





ABSTRACT
Mutations in SQSTM1 are strongly associated with Paget's disease of bone (PDB), but little is known about the clinical characteristics of those with early disease. Radionuclide bone scans, biochemical markers of bone turnover, and clinical characteristics were analyzed in SQSTM1 mutation carriers who took part in the Zoledronic acid in the Prevention of Paget's disease (ZiPP) study. We studied 222 individuals, of whom 54.9% were female, with mean ± SE age of 50.1 ± 0.6 years. Twelve SQSTM1 mutations were observed, including p.Pro392Leu, which was present in 141 of 222 (63.5%) subjects. Bone scan examination revealed evidence of PDB in 20 subjects (9.0%), ten of whom (50%) had a single affected site. Participants with lesions were older than those without lesions but the difference was not significant (53.6 ± 9.1 versus 49.8 ± 8.9; p = .07). The mean age of participants with lesions was not significantly different from the age at which their parents were diagnosed with PDB (55 years versus 59 years, p = .17). All individuals with lesions were asymptomatic. Serum concentrations of total alkaline phosphatase (ALP) normalized to the upper limit of normal in each center were higher in those with lesions (0.75 ± 0.69 versus 0.42 ± 0.29 arbitary units; p < .0001). Similar findings were observed for other biochemical markers of bone turnover, but the sensitivity of ALP and other markers in detecting lesions was poor. Asymptomatic PDB is present in about 9% of SQSTM1 mutation carriers by the fifth decade. Further follow‐up of this cohort will provide important information on the natural history of early PDB and its response to treatment. © 2020 The Authors. Journal of Bone and Mineral Research published by American Society for Bone and Mineral Research.
Introduction
Paget's disease of bone (PDB) is a skeletal disorder characterized by focal increases in bone remodeling at one or more sites throughout the skeleton.1 Genetic factors play a key role in PDB and the most important susceptibility gene is SQSTM1.2, 3 Protein coding mutations of SQSTM1, which predominantly affect the ubiquitin‐associated (UBA) domain, have been detected in between 40% and 50% of people with a family history of PDB and up to 15% of those who are unaware of a family history.4 Cross‐sectional studies suggest that SQSTM1 mutations are highly penetrant such that up to 80% of carriers develop the disease by the seventh decade.4 Furthermore, people with PDB that carry SQSTM1 mutations have more severe disease with an earlier age at onset than those that do not have SQSTM1 mutations.5 Although the causal link between SQSTM1 mutations and PDB is beyond doubt, there is limited information on the natural history of PDB in people who carry SQSTM1 mutations and the acceptability of genetic testing for PDB in routine clinical practice. In order to gain an insight into these issues, we investigated the clinical and biochemical characteristics of SQSTM1 mutation carriers with evidence of PDB‐like bone lesions who were enrolled into the Zoledronate in the Prevention of Paget's disease (ZiPP) study, which is a large‐scale prospective study that aims to investigate the acceptability of genetic testing for PDB, coupled with offer of targeted intervention with zoledronic acid or placebo in SQSTM1 mutation carriers.
Patients and Methods
Study design
The ZiPP study (ISRCTN Registry; http://www.isrctn.com/; Registration number: ISRCTN11616770) is a multicenter, multinational, randomized, placebo‐controlled trial involving a large scale program of genetic testing for SQSTM1 mutations, coupled with the offer of targeted intervention in the form of a randomized trial, to determine whether treatment with a single infusion of intravenous zoledronic acid can modify the development of PDB‐related lesions in asymptomatic carriers of SQSTM1 mutations.6 The study commenced in 2009 and is still ongoing. The data presented here are derived from untreated individuals who consented to take part in the study and completed the baseline assessment. The ZiPP study involved an initial phase of genetic testing for SQSTM1 mutations in probands known to have PDB followed by testing of first‐degree relatives (who in almost all cases were children of probands). If the relative tested positive for SQSTM1 mutations they were then invited to participate in the ZiPP study. All individuals gave written informed consent and the study received ethical approval in all participating centers.
Participants
The selection process for inclusion was broadly similar in each center. The local investigator invited patients with PDB attending outpatient clinics to undergo genetic testing for SQSTM1 mutations and if the result was positive to pass on the information to first‐degree relatives about the study. Relatives who consented to undergo testing and were found to be positive for SQSTM1 mutations were then invited to participate in the interventional phase of the ZiPP study. There were two exceptions; in Auckland and in Oswestry, relatives underwent genetic testing directly without their parents having undergone testing as part of this study. An overview of disposition of individuals who took part in the genetic testing phase is provided in Fig. 1. We invited 2394 probands to undergo testing, and of the 1428 (59.5%) who agreed, 247 (17.2%) were found to be carriers of SQSTM1 mutations. We then invited 1307 relatives of SQSTM1 mutation carriers to undergo testing and 750 (57.3%) agreed. Of these individuals, 350 (46.6%) tested positive for SQSTM1 mutations and 222 (63.4%) agreed to take part in the randomized trial. Supplementary Table S1 provides full details of the numbers of probands and relatives screened in each study center. Participants that entered the trial were derived from the UK (n = 134, 60.4%), Australia (n = 34, 15.3%), Italy (n = 19; 8.5%), Spain (n = 18; 8.1%), Ireland (n = 10; 4.5%), New Zealand (n = 4; 1.8%), and Belgium (n = 3, 1.3%). The participants were children of 129 probands diagnosed with PDB, giving an average recruitment rate of 1.72 subjects per family.
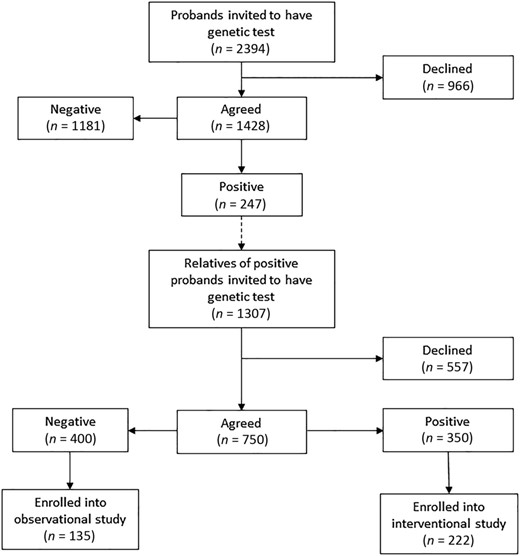
Recruitment into the ZiPP study. Disposition of probands and relatives of probands undergoing genetic testing for SQSTM1 mutations with the ZiPP study population. The numbers and proportion of subjects at each stage is indicated.
The inclusion and exclusion criteria for participation in the trial have been published previously, but in essence, relatives who carried SQSTM1 mutations were eligible provided that they were age >30 years, had not been diagnosed with PDB, and had no contraindication to receiving zoledronic acid. Recruitment to the trial commenced in May 2009 and closed in April 2015; the end‐of‐study visits are currently in progress and the results of the study are expected to be available in 2021.
Genetic testing
Testing for SQSTM1 mutations was carried out by Sanger sequencing of exons 7 and 8 and the intron‐exon boundaries of the SQSTM1 gene according to standard techniques. If a previously unidentified mutation was detected, bioinformatic and functional studies were conducted to determine if it was likely to be pathogenic.
Biochemical studies
Samples were sent for routine biochemistry at participating centers and measurements of serum creatinine (Cr), serum total alkaline phosphatase (ALP), calcium, 25(OH)D, albumin, and liver function, and were performed by the local chemical pathology laboratory according to standard techniques.
Measured ALP values were adjusted to the local reference range (RR) to give an adjusted ALP value that was used in statistical analysis. The formula used for calculation was as follows: Adjusted ALP = (measured ALP – ALP lower limit of RR)/(ALP upper limit of RR – ALP lower limit of RR). According to this methodology, a value of 1.0 would equate to an adjusted ALP at the upper limit of the reference range, a value of 2.0 to twice the upper limit, a value of 0.5 to the midpoint of the reference range, and a value of 0 to the lower limit of the reference range.
Additional markers of bone resorption and formation were measured by enzyme‐linked immunosorbent assay (ELISA)‐based techniques according to the manufacturer's instructions at the University of East Anglia. Crosslinked N‐telopeptides of type I collagen (NTX) were measured in urine by ELISA (Osteomark; Ostex International, Inc., Seattle, WA, USA). The values were corrected for Cr and were expressed in nanomoles of bone collagen equivalents per liter (BCE; nM) per millimole Cr per liter (mM). Interassay coefficient of variation (CV) for NTX was ≤7.3% up to the concentration of 3000nM BCE with the lower limit of sensitivity at 20nM BCE. The reference ranges were 5 to 65 nmol/mmol in women and 3 to 63 nmol/mmol in men. Plasma concentrations of type I collagen C‐telopeptides (CTX) were measured using electrochemiluminesence immunoassay (ECLIA) on a Cobas e601 analyzer (Roche Diagnostics, Mannheim, Germany). The interassay CV for CTX was ≤3% between 0.2 and 1.5 μg/L with the sensitivity of 0.01 μg/L. The reference ranges in women were 0.16 to 0.57 μg/L (age <50 years); 0.25 to 1.02 μg/L (age >50 years), and in men were 0.19 to 0.58 μg/L (age 30 to 50 years), 0.19 to 0.70 μg/L (age 50 to 70 years), and 0.19 to 0.85 μg/L (age >70 years). Serum bone‐specific alkaline phosphatase (BAP) was measured using the MicroVue enzyme immunoassay (Quidel, Athens, OH, USA). Interassay CV for BAP was ≤2.4% up to the concentration of 140 U/L with the lower limit of sensitivity at 0.7 U/L. The reference ranges in premenopausal women were 11.6 to 29.6 U/L, in postmenopausal women 14.2 to 42.7 U/L, and in men 15 to 41.2 U/L. Plasma concentrations of Procollagen type I amino‐terminal propeptide (P1NP) were measured using ECLIA on a Cobas e601 analyzer (Roche Diagnostics, Mannheim, Germany). P1NP interassay CV was ≤3% between 20 to 600 μg/L with the sensitivity of 8 μg/L. The reference range in premenopausal women was 15 to 58.6 μg/L, in postmenopausal women 20.3 to 76.3 μg/L, and in men 20 to 76 μg/L. All samples were collected after an overnight fast between 9:00 a.m. and 12:00 p.m., and urine samples were from the second void of the day.
Radionuclide bone scans
Radionuclide scintigraphy was performed to detect evidence of PDB‐like bone lesions according to standard techniques in all subjects following an injection of 99mTc‐labeled bisphosphonate at the baseline visit. Anonymized images were uploaded onto the study database and assessed independently by two imaging experts (DS and SHR) for evidence of PDB‐like bone lesions. Lesions were coded as being present only when they were independently identified by both observers. The location and number of confirmed lesions was recorded in the study database. The reason for using radionuclide bone scans as the method of assessment is that this technique is more sensitive than plain X‐rays at detecting bone lesions in PDB.7, 8, 9 Although many conditions can cause focal increases in tracer uptake on bone scan, the appearances in PDB almost always allow differentiation from other conditions.10 Nonetheless, further imaging could be performed at the discretion of the local investigator with plain X‐ray, CT, or MRI if clinically indicated or if the pattern of uptake on the bone scan was thought not to be typical of PDB.
Statistical analysis
Descriptive and inferential statistics were generated using Minitab Express version 1.5.1 (Minitab, LLC, State College, PA, USA). Differences between groups for continuous variables were assessed by one‐way analysis of variance (ANOVA) and for categorical variables by chi‐square testing. Kaplan‐Meier analysis and sensitivity and specificity testing, negative predictive (NPV), and positive predictive values (PPV) were performed using SPSS version 24 (IBM Corp., Armonk, NY, USA). The significance level was set at p < .05 and all tests were two‐tailed.
Results
Clinical characteristics, demographics and spectrum of SQSTM1 mutations
Of the 222 SQSTM1 positive individuals enrolled into the study, 122 (54.9%) were female. The mean ± SD age participants was 50.2 ± 9.1 years. The demographics of these subjects were similar to the 128 relatives who tested positive and who decided not to enroll in the study. The average age of these individuals was 53.1 ± 13.3 years and 79 (61.7%) were female. The spectrum of mutations in subjects who were enrolled into the trial is shown in Table 1. Twelve mutations were identified, the most common of which was p.Pro392Leu, along with a variety of other missense and truncation mutations that have all been previously reported. The average adjusted ALP value was 0.44 ± 0.35, and in 20 of 222 individuals (9.0%) the ALP was above the upper limit of the local reference range. None of the participants had symptoms or signs of PDB at the time of recruitment or the baseline visit.
Spectrum of Mutations in the ZiPP Study
| SQSTM1 variant | Mutations n (%) | Type of mutation |
| p.Pro392Leu | 141 (63.5) | Missense |
| p.Gly425Arg | 24 (10.8) | Missense |
| p.Met404Val | 25 (11.3) | Missense |
| p.Gly411Ser | 9 (4.1) | Missense |
| aAla390* | 8 (3.6) | Truncating |
| p.Glu396* | 3 (1.4) | Truncating |
| p.Thr350GInfsX28 | 3 (1.4) | Truncating |
| p.Phe406Val | 2 (0.9) | Truncating |
| p.Lys378* | 2 (0.9) | Truncating |
| p.Gln371* | 2 (0.9) | Truncating |
| p.Ile424Ser | 2 (0.9) | Missense |
| p.Glu396* | 1 (0.46) | Truncating |
| SQSTM1 variant | Mutations n (%) | Type of mutation |
| p.Pro392Leu | 141 (63.5) | Missense |
| p.Gly425Arg | 24 (10.8) | Missense |
| p.Met404Val | 25 (11.3) | Missense |
| p.Gly411Ser | 9 (4.1) | Missense |
| aAla390* | 8 (3.6) | Truncating |
| p.Glu396* | 3 (1.4) | Truncating |
| p.Thr350GInfsX28 | 3 (1.4) | Truncating |
| p.Phe406Val | 2 (0.9) | Truncating |
| p.Lys378* | 2 (0.9) | Truncating |
| p.Gln371* | 2 (0.9) | Truncating |
| p.Ile424Ser | 2 (0.9) | Missense |
| p.Glu396* | 1 (0.46) | Truncating |
Spectrum of Mutations in the ZiPP Study
| SQSTM1 variant | Mutations n (%) | Type of mutation |
| p.Pro392Leu | 141 (63.5) | Missense |
| p.Gly425Arg | 24 (10.8) | Missense |
| p.Met404Val | 25 (11.3) | Missense |
| p.Gly411Ser | 9 (4.1) | Missense |
| aAla390* | 8 (3.6) | Truncating |
| p.Glu396* | 3 (1.4) | Truncating |
| p.Thr350GInfsX28 | 3 (1.4) | Truncating |
| p.Phe406Val | 2 (0.9) | Truncating |
| p.Lys378* | 2 (0.9) | Truncating |
| p.Gln371* | 2 (0.9) | Truncating |
| p.Ile424Ser | 2 (0.9) | Missense |
| p.Glu396* | 1 (0.46) | Truncating |
| SQSTM1 variant | Mutations n (%) | Type of mutation |
| p.Pro392Leu | 141 (63.5) | Missense |
| p.Gly425Arg | 24 (10.8) | Missense |
| p.Met404Val | 25 (11.3) | Missense |
| p.Gly411Ser | 9 (4.1) | Missense |
| aAla390* | 8 (3.6) | Truncating |
| p.Glu396* | 3 (1.4) | Truncating |
| p.Thr350GInfsX28 | 3 (1.4) | Truncating |
| p.Phe406Val | 2 (0.9) | Truncating |
| p.Lys378* | 2 (0.9) | Truncating |
| p.Gln371* | 2 (0.9) | Truncating |
| p.Ile424Ser | 2 (0.9) | Missense |
| p.Glu396* | 1 (0.46) | Truncating |
The characteristics of participants with lesions compared to those without lesions are shown in Table 2. The most commonly involved anatomical sites were the pelvis and spine. Most participants with lesions had monostotic disease (10/20; 50%). Representative examples of lesions are shown in Fig. 2A‐D. Participants with lesions were on average about 5 years older than those without lesions but this difference was not significant (p = .07). Lesions were equally common in men and women. There was no difference in the proportion of subjects with lesions depending on whether the mutation was missense or truncating. All of the biochemical markers of bone turnover tested were significantly higher in those with lesions as compared with those who did not have lesions (Table 2). The sensitivity of ALP and other biochemical markers in detecting individuals lesions was generally poor, although specificity was above 80% for all of the markers except urinary NTX (uNTX)/Cr (Table 3).
Characteristics of Participants With and Without Lesions
| Variable | Lesion (n = 20) | No lesion (n = 202) | p |
| Age (years), mean ± SD | 53.6 ± 9.1 | 49.8 ± 9.0 | .07 |
| Gender | .95 | ||
| Male, n (%) | 10 (50.0) | 90 (44.5) | |
| Female, n (%) | 10 (50.0) | 112 (55.4) | |
| Serum calcium (mmol/L), mean ± SD | 2.41 ± 0.16 | 2.40 ± 0.11 | .63 |
| Serum 25(OH)D (nmol/L), mean ± SD | 50.1 ± 26.2 | 52.2 ± 29.7 | .76 |
| Adjusted ALP (AU), mean ± SD | 0.75 ± 0.69 | 0.42 ± 0.29 | <.0001 |
| Elevated ALP, n (%) | 4/20 (20.0) | 16/198 (8.0) | .007 |
| uNTX/Cr (nM/mM), mean ± SD | 305.5 ± 808.6 | 51.9 ± 51.9 | <.0001 |
| Elevated uNTX/Cr, n (%) | 10/15 (66.6) | 5/132 (3.8) | .0075 |
| BAP (U/L), mean ± SD | 15.1 ± 12.8 | 10.4 ± 7.0 | .015 |
| Elevated BAP, n (%) | 1/17 (5.8) | 1/186 (0.5) | .16 |
| CTX (μg/L), mean ± SD | 0.44 ± 0.26 | 0.32 ± 0.15 | .004 |
| Elevated CTX, n (%) | 2/17 (11.7) | 15/187 (8.0) | .17 |
| P1NP (μg/L), mean ± SD | 99.9 ± 84.9 | 53.4 ± 22.3 | <.0001 |
| Elevated P1NP, n (%) | 10/17 (58.8) | 35/187 (19.2) | .0001 |
| Mutation type, n (%) | 1.0 | ||
| Missense | 19 (94.4) | 184 (91.0) | |
| Truncating | 1 (5.6) | 18 (8.9) | |
| Sites affected, n (%) | |||
| 1 | 10 (50.0) | – | |
| 2 | 6 (30.0) | – | |
| ≥3 | 4 (20.0) | – |
| Variable | Lesion (n = 20) | No lesion (n = 202) | p |
| Age (years), mean ± SD | 53.6 ± 9.1 | 49.8 ± 9.0 | .07 |
| Gender | .95 | ||
| Male, n (%) | 10 (50.0) | 90 (44.5) | |
| Female, n (%) | 10 (50.0) | 112 (55.4) | |
| Serum calcium (mmol/L), mean ± SD | 2.41 ± 0.16 | 2.40 ± 0.11 | .63 |
| Serum 25(OH)D (nmol/L), mean ± SD | 50.1 ± 26.2 | 52.2 ± 29.7 | .76 |
| Adjusted ALP (AU), mean ± SD | 0.75 ± 0.69 | 0.42 ± 0.29 | <.0001 |
| Elevated ALP, n (%) | 4/20 (20.0) | 16/198 (8.0) | .007 |
| uNTX/Cr (nM/mM), mean ± SD | 305.5 ± 808.6 | 51.9 ± 51.9 | <.0001 |
| Elevated uNTX/Cr, n (%) | 10/15 (66.6) | 5/132 (3.8) | .0075 |
| BAP (U/L), mean ± SD | 15.1 ± 12.8 | 10.4 ± 7.0 | .015 |
| Elevated BAP, n (%) | 1/17 (5.8) | 1/186 (0.5) | .16 |
| CTX (μg/L), mean ± SD | 0.44 ± 0.26 | 0.32 ± 0.15 | .004 |
| Elevated CTX, n (%) | 2/17 (11.7) | 15/187 (8.0) | .17 |
| P1NP (μg/L), mean ± SD | 99.9 ± 84.9 | 53.4 ± 22.3 | <.0001 |
| Elevated P1NP, n (%) | 10/17 (58.8) | 35/187 (19.2) | .0001 |
| Mutation type, n (%) | 1.0 | ||
| Missense | 19 (94.4) | 184 (91.0) | |
| Truncating | 1 (5.6) | 18 (8.9) | |
| Sites affected, n (%) | |||
| 1 | 10 (50.0) | – | |
| 2 | 6 (30.0) | – | |
| ≥3 | 4 (20.0) | – |
Values are mean ± SD or numbers and percentages. The p values refer to differences between groups assessed by t test for continuous variables or Fisher's exact test for categorical variables. Serum calcium values were adjusted for albumin.
ALP = total alkaline phosphatase; AU = arbitary units; BAP = bone‐specific alkaline phosphatase; CTX = C‐terminal collagen crosslinks; P1NP = procollagen type‐I N‐terminal propeptide fragment; uNTX/Cr = urinary N‐telopeptide collagen crosslinks/urine creatinine.
Characteristics of Participants With and Without Lesions
| Variable | Lesion (n = 20) | No lesion (n = 202) | p |
| Age (years), mean ± SD | 53.6 ± 9.1 | 49.8 ± 9.0 | .07 |
| Gender | .95 | ||
| Male, n (%) | 10 (50.0) | 90 (44.5) | |
| Female, n (%) | 10 (50.0) | 112 (55.4) | |
| Serum calcium (mmol/L), mean ± SD | 2.41 ± 0.16 | 2.40 ± 0.11 | .63 |
| Serum 25(OH)D (nmol/L), mean ± SD | 50.1 ± 26.2 | 52.2 ± 29.7 | .76 |
| Adjusted ALP (AU), mean ± SD | 0.75 ± 0.69 | 0.42 ± 0.29 | <.0001 |
| Elevated ALP, n (%) | 4/20 (20.0) | 16/198 (8.0) | .007 |
| uNTX/Cr (nM/mM), mean ± SD | 305.5 ± 808.6 | 51.9 ± 51.9 | <.0001 |
| Elevated uNTX/Cr, n (%) | 10/15 (66.6) | 5/132 (3.8) | .0075 |
| BAP (U/L), mean ± SD | 15.1 ± 12.8 | 10.4 ± 7.0 | .015 |
| Elevated BAP, n (%) | 1/17 (5.8) | 1/186 (0.5) | .16 |
| CTX (μg/L), mean ± SD | 0.44 ± 0.26 | 0.32 ± 0.15 | .004 |
| Elevated CTX, n (%) | 2/17 (11.7) | 15/187 (8.0) | .17 |
| P1NP (μg/L), mean ± SD | 99.9 ± 84.9 | 53.4 ± 22.3 | <.0001 |
| Elevated P1NP, n (%) | 10/17 (58.8) | 35/187 (19.2) | .0001 |
| Mutation type, n (%) | 1.0 | ||
| Missense | 19 (94.4) | 184 (91.0) | |
| Truncating | 1 (5.6) | 18 (8.9) | |
| Sites affected, n (%) | |||
| 1 | 10 (50.0) | – | |
| 2 | 6 (30.0) | – | |
| ≥3 | 4 (20.0) | – |
| Variable | Lesion (n = 20) | No lesion (n = 202) | p |
| Age (years), mean ± SD | 53.6 ± 9.1 | 49.8 ± 9.0 | .07 |
| Gender | .95 | ||
| Male, n (%) | 10 (50.0) | 90 (44.5) | |
| Female, n (%) | 10 (50.0) | 112 (55.4) | |
| Serum calcium (mmol/L), mean ± SD | 2.41 ± 0.16 | 2.40 ± 0.11 | .63 |
| Serum 25(OH)D (nmol/L), mean ± SD | 50.1 ± 26.2 | 52.2 ± 29.7 | .76 |
| Adjusted ALP (AU), mean ± SD | 0.75 ± 0.69 | 0.42 ± 0.29 | <.0001 |
| Elevated ALP, n (%) | 4/20 (20.0) | 16/198 (8.0) | .007 |
| uNTX/Cr (nM/mM), mean ± SD | 305.5 ± 808.6 | 51.9 ± 51.9 | <.0001 |
| Elevated uNTX/Cr, n (%) | 10/15 (66.6) | 5/132 (3.8) | .0075 |
| BAP (U/L), mean ± SD | 15.1 ± 12.8 | 10.4 ± 7.0 | .015 |
| Elevated BAP, n (%) | 1/17 (5.8) | 1/186 (0.5) | .16 |
| CTX (μg/L), mean ± SD | 0.44 ± 0.26 | 0.32 ± 0.15 | .004 |
| Elevated CTX, n (%) | 2/17 (11.7) | 15/187 (8.0) | .17 |
| P1NP (μg/L), mean ± SD | 99.9 ± 84.9 | 53.4 ± 22.3 | <.0001 |
| Elevated P1NP, n (%) | 10/17 (58.8) | 35/187 (19.2) | .0001 |
| Mutation type, n (%) | 1.0 | ||
| Missense | 19 (94.4) | 184 (91.0) | |
| Truncating | 1 (5.6) | 18 (8.9) | |
| Sites affected, n (%) | |||
| 1 | 10 (50.0) | – | |
| 2 | 6 (30.0) | – | |
| ≥3 | 4 (20.0) | – |
Values are mean ± SD or numbers and percentages. The p values refer to differences between groups assessed by t test for continuous variables or Fisher's exact test for categorical variables. Serum calcium values were adjusted for albumin.
ALP = total alkaline phosphatase; AU = arbitary units; BAP = bone‐specific alkaline phosphatase; CTX = C‐terminal collagen crosslinks; P1NP = procollagen type‐I N‐terminal propeptide fragment; uNTX/Cr = urinary N‐telopeptide collagen crosslinks/urine creatinine.
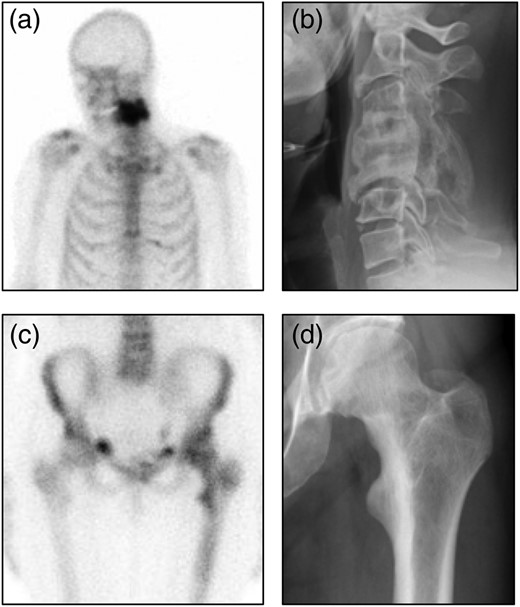
Representative images from participants with bone lesions. (A) Radionuclide bone scan image showing intense tracer uptake in cervical vertebrae 4 and 5. (B) Radiograph from the same patient showing typical features of Paget's disease with expansion and fusion of C4 and C5. The patient was asymptomatic and had no neurological abnormalites. (C) Radionuclide bone scan image showing increased tracer uptake in the upper femur on the left side. (D) Radiograph from the same patient showing coarsening of the trabecular pattern in the upper femur and cortical thickening of the medial aspect of the upper femur.
Predictive Values of Biochemical Markers in Detecting Individuals With Lesions
| Biomarker | Sample | Units | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Samples (N) |
| ALP | Serum | AU | 20.0 | 97.0 | 40.0 | 92.4 | 222 |
| uNTX/Cr | Urine | nM/mM | 66.7 | 67.5 | 14.1 | 96.2 | 203 |
| CTX | Plasma | ug/L | 11.7 | 92.3 | 22.2 | 92.3 | 204 |
| P1NP | Plasma | ug/L | 58.8 | 81.3 | 22.2 | 95.6 | 204 |
| BAP | Serum | U/L | 5.9 | 99.4 | 50.0 | 92.0 | 203 |
| Biomarker | Sample | Units | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Samples (N) |
| ALP | Serum | AU | 20.0 | 97.0 | 40.0 | 92.4 | 222 |
| uNTX/Cr | Urine | nM/mM | 66.7 | 67.5 | 14.1 | 96.2 | 203 |
| CTX | Plasma | ug/L | 11.7 | 92.3 | 22.2 | 92.3 | 204 |
| P1NP | Plasma | ug/L | 58.8 | 81.3 | 22.2 | 95.6 | 204 |
| BAP | Serum | U/L | 5.9 | 99.4 | 50.0 | 92.0 | 203 |
ALP = total alkaline phosphatase; AU = arbitary units; BAP = bone‐specific alkaline phosphatase; CTX = C‐terminal collagen crosslinks; N = number of samples where results were available; NPV = negative predictive value; P1NP = procollagen type‐I N‐terminal propeptide fragment; PPV = positive predictive value; uNTX/Cr = urinary N‐telopeptide collagen cross links/urine creatinine.
Predictive Values of Biochemical Markers in Detecting Individuals With Lesions
| Biomarker | Sample | Units | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Samples (N) |
| ALP | Serum | AU | 20.0 | 97.0 | 40.0 | 92.4 | 222 |
| uNTX/Cr | Urine | nM/mM | 66.7 | 67.5 | 14.1 | 96.2 | 203 |
| CTX | Plasma | ug/L | 11.7 | 92.3 | 22.2 | 92.3 | 204 |
| P1NP | Plasma | ug/L | 58.8 | 81.3 | 22.2 | 95.6 | 204 |
| BAP | Serum | U/L | 5.9 | 99.4 | 50.0 | 92.0 | 203 |
| Biomarker | Sample | Units | Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Samples (N) |
| ALP | Serum | AU | 20.0 | 97.0 | 40.0 | 92.4 | 222 |
| uNTX/Cr | Urine | nM/mM | 66.7 | 67.5 | 14.1 | 96.2 | 203 |
| CTX | Plasma | ug/L | 11.7 | 92.3 | 22.2 | 92.3 | 204 |
| P1NP | Plasma | ug/L | 58.8 | 81.3 | 22.2 | 95.6 | 204 |
| BAP | Serum | U/L | 5.9 | 99.4 | 50.0 | 92.0 | 203 |
ALP = total alkaline phosphatase; AU = arbitary units; BAP = bone‐specific alkaline phosphatase; CTX = C‐terminal collagen crosslinks; N = number of samples where results were available; NPV = negative predictive value; P1NP = procollagen type‐I N‐terminal propeptide fragment; PPV = positive predictive value; uNTX/Cr = urinary N‐telopeptide collagen cross links/urine creatinine.
Information on age at clinical diagnosis of PDB in the probands was available for 12 of the subjects with bone lesions. Kaplan‐Meier analysis showed that the estimated mean age at clinical diagnosis in probands was 57.3 years (95% CI, 52.9 to 61.6) as compared with 53.6 years (95% CI, 49.6 to 57.6) for the children who had PDB lesions on bone scan, a difference that was not significant (p = .17) (Fig. 3).
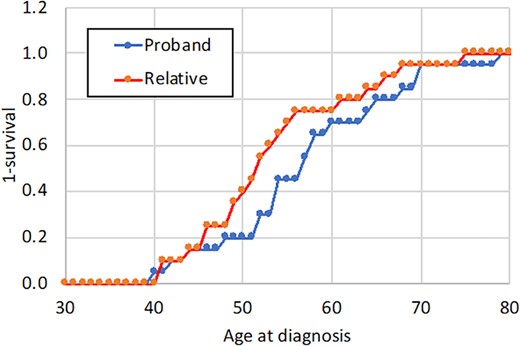
Comparison of age at diagnosis in probands with Paget's disease and their children. The age at clinical diagnosis of Paget's disease in probands is compared with the age at which their children were diagnosed with Paget's disease through radionuclide bone scan imaging in the ZiPP study.
Discussion
Genetic factors play a key role in the pathogenesis of PDB, and it has previously been suggested that genetic testing for SQSTM1 mutations and other susceptibility alleles might be used clinically to detect people with the disease.11 The ZiPP study is unique in that it comprised a large‐scale program of genetic testing for SQSTM1 mutations in both probands and relatives, coupled with the offer of targeted intervention to prevent the development of PDB in relatives that tested positive. To our knowledge the acceptability of genetic testing for susceptibility to PDB has not previously been explored in this way. Our study showed that uptake of the offer of genetic testing was reasonably good in both probands (59%) and relatives (57%), indicating that this approach is acceptable to the target population and could be implemented in clinical practice in the future, should the ZiPP study show evidence of benefit of zoledronic acid in slowing or preventing the emergence of PDB in this high risk group.
Analysis of the baseline data confirmed the relevance of SQSTM1 mutations as a risk factor for PDB reflected by the fact that 20 of 222 (9.0%) of participants in the study already had evidence of early PDB on bone scan at the baseline visit. In most cases, the imaging findings were consistent with mild disease but one individual had already developed a striking deformity of two cervical vertebrae at the time of enrolment into the study (Fig. 2B). Interestingly, this participant, in common with other participants, had not experienced symptoms related to PDB.
Measurements of ALP were performed in all subjects at the baseline visit along with other markers of bone turnover. All biochemical markers of bone turnover that we measured were significantly higher in people with lesions as compared with those without lesions but the sensitivity of these markers in detecting people with lesions was poor. Although previous studies have shown that markers such as ALP, BAP, and P1NP are elevated in a high proportion of individuals with established PDB, most of the participants in this study had very early disease, which could explain why only a modest proportion of people with lesions had elevated marker levels. Whatever the reason it is clear that screening with biomarkers is not an effective method of detecting lesions in SQSTM1 mutation carriers.
Our findings are of interest in relation to the previous study by Bolland and colleagues12 and Cundy and colleagues,13 who screened for evidence of PDB by scintigraphy in the children of individuals from 10 kindreds with familial PDB caused by SQSTM1 mutations. In their initial study four of 23 (17%) offspring with SQSTM1 mutations had evidence of PDB on bone scintigraphy at an average age of 45 years.12 Subsequent follow up of this cohort 5 years later13 revealed the emergence of PDB in two carriers who did not have evidence of the disease at baseline. Another study by Peeters and colleagues14 evaluated 14 SQSTM1 mutation carriers from the Netherlands who were followed for periods of up to 15 years. The design of this study differed from that reported here because scintigrams were only performed in participants who had raised levels of ALP or P1NP or both. Of the eight subjects who underwent scintigraphy, new onset PDB was identified in one case (12.5%). The data presented here show that PDB lesions can be present on scintigraphy even though ALP and P1NP values are normal and so it is possible some of the subjects studied by Peeters and colleagues14 who did not undergo scintigraphy could have had occult lesions in the absence of biochemical abnormalities.
In contrast to the findings reported by Bolland and colleagues12 and Cundy and colleagues13 who used similar methodology to that reported here, we did not observe a delayed onset of PDB in the children of patients who had SQSTM1 mutations. It is important to point out, however, that in this study and previous studies, individuals were actively screened for PDB by radionuclide bone scanning, and it is quite likely that several years may have elapsed before the diagnosis may have been picked up in routine clinical care. We think that the lower proportion of individuals with lesions observed in this study as compared with Bolland and colleagues’ study12 was that we enrolled individuals who tested positive for SQSTM1 mutations irrespective of whether other family members were affected,whereas Bolland and colleagues12 focused exclusively on nuclear families with SQSTM1 mutations.
The present study attests to the fact that PDB is a clinically silent condition in the early stages and may remain so for many years. This is in keeping with the findings of another study which revealed that in about 40% of individuals, PDB first presents with a complication of the disease such as deformity, fracture, or deafness.15 Although nitrogen‐containing bisphosphonates are highly effective at supressing elevated bone turnover in PDB16, 17 and are effective at treating bone pain associated with PDB,18, 19 the PRISM and PRISM‐EZ trials showed that long‐term supression of bone turnover with bisphosphonates carried no advantage over symptom‐directed treatment in preventing complications of PDB such as progression of deafnes, osteoarthritis, quality of life, or pathological fractures.20, 21 The results of these studies provided the impetus for undertaking the ZiPP trial, which seeks to determine if intervening at a very early stage may favorably alter the natural history of the disease.
The ZiPP study has confirmed the importance of SQSTM1 mutations as a genetic risk factor for PDB and has shown that a programme of genetic testing coupled to therapeutic intervention is both acceptable and feasible in the target population. Given that most participants with lesions had early‐stage disease, it is likely that longer‐term follow‐up of this cohort will be required to further investigate the effects of the intervention on symptoms and complications. This is being achieved through the ZiPP–long‐term extension (ZiPP‐LTE) study (NCT03859895), in which participants who completed ZiPP will be offered follow up for a further 5 years.
Disclosures
No potential conflicts of interest were disclosed.
Acknowledgments
This ZiPP study (project reference 09/800/05) was funded by the Efficacy and Mechanism Evaluation (EME) Programme, a MRC and NIHR partnership. The views expressed in this publication are those of the authors and not necessarily those of the MRC, NIHR or the Department of Health and Social Care. The ZiPP study also received financial support from Arthritis Research UK (reference 18163) and non‐financial support from the UK Paget's Association. We thank Novartis for kindly donating the investigational medicinal product and placebo. We acknowledge the contribution of the late Professor Ignac Fogelman to design of the ZiPP study and analysis of the baseline radionuclide bone scans. We also thank the many individuals who took part in the study for their support, the UK Paget's Association for supporting and publicizing the study; the research nurses in study centers and Ms Lynsey Milne from Edinburgh Clinical Trials Unit for her support with data management. The results of this study were presented in part as an abstract at the Annual Meeting of the American Society of Bone and Mineral Research in September 2019.
Authors’ roles: First draft of the manuscript: OC and SHR; study concept and design: SHR; obtaining funding: SHR MP and WDF; trial management during study setup and recruitment: LF, KG and SHR; genetic analysis and training of research staff in genetic counselling: MP and RC; Development of statistical analysis plan and sample size calculations: SCL and CK; design and maintenance of study database: AW; participant recruitment and study assessments: SHR, LF, KG, LRR, PLS, GH, RC, SH, JHT, SAY‐M, MJM, RKC, WDF, JT, LG, RN, M‐LB, JDP‐M, J‐PD, AD, GCI, MDS, JBR, NG, MJS, JPW, MAK, GCN, ELD, GM, AH and NLG; supervision of conduct of the trial. SHR, LF, KG, LRR, PLS, GH, RC, SH, JHT, SAY‐M, MJM, RKC, LG, RN, M‐LB, JDP‐M, J‐PD, AD, GCI, MDS, JBR, NG, MJS, JPW, MAK, GCN, ELD, GM, AH and NLG. All authors commented on and revised the manuscript for intellectual content and approved the final version of the manuscript.
References
Author notes
Current address: Kirsteen Goodman, NMAHP Research Unit, Glasgow Caledonian University, Glasgow, UK.
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4007.

{kind=link}
{kind=link}
{kind=link}