data interpretation: AH, HPH, TR, UK; Writing manuscript: AFH, AH, HPH, TR, UK; Revising manuscript content: AH, MA, RO, TR, UK; Approving final version of manuscript: AFH, AH, AK, HPH, JZ, MA, NS, RO, SB, SG, TR, UK, UR; The following individuals take responsibility for the integrity of the data analysis: HPH, RO, UK.
The URLs for data presented herein are as follows: Ensembl Genome Browser (http://www.emsembl.org), Mutation Taster (http://www.mutationtaster.org/), Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/omim), Human Splicing Finder (http://www.umd.be/HSF3/HSF.shtml); Geneious (https://www.geneious.com/), and The Protein Model Portal (https://www.proteinmodelportal.org).
References
Author notes
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.3995.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}