





Abstract
The readthrough of premature termination codon (PTC) by ribosome sometimes produces full-length proteins. We previously reported a readthrough of PTC of glycosyltransferase gene B4GALNT1 with hereditary spastic paraplegia (HSP). Here we featured the readthrough of B4GALNT1 of two mutants, M4 and M2 with PTC by immunoblotting and flow cytometry after transfection of B4GALNT1 cDNAs into cells. Immunoblotting showed a faint band of full-length mutant protein of M4 but not M2 at a similar position with that of wild-type B4GALNT1. AGC sequences at immediately before and after the PTC in M4 were critical for the readthrough. Treatment of cells transfected with mutant M4 cDNA with aminoglycosides resulted in increased readthrough of PTC. Furthermore, treatment of transfectants of mutant M2 cDNA with G418 also resulted in the induction of readthrough of PTC. Both M4 and M2 cDNA transfectants showed increased/induced bands in immunoblotting and GM2 expression in a dose-dependent manner of aminoglycosides. Results of mass spectrometry supported this effect. Here, we showed for the first time the induction and/or enhancement of the readthrough of PTCs of B4GALNT1 by aminoglycoside treatment, suggesting that aminoglycosides are efficient for patients with HSP caused by PTC of B4GALNT1, in which gradual neurological disorders emerged with aging.
Gangliosides, sialic acid-containing glycosphingolipids, have been considered to play important roles in the regulation of nervous tissues (1). Since cDNA of B4GALNT1 responsible for the synthesis of complex gangliosides was isolated, various glycosyltransferase genes were cloned, and their knockout (KO) mice have been generated, leading to advanced understanding of functions of their products (2–4). Complex gangliosides are now considered to be essential for the maintenance and repair of nervous tissues (2).
Although patients having mutations in ganglioside synthase genes had long been not reported, ST3GAL5 deficiency was first reported as a ganglioside synthase deficiency (5). Subsequently, congenital deficiency of B4GALNT1 was reported (6), and similar cases have been reported (7, 8), forming totally 11 family cases of patients with hereditary spastic paraplegia (HSP) based on B4GALNT1 mutation. Clinical features of these patients are similar to those detected in B4galnt1-KO mice (9).
Recently, we analysed the enzyme activity of those mutants, revealing that majority of them except two cases lack the enzyme activity (9). During these investigations, we found that readthrough of a premature termination codon (PTC) occurred in transfectant cells of mutant M4 cDNA of B4GALNT1, in which a termination codon was present at amino acid (AA) 228 due to nonsense mutation.
The other mutant M2 of B4GALNT1 containing a PTC at AA 120 (caused by nonsense mutation) showed no readthrough of the PTC. Here, we demonstrated for the first time that the addition of aminoglycosides to the culture medium of the transfectants resulted in the induction or increase of GM2-expressing cells after transfection of M2 or M4 mutant B4GALNT1 cDNAs, respectively. We discussed on the possibility for the application of these findings in the therapeutics of HSP with PTC of B4GALNT1 that exhibit gradual progression of neurological disorders with ageing.
Materials and Methods
Antibodies and reagents
Anti-GM2 monoclonal antibody (mAb), 10–11 was provided by L.J. Old at Memorial Solan-Kettering Cancer Center (New York). The other antibodies and reagents were purchased as follows: anti-B4GALNT1 mouse mAb (sc-376505) from Santa Cruz Biotechnology (Santa Cruz, CA), fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (H + L) from Cappel (Durham, NC), horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody from Amersham Biosciences, thymidine and gentamicin from Sigma-Aldrich (St. Louis, MO), G418 from Wako Pure Chem. (Osaka, Japan) and KOD–Plus-MutagenesisTM kit from TOYOBO (Osaka, Japan).
Cell lines
A murine melanoma cell line, B78, and a Chinese hamster ovary cell line, CHO were cultured in Dulbecco’s minimal essential medium (DMEM) containing 7.5% foetal calf serum (FCS) and penicillin and streptomycin at 37°C in 5% CO2.
Transfection of expression vectors
B78 cells and CHO cells were plated in 6-cm dishes prior to 24 h of transfection. Next day, cells were transfected with WT or mutant cDNAs of pMIKneo/B4GALNT1 using Lipofectamine 2000TM (Invitrogen, Carlsbad, CA), according to the manufacturer’s instruction.
Flow cytometry
Expression of GM2 on the cell surface was analysed using Accuri C6 CytometerTM (Accuri Flow cytometers Inc., Parkland Plaza, MI) as described (10). Briefly, after 48 h of transfection with cDNAs, cells were trypsinized and used for flow cytometry. Cells were incubated with 100-fold-diluted ascites of anti-GM2 mAb and then stained with FITC-conjugated goat anti-mouse IgG. Cells incubated with secondary antibody alone were used for a control. CELLQuestTM or CFlow PlusTM software was used to analyse GM2 expression.
Immunoblotting
B78 cells were lysed at 48 h after transfection with B4GALNT1 cDNAs using a lysis buffer (20 mM Tris–HCl, 1 mM Na2EDTA, 150 mM NaCl, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin) (Cell Signaling Technology, Danvers, MA) containing 1 mM PMSF and Protease Inhibitor MixtureTM (Calbiochem, San Diego, CA). After preparing cell lysates, SDS-PAGE was performed to separate proteins using 10% gel. Then, separated proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA). After blocking with PBS containing 0.05% Tween 20 and 3% skim milk, the membranes were incubated with 500-fold-diluted anti-B4GALNT1 monoclonal antibody (mouse IgG1 reactive with N-terminus 1-180 AA of B4GALNT1). After washing, the membrane was incubated with HRP-conjugated anti-mouse IgG (1:1000) (Cell Signaling). Protein bands were detected by Amersham Imager 680TM (GE Healthcare Bio-Sciences AB, Tokyo, Japan) using an ImmunoStarTM LD (Wako) luminescence reagent.
Construction of cDNAs of mutant M2 and M4 including those for converting nucleotide C to T in AGC sequences
Expression plasmids of B4GALNT1 cDNA of mutant M2 and M4 were constructed as described (9) by inverse polymerase chain reaction (PCR) using oligonucleotides with KOD-Plus-MutagenesisTM kit (TOYOBO) according to manufacturer’s instructions. These methods were also used to make mutant constructs to convert C to T in AGC sequence of immediate before and after PTC in M4. In brief, template of B4GALNT1 of mutant M4 and oligonucleotides with specific mutation (C to T) were used for inverse PCR. Then Dpn I was added to PCR products for digestion of methylated template cDNA. PCR products were transformed into competent cells after self-ligation with T4 polynucleotide kinase and ligase. Oligonucleotides used for the construction of mutant M4 cDNAs to convert C to T in AGC sequences before and after the termination codon were as follows: before (forward): 5′-CAGCAGTTGAAGCTACCAGACCAAC-3′/(reverse): 5′-TAAG TGACCAGTTGTAGTTGCCTGTTGA G-3′; after (forward): 5′-CTGAAGTTACC AGACCAACACAGCAG-3′/(reverse):5′-CTGCTGTAAGTGACCAGTTGTGTTGC-3′; before and after (forward):5′-AAGTTACCAGACCAACACAGCAGACAC-3′/(reverse):5′-CAACTGCTGTAAGTGACCAGTTGTAGTTGC-3′. Mutated nucleotides were indicated in bold.
Construction of pM2T1-1(M4)/PROTA for producing soluble protein of B4GALNT1 fused with Protein A
For substitution of specific nucleotide at c.682C>T in cDNA of B4GALNT1 fused with Protein A in pM2T1-1(M4)/PROTA was performed using previously constructed pM2T1-1(WT)/PROTA (11) by inverse PCR as described (9) using oligonucleotides with KOD-Plus-MutagenesisTM kit (TOYOBO) according to manufacturer’s instructions. In brief, template of B4GALNT1 of pM2T1-1(WT)/PROTA and oligonucleotides with specific mutation at (c.682C>T) were used for inverse PCR. Then Dpn I was added to PCR products for digestion of methylated template cDNA. PCR products were transformed into competent cells after self-ligation with T4 polynucleotide kinase and ligase. Oligonucleotides used for the construction of mutant M4 with C to T conversion (c.682C>T) were as follows: forward: 5′-CAGCAGTTGAAGCTACCAGACCAAC-3′/reverse: 5′-CTGTAAGTGACCAGTTGT AGTTGCCTG-3′. Mutated nucleotide was indicated in bold.
Expression and purification of soluble M4 or WT B4GALNT1 fused with Protein A
B78 cells were transfected with pM2T1-1 (M4/WT)/PROTA for producing soluble protein of B4GALNT1 (M4 or WT) fused with Protein A using Lipofectamine 2000TM (Invitrogen, Carlsbad, CA), according to the manufacturer’s instruction. After 6 h, transfection medium was replaced by serum-free ITS medium (insulin, transferrin and selenious acid) in the presence or absence of 8 μg/ml G418, and culture medium was collected at days 2–5 after transfection. Collected medium was concentrated to 100-fold with AmiconTM Ultra centrifuge filter (Millipore). A column was prepared using a nonrelevant purified mouse IgG (mAb 220-51 reactive with glycosphingolipid GD2, IgG1 class) conjugated with Sepharose 4BTM (Pharmacia, Uppsala Sweden), according to the manufacturer’s instruction, and packed in a small column. The concentrated medium was applied twice into the column, and unbound proteins were removed from column by washing rigorously with a washing buffer. Bound proteins were eluted, and portions of eluates were applied to SDS-PAGE for Coomassie Brilliant Blue (CBB) staining as well as for immunoblotting of B4GALNT1. Excised portions of gels corresponding to the bands of B4GALNT1 in western blotting were used for mass spectrometry (MS) analysis.
MS analysis
CBB-stained gels were treated by reducing and alkylating reagents after destaining, then served for in-gel digestion by trypsin at 37°C overnight. Peptides were extracted by adding 50% acetonitrile (ACN)/0.1% trifluoroacetic acid (TFA) with shaking, and subsequently by 100% ACN/0.1% TFA and 0.1% TFA. After drying, peptides were solved in 4% ACN/0.1% TFA, and z applied for LC/MS/MS. HPLC was performed by Easy-nLC 1200TM (Thermo Scientific), and mass spectrum was measured by Orbitrap EclipseTM (Thermo Scientific). Data analysis was performed using Proteome Discoverer 2.4 to identify peptides by database search. To examine the possibility of the incorporation of six kinds of AA at the PTC (AA228), supposed mutant AA sequences were added to the database to form ‘Target Mass List’ based on the theoretical m/. Details of this list were presented in Supplementary Material.
DNA sequencing
Sequences of mutant cDNAs were checked with a focus on mutated nucleotides by a sequencer (Applied BiosystemsTM 3130 genetic analyzer, Foster City, CA).
Effects of aminoglycoside treatment on the readthrough
B78 and CHO cells were plated in 6-cm dishes prior to 24 h of transfection. Next day, cells were transfected with B4GALNT1 cDNAs of the mutant M4 or M2, and cultured in the presence or absence of G418 for CHO or B78 cells after transfection. We also cultured these cells in the presence of gentamicin.
Statistics analysis
Data were presented as means ± standard deviation (SD). Student’s t-test was used to analyse statistical significance.
Results
Readthrough of PTC produced full-length active protein of mutant M4 B4GALNT1
Enzyme reactions catalysed by B4GALNT1 were shown in Fig. 1A. Since B78 and CHO cells mainly express GM3 among gangliosides, GM2 expression reflects the enzyme activities in the transfectant cells of B4GALNT1 cDNA. Therefore, we analysed enzyme activity of B4GALNT1 via GM2 expression using flow cytometry after the cDNA transfection. The results revealed that GM2 was expressed in small population in both B78 and CHO cells transfected by mutant M4 B4GALNT1 cDNA compared with those by WT cDNA (Fig. 1B). No peak of GM2 was observed in B78 and CHO cells transfected by mutant M2 B4GALNT1 cDNA. The mean fluorescence intensity of the GM2 peak in M4 mutant transfectants was almost equal to or even higher than that of WT transfectants, suggesting that normal levels of B4GALNT1 enzyme is expressed in some restricted cell population of M4 cDNA transfectants, or the efficiency of the transfection might be different among cDNAs. Some other reasons may also exist, and this point remains to be investigated.
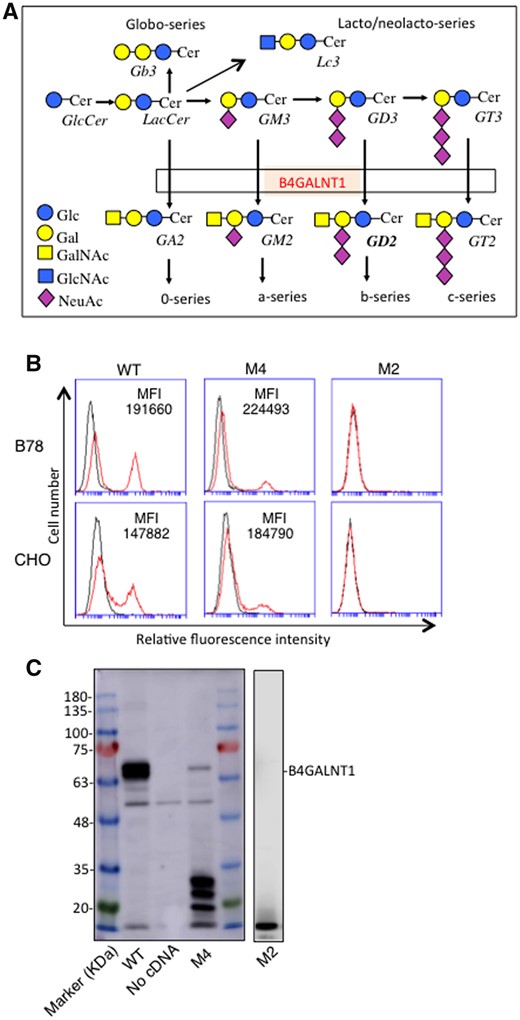
Readthrough of PTC produced full-length B4GALNT1 and resulting product GM2. (A) B4GALNT1 catalyses the transfer of GalNAc to LacCer/GM3/GD3, generating GA2/GM2/GD2, respectively in glycosphingolipid synthetic pathway. (B) Flow cytometry of B78 and CHO cells transiently transfected with cDNAs of B4GALNT1 of WT, mutant M4 or M2. Cells were trypsinized after 48 h of transfection, and incubated with primary antibody. After washing, cells were stained with FITC-conjugated goat antimouse IgG as a secondary antibody. Control cells were prepared with secondary antibody alone. Mean fluorescence intensity (MFI, GM2 peak-negative peak) of each transfectant was shown. (C) Immunoblotting of cell lysates from B78 transfectants with B4GALNT1 cDNAs of WT, mutant M4 or M2. Cell lysates were applied for SDS-PAGE and then blotted onto a PVDF membrane. After blocking, the membranes were incubated with 1:500 fold-diluted anti-B4GALNT1 antibody. Then, the membranes were incubated with HRP-conjugated antimouse IgG (1:2000). Protein bands on the membrane were detected using ImmunoStarTM LD (Wako, Osaka, Japan) luminescence reagent.
Then, we have performed immunoblotting using cell lysates from B78 cells transfected with B4GALNT1 cDNAs of WT, M4 and M2. Immunoblotting revealed B4GALNT1 proteins of WT at ca. 67 kDa and a weak band of B4GALNT1 mutant M4 at a similar size of WT, suggesting it is a product from readthrough of PTC codon. Several low MW bands with stronger intensity were also observed in the lysates from mutant M4 (Fig. 1C). On the other hand, no definite bands were detected in the lysates from M2 mutant (Fig. 1C).
AGC sequence is critical for readthrough in mutant M4
To examine whether the readthrough in M4 depends on AGC sequences immediately before and after the PTC, we constructed cDNAs of B4GALNT1 from M4 by converting the nucleotide from C to T in AGC before or after or both sides of the PTC (Fig. 2A). Then, we transfected these expression vectors into B78, and performed flow cytometry, revealing that the transfectants of nucleotide-modified cDNA from C to T in both sides of the PTC did not produce GM2, and those with C to T change either before or after the PTC showed very low expression (Fig. 2B). These results indicated that the readthrough in M4 mutant depends on the sequences immediately before and after the PTC. The nonsense mutation at nucleotide 358 in M2 leading to conversion from Gln to stop codon at AA120 was shown in Fig. 2C.
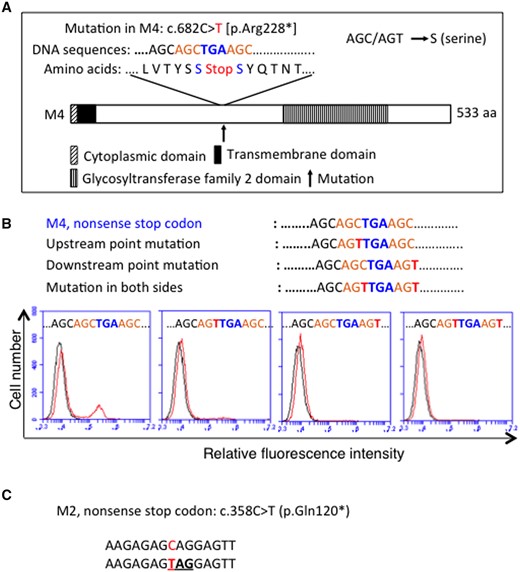
Readthrough of mutant M4 depends on AGC sequence around the PTC. (A) Schematic presentation of B4GALNT1 enzyme of mutant M4, showing nucleotides and amino acids around the PTC, and also a point mutation in the constructed cDNAs by changing C to T in AGC (as AGC/AGT = serine) before and after or both sides of the PTC. (B) At 48 h after transfection into B78 with B4GALNT1 cDNAs, cells were analysed by flow cytometry using anti-GM2 mAb. Expression of GM2 was examined by Accuri C6 CytometerTM. (C) As for M2, CAG was replaced with a stop codon TAG.
Induction and enhancement of the expression of full-length B4GALNT1 proteins and its product GM2 in the transfectants of M2 or M4 cDNA by aminoglycoside treatment
To examine the induction and/or enhancement of readthrough of PTC leading to full-length enzyme protein B4GALNT1 and its product GM2 in the transfectants of M2 or M4 cDNA by aminoglycoside treatment, we performed western blotting and flow cytometry. M2 or M4 transfectants of B78 cells were treated with G418 at 200 μg/ml or 8 μg/ml, respectively after transfection, and lysates of the transfectants were applied for western blotting. As shown in Fig. 3A, the lysates from mutant M2 cDNA transfectants showed a new band at the same level as the WT sample after G418 treatment, although no band could be seen when not treated by G418 (Fig. 3A). As for M4 cDNA transfectants, the G418 treatment resulted in increased intensity of the band. These results suggested that aminoglycosides can induce/enhance B4GALNT1 enzyme activity based on the readthrough of PTC.
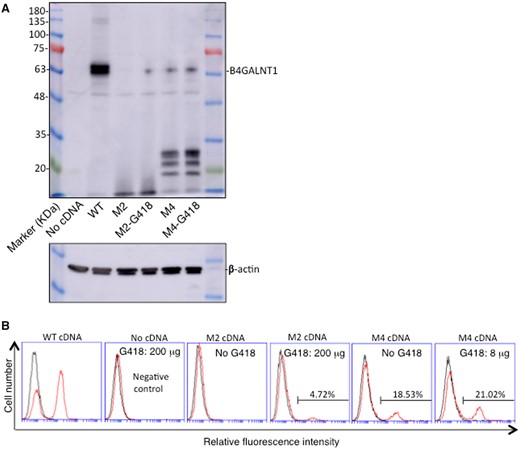
Readthrough of the PTC was induced/increased as full-length proteins of B4GALNT1 and its product GM2 by treatment with aminoglycosides. (A) Immunoblotting of lysates from B78 cells after transfection with B4GALNT1 cDNAs of WT, mutant M4 or M2, and cultured in the presence or absence of G418 (M4: 8 μg/ml and M2: 200 μg/ml) for 48 h after transfection. Cell lysates were applied for SDS-PAGE and then blotted onto a PVDF membrane. After blocking, the membranes were incubated with 1:500 fold-diluted anti-B4GALNT1 antibody, then incubated with HRP-conjugated anti-mouse IgG (1:2000) after washing. Protein bands on the membrane were detected by Amersham Imager 680TM (GE Healthcare Bio-Sciences AB, Tokyo, Japan) using ImmunoStarTM LD (Wako, Osaka, Japan) luminescence reagents. β-actin was examined to confirm equal loading of samples. (B) Flow cytometry was performed using B78 cells transfected with B4GALNT1 cDNAs of WT, mutant M4 or M2, and cultured in the presence or absence of G418 as described in (A). Cells were incubated with the primary antibody, and then stained with FITC-conjugated goat anti-mouse IgG as a secondary antibody after washing. Control cells were prepared with secondary antibody alone. Percent of positive cells in each transfectant was shown.
To analyse whether induced and/or enhanced full-length B4GALNT1 enzyme proteins are functional to produce GM2, flow cytometry was performed using these transfectant cells, revealing that M2 cDNA transfectants newly expressed GM2 after the aminoglycoside treatment (G418: 200 µg/ml), and M4 cDNA transfectants showed increased expression of GM2 by the treatment of aminoglycoside (G418: 8 µg/ml) (Fig. 3B). The treatment of B78 cells by G418 (200 µg/ml) resulted in no expression of GM2 as expected.
Dose-dependent enhancement/induction of readthrough in mutant cDNA transfectants of M4 or M2 by aminoglycosides
To examine whether expression of GM2 increases by aminoglycoside treatment in a dose-dependent manner, CHO cells were transfected with the mutant M4 B4GALNT1 cDNA and cultured with aminoglycoside (G418 at 0–24 µg/ml for 48 h). Results of flow cytometry showed that expression levels of GM2 in CHO cells were increased from 13.4% to 20.8% (average) by treatment with 0–24 µg/ml of G418 (Fig. 4A-b), while transfectants of WT cDNA showed a positive peak of more than 50% (Fig. 4A-c). These results were presented in graphs of Fig. 4B-b. As for B78 cells, B78 transfectants were treated with G418 at 0–8 µg/ml after transfection with a mutant cDNA of M4. Flow cytometry revealed that expression levels of GM2 were also increased from 18.7% to 26.5% after treatment. GM2 expression was also increased from 16.9% to 24.9% after treatment with gentamicin (50–300 µg/ml) (Supplementary Fig. S1A).
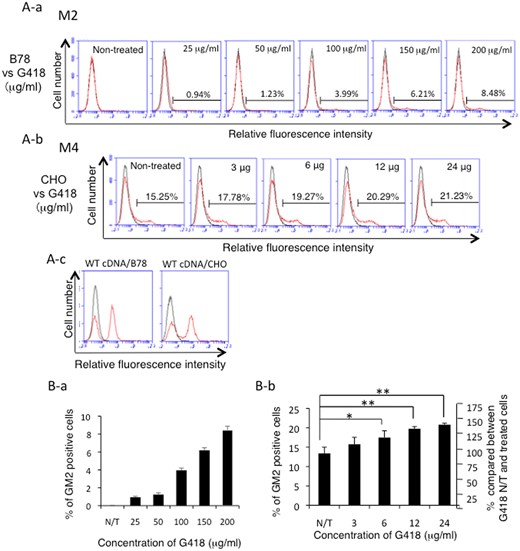
Readthrough of the PTC was induced/increased in a dose-dependent manner by treatment with G418 in M2 or M4 cDNA-transfectant cells. (A) Expression patterns of GM2 as analysed by flow cytometry of B78 cells transfected with mutant M2 cDNA (A-a), or CHO cells transfected with mutant M4 cDNA (A-b). GM2 expression on the transfectants of WT cDNA was also presented for comparison (A-c). After transfection, cells were treated with G418 with various concentrations at 25–200 μg/ml for M2-transfectant cells, and at 3–24 μg/ml for M4 transfectants, and incubated for 48 h. Then, GM2 expression was analysed as described in Fig. 1. Results of representing one of three experiments were shown. (B) Percentages of average GM2-expressing cells from three individual experiments were presented in graphs (B-a, M2 cDNA; B-b, M4 cDNA). *P < 0.05; **P < 0.01.
To examine whether expression of GM2 is also induced dose dependently by aminoglycoside treatment, B78 cells were transfected with the mutant M2 B4GALNT1 cDNA and cultured with G418 (0–200 µg/ml) for 48 h. Flow cytometry revealed that expression of GM2 was induced and increased depending on the dose of aminoglycoside (G418: 25–200 µg/ml) (Fig. 4A-a). These results were presented in the graph of Fig. 4B-a.
During the analysis of the enzyme activity of B4GALNT1 mutant cDNAs, we realized that the intensity of GM2 expression was almost equivalent with that of WT cDNA although GM2+ population was apparently low (Fig. 1). These results suggested that the B4GALNT1 enzyme was expressed in a restricted cell population, or the transfection efficiency was not same among cDNAs. Expression analysis of B4GALNT1 in cell-cycle-modulated cells revealed that S-phase cells might be advantageous for the readthrough of the PTC (Supplementary Fig. S2).
A full-length B4GALNT1 protein derived from M4 mutant cDNA was identified by MS
In order to increase the levels of readthrough M4 product, we treated transfectant cells of M4 cDNA by G418, and purified the fusion protein, ProtA-B4GALNT1 (WT and M4). As shown in Fig. 5A, six kinds of AA can be converted from the nonsense codon at AA228. Immunoblotting of the purified proteins revealed that the fusion proteins purified from culture supernatants contained large amounts of WT B4GALNT1 and very small amount of M4 protein as products of readthrough. From Coomassie-stained gel, –100 kDa portions were excised from both WT and M4-G418 samples and served for MS analysis (Fig. 5B). As shown in Fig. 5A (green zone), MS revealed many peptide fragments derived from ProtA-B4GALNT1 proteins. The cover maps of the identified fragments from WT and M4 mutants were shown in Fig. 5C. Cover rates of them were also shown, i.e. more than 60% in WT and 14–26% of M4-derived protein. Even by Ion Mobility system (FAIMS ProTM) that can identify further minor components, the peptide containing the nonsense mutation site could not be detected, while M4 product showed 26% cover rate. Despite that we tried to focus on possible AA as a replaced one at the stop codon as shown in Fig. 5A by registering supposed sequence information as ‘Target Mass List’ (Supplementary Table S1), it was hard to identify the AA sequence. However, there were many peptide fragments located at C′-terminus side of the nonsense mutation identified by MS (Fig. 5C). These results indicated that mutant M4 mRNA actually underwent readthrough, leading to the expression of functional full-length B4GALNT1 protein(s).
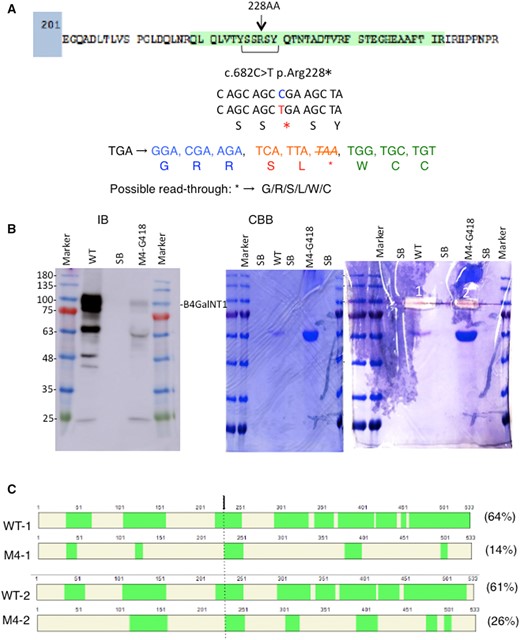
Identification of the readthrough full-length B4GALNT1 protein derived from M4 mutant cDNA. (A) A peptide of B4GALNT1 containing the nonsense mutation at AA228 was shown. Green-coloured sequence is actually identified in MS of WT ProtA-B4GALNT1 fusion protein. This figure shows nonsense mutation at 682nd nucleotide from C to T, and possible readthrough sequences by replacing a new AA with the termination codon, TGA. (B) ProtA-B4GALNT1 fusion proteins consisting of WT or M4 mutated products were purified from culture supernatants and served for western blotting as described in Materials and Methods section. A faint band of M4 protein could be detected, while WT protein was stained with much more intensity. A CBB-stained gel and excision sites (1, 2) were shown. (C) MS analysis was performed by in-gel digestion with trypsin as described in Materials and Methods section. Cover maps of the identified fragments from WT and M4 mutant were presented. Cover rates of the individual samples were also attached. An arrow indicates the nonsense mutation site (AA228). Upper two panels are from measurement by Easy-nLC 1200TM and Orbitrap EclipseTM (Thermo Scientific), and lower two are from Ion Mobility system (FAIMS ProTM) (Thermo Scientific) that can detect further minor components. Note that a fragment immediately following the PTC was identified. Green zones indicate individual peptide fragments identified. Detailed peptide sequences identified are presented in Supplementary Fig. S3.
Furthermore, we found a peptide AA229-252 that located just down-stream of the PTC in both regular Orbitrap Eclipse analysis and also in FAIMS Pro data as shown in the cover map of M4 sample in Fig. 5C. This result strongly suggested that the PTC was replaced with R since trypsin cleaves proteins at C′-side of R or K.
Clinical features of patients with M4 or M2 mutations compared with other mutants
Neurological disorders and abnormalities in clinical examinations in HSP patients were compared between M2, M4 cases and other family cases except M1 (Table I). The onset ages of M4 family are older than those of others, while onset of M2 occurs at similar time points with other groups. In neurological disorders, such as intellectual disorders were milder in M2 and M4 than those in other families. On the other hand, results of ENMG and MRI of brain and spinal cord showed normal ranges in M4, while those in others tended to exhibit features of neuropathy or atrophy. Those in M2 showed approximately intermediate between M4 and others. Details of the clinical features in the individual families were reported in Ref. (9).
Clinical features of M2, M4 and other patients with HSPa
| Onset (y) | Neurological disorders | Examination abnormalities | |
|---|---|---|---|
| M2 | 2/2/3/2 | #Intellectual disability: mild | #ENMG: ND/normal |
| M49/M45/M43/F39 | #Brain and spinal MRI: ND/subcortical periventricular WMH/normal | ||
| c.358C>T (p.Gln120) | |||
| M4 | 9/14 | #Intellectual disability: mild | #ENMG: normal |
| M15/F24 | #Brain and spinal MRI: normal | ||
| c.682C > T (p.Arg228) | |||
| Others (except M1) | 1.6–11 | #Intellectual disability:mod (partly mild) | #ENMG: axonal sensory neuropathy, mild axonal motor neuropathy, axonal sensitive neuropathy, normal |
| #Brain and spinal MRI: ND/mild cortical atrophy, subcortical FLAIR WMH T2, cerebral atrophy, cortical and subcortical atrophy, congenital spinal stenosis, no |
| Onset (y) | Neurological disorders | Examination abnormalities | |
|---|---|---|---|
| M2 | 2/2/3/2 | #Intellectual disability: mild | #ENMG: ND/normal |
| M49/M45/M43/F39 | #Brain and spinal MRI: ND/subcortical periventricular WMH/normal | ||
| c.358C>T (p.Gln120) | |||
| M4 | 9/14 | #Intellectual disability: mild | #ENMG: normal |
| M15/F24 | #Brain and spinal MRI: normal | ||
| c.682C > T (p.Arg228) | |||
| Others (except M1) | 1.6–11 | #Intellectual disability:mod (partly mild) | #ENMG: axonal sensory neuropathy, mild axonal motor neuropathy, axonal sensitive neuropathy, normal |
| #Brain and spinal MRI: ND/mild cortical atrophy, subcortical FLAIR WMH T2, cerebral atrophy, cortical and subcortical atrophy, congenital spinal stenosis, no |
Clinical features of M2, M4 and other patients with HSPa
| Onset (y) | Neurological disorders | Examination abnormalities | |
|---|---|---|---|
| M2 | 2/2/3/2 | #Intellectual disability: mild | #ENMG: ND/normal |
| M49/M45/M43/F39 | #Brain and spinal MRI: ND/subcortical periventricular WMH/normal | ||
| c.358C>T (p.Gln120) | |||
| M4 | 9/14 | #Intellectual disability: mild | #ENMG: normal |
| M15/F24 | #Brain and spinal MRI: normal | ||
| c.682C > T (p.Arg228) | |||
| Others (except M1) | 1.6–11 | #Intellectual disability:mod (partly mild) | #ENMG: axonal sensory neuropathy, mild axonal motor neuropathy, axonal sensitive neuropathy, normal |
| #Brain and spinal MRI: ND/mild cortical atrophy, subcortical FLAIR WMH T2, cerebral atrophy, cortical and subcortical atrophy, congenital spinal stenosis, no |
| Onset (y) | Neurological disorders | Examination abnormalities | |
|---|---|---|---|
| M2 | 2/2/3/2 | #Intellectual disability: mild | #ENMG: ND/normal |
| M49/M45/M43/F39 | #Brain and spinal MRI: ND/subcortical periventricular WMH/normal | ||
| c.358C>T (p.Gln120) | |||
| M4 | 9/14 | #Intellectual disability: mild | #ENMG: normal |
| M15/F24 | #Brain and spinal MRI: normal | ||
| c.682C > T (p.Arg228) | |||
| Others (except M1) | 1.6–11 | #Intellectual disability:mod (partly mild) | #ENMG: axonal sensory neuropathy, mild axonal motor neuropathy, axonal sensitive neuropathy, normal |
| #Brain and spinal MRI: ND/mild cortical atrophy, subcortical FLAIR WMH T2, cerebral atrophy, cortical and subcortical atrophy, congenital spinal stenosis, no |
Discussion
Since KO mice of B4galnt1 were reported (3), no reports on human disease based on the genetic defects of the gene have been reported. Mutations in ST3GAL5 (GM3 synthase) were, however, reported as a hereditary disease of glycolipid synthetic enzymes (5).
HSP constitutes a clinically and genetically heterogenous group of neurodegenerative disorders (12). Major abnormal features exhibited by patients with HSP with mutaions in B4GALNT1 were very similar with phenotypes observed in B4galnt1 KO mice (13, 14). Interestingly, the mutant M4 showed residual enzyme activity in its transfectant cells, while it has a termination codon at upstream of the glycosyltransferase family 2 domain at AA228 (9), leading to the production of a truncated form of enzyme lacking the catalytic domain. As for mutant M2, there was no enzyme activity in its transfectant cells due to the PTC at AA120 located at the stem region of B4GALNT1 enzyme.
All amino acids including the termination of translation are coded by triplets of bases, A, G, T and C. Therefore, the fact that M4 mutant cDNA expressed some levels of B4GALNT1 activity was surprising (9). However, there were some previous reports on ‘readthrough’ events in lower-grade organisms such as virus (15), saccharomyces (16), euplotes (17) and drosophila (18). There were also reports on readthrough of PTC in the human genes involved in several diseases (19, 20), such as Duchenne muscular dystrophy (DMD) (21, 22), mucopolysaccharidosis type I (MPS I) (23), cystic fibrosis (24) and ataxia-telangiectasia (A-T) (25). Studies of efficient stop codon readthrough in mammalian genes indicated that UGA immediately followed by CUAG underwent high levels of readthrough (19). Although M4 or M2 mutations analysed in this study were not the case, it was demonstrated that immediately upstream and downstream of the stop codon are critical for the readthrough (Fig. 2).
The efficiency of translational readthrough depends on a variety of factors, including the identity of the stop codon, the surrounding mRNA sequence context and the presence of stimulating compounds (26). Stop codon readthrough relies on competition between two distinct phenomena, i.e. the recognition of a termination codon by a release factor, eRF1 to trigger the proper termination of translation (27) and accommodation of a near-cognate tRNA in the A-site of the ribosome, leading to erroneous decoding of the stop codon (26). Namely, complex interactions take place between the mRNA and the various components of the translation termination machinery such as eRF1 (27). As reported previously, termination mutations in CFTR were efficiently suppressed by near-cognate tRNA mispairing (28), suggesting that reagents that compromise termination fidelity by alternative base pairing could be used for the therapeutic nonsense suppression (29). Whatever the mechanism, it was strongly suggested that readthrough of the PTC occurred and was enhanced by aminoglycosides in M4 sample as shown in the results of MS as well as of western blotting.
As for effects of aminoglycosides (25) and other compounds (30, 31) on the induction of readthrough of PTCs, a number of studies have been performed (32) using cultured cells from patients carrying PTC mutations (25, 31), and in clinical cases. Gentamicin or PTC124 (ataluren) brought about some effects in DMD cases and Hurler syndrome (mutated MSP I) (23). Since PTC124 selectively induces ribosomal readthrough of premature but not normal termination codons (30), it has been applied for treatment of DMD (22) and cystic fibrosis (24) with time-dependent improvement of clinical signs and/or good tolerability. During the trials of gentamicin-induced readthrough of stop codons in DMD patients, the best termination codon for the readthrough, and the best adjacent sequences around the termination codons were demonstrated (27). The frequency of the inserted amino acids was distinct for specific nonsense codons and readthrough–inducing agents (27, 29).
This is the first report for induction and enhancement of reafthrough of PTC of glycosyltransferase genes by treatment with aminoglycosides. In the case of M4 here, both gentamicin and G418 brought about significant increase of the enzyme activity of B4GALNT1 detected via GM2 expression. As for M2, it seems surprising and encouraging that B4GALNT1 enzyme activity could be induced as GM2 neo-expression from completely null levels. These results indicate that administration of aminoglycosides might be tried not only at earlier stage of life, but also at relatively later stage, since the progression of neurological disorders are slow and age-dependent in HSP patients with B4GALNT-1 mutation (6–8). All clinical features and pathological findings indicated milder and slower progression of neurological disorders in M4 families than majority of other families (Table I) (6–8). Furthermore, M2 families also showed relatively milder clinical features of neurological disorders.
Since genetic mutations in synthetic enzymes of glycosphingolipids have been discovered recently (5–8), there might be more neurological disorders with genetic mutations in other glycosyltransferase genes involved in the synthesis of glycosphingolipids, probably showing relatively mild clinical features with gradual and age-dependent progression (2). Therefore, all or some of those diseases derived from PTC in the individual enzyme genes might be applied for the therapy to induce/enhance readthrough termination codons.
Conclusion
There have been no reports on readthrough of PTCs depending on a specific cell cycle so far. This article should be the first report to indicate the possibility that cell cycle S-phase might be advantageous for readthrough of PTCs whatever the mechanisms are. This finding might be helpful for therapeutics of HSP patients with PTC together with treatment with aminoglycosides. Then, we need to consider efficient administration route and time for these drugs in the future.
Supplementary Data
Supplementary Data are available at JB Online.
Acknowledgements
The authors thank Satoko Yamamoto and Hiroko Kosuge for technical assistance.
Funding
This study was supported by Grants-in-aid for Scientific Research (17K19616, 18H02628 and 19K22518) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).
Conflict of Interest
None declared.
References
Abbreviations
- AA
amino acid
- CAN
acetonitrile
- CBB
Coomassie Brilliant Blue
- DMD
Duchenne muscular dystrophy
- DMEM
Dulbecco’s minimal essential medium
- FCS
foetal calf serum
- GM2
GalNAcβ1,4(Neu5Acα2,3)Galβ1,4Glc-cer
- HSP
hereditary spastic paraplegia
- KO
knockout
- MS
mass spectrometry
- PBS
phosphate-buffered saline
- PTC
premature termination codon
- TFA
trifluoroacetic acid
- WT
wild type.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}