





Abstract
Anxiety and trauma-related disorders are among the most prevalent and disabling medical conditions in the United States, and posttraumatic stress disorder in particular exacts a tremendous public health toll. We examined the tolerability and anxiolytic efficacy of neuropeptide Y administered via an intranasal route in patients with posttraumatic stress disorder.
Twenty-six individuals were randomized in a cross-over, single ascending dose study into 1 of 5 cohorts: 1.4 mg (n=3), 2.8 mg (n=6), 4.6 mg (n=5), 6.8 mg (n=6), and 9.6 mg (n=6). Each individual was dosed with neuropeptide Y or placebo on separate treatment days 1 week apart in random order under double-blind conditions. Assessments were conducted at baseline and following a trauma script symptom provocation procedure subsequent to dosing. Occurrence of adverse events represented the primary tolerability outcome. The difference between treatment conditions on anxiety as measured by the Beck Anxiety Inventory and the State-Trait Anxiety Inventory immediately following the trauma script represented efficacy outcomes.
Twenty-four individuals completed both treatment days. Neuropeptide Y was well tolerated up to and including the highest dose. There was a significant interaction between treatment and dose; higher doses of neuropeptide Y were associated with a greater treatment effect, favoring neuropeptide Y over placebo on Beck Anxiety Inventory score (F1,20=4.95, P=.038). There was no significant interaction for State-Trait Anxiety Inventory score.
Our study suggests that a single dose of neuropeptide Y is well tolerated up to 9.6 mg and may be associated with anxiolytic effects. Future studies exploring the safety and efficacy of neuropeptide Y in stress-related disorders are warranted.
The reported study is registered at: http://clinicaltrials.gov (ID: NCT01533519).
Anxiety and trauma-related disorders are among the most prevalent medical conditions in the United States. Posttraumatic stress disorder (PTSD) in particular is a debilitating disorder that develops in a subset of individuals exposed to extreme stress. Drug discovery for PTSD and anxiety disorders has been largely stagnant, contributing to a substantial disease burden and continued patient suffering. A large body of evidence implicates neuropeptide Y (NPY) in the regulation of stress-related behaviors, and preclinical data suggest that enhancing NPY signaling may reduce anxiety and symptoms of PTSD. Herein, we conducted phase Ib double-blind, randomized, placebo-controlled, dose-ranging study of intranasal administration of NPY in subjects with PTSD. We found that NPY was well tolerated at all tested doses and that high, but not low, doses of NPY were associated with reduced anxiety on some measures. The NPY system may represent a promising target for treatment development for anxiety and trauma-related disorders.
Introduction
Posttraumatic stress disorder (PTSD) is a debilitating disorder that develops in a subset of individuals exposed to extreme psychological stress (Kessler et al., 2005). The disorder is characterized by intrusive reexperiencing of traumatic memories along with symptoms of increased arousal and avoidance of stimuli associated with the trauma (Zelazny and Simms, 2015). Current treatment options are inadequate to meet this large public health challenge. For example, evidence-based psychotherapies lead to clinically meaningful change in one-third to two-thirds of treated patients, leaving many with unremitting symptoms (Steenkamp et al., 2015). Two medications are approved for the treatment of PTSD in the United States (the serotonin selective reuptake inhibitors sertaline and paroxetine), but here too efficacy is modest at best (Berg et al., 2007). It is likely that serotonin selective reuptake inhibitors have limited efficacy in PTSD, because they were chosen for their efficacy in treating mood and anxiety disorders, which share a number of common symptoms with PTSD, rather than for their effects on known trauma-related neurobiological alterations. Currently, there is an urgent need to investigate pharmacologic agents that specifically target the known pathophysiology of PTSD and trauma-related disorders.
An accumulating body of evidence supports the role of neuropeptide Y (NPY) in the regulation of stress and anxiety-related behaviors (Charney, 2004; Wu et al., 2011; Sah and Geracioti, 2013). NPY is the most abundant neuropeptide in the brain and is highly conserved across species (Griebel and Holmes, 2013). Major sites of NPY and NPY receptor expression in the brain include the hypothalamus, locus coeruleus (LC), cerebral cortex, amygdala, and hippocampus (Adrian et al., 1983; Kask et al., 2002a). Rodent studies consistently show that NPY enhances resilience to stress exposure (Cohen et al., 2012, 2015). The effects of NPY are mediated by at least 5 different G-protein coupled receptors (Y1, Y2, Y4, Y5, and Y6), wherein activation of Y1 via NPY or an exogenous ligand is most consistently associated with an anxiolytic effect in animals (for review, see Kautz et al., 2017). In line with preclinical data, studies in humans have demonstrated that high levels of plasma NPY are associated with resilience in the face of extreme psychological stress (Morgan et al., 2000; Russo et al., 2012), while low levels of NPY in plasma and cerebrospinal fluid (CSF) are associated with PTSD (Rasmusson et al., 2000; Yehuda et al., 2006; Sah et al., 2009). NPY is inversely related to PTSD symptoms, with low CSF NPY correlating specifically with the presence of intrusive traumatic memory (Sah et al., 2014) and lower haplotype-driven NPY expression in humans with a heightened amygdala response to threat (Zhou et al., 2008). Taken together, these data support the hypothesis that increasing NPY signaling within the CNS represents a promising therapeutic strategy for PTSD or other stress-related disorders.
Achieving adequate brain exposure is a primary hurdle for the development of neuropeptide-based therapeutics due to the susceptibility of peptides to degradation in the gastrointestinal tract and to lack of transport across the blood brain barrier. Peptide delivery via an intranasal direct nose-to-brain route may be a viable strategy for CNS disorders (Born et al., 2002; Craft, 2012). Preclinical models of direct nose-to-brain delivery indicate the rapid transport of peptides into the CNS via intracellular neuronal olfactory and extracellular trigeminal-associated pathways (Thorne et al., 2004; Jiang et al., 2015). Recently, in the single prolonged stress (SPS) animal model of PTSD, NPY delivered intranasally displayed CNS penetration to key brain regions, including the amygdala and ventral hippocampus (Serova et al., 2013, 2014; Laukova et al., 2014) and prevented stress-triggered dysregulation of the HPA-axis. In human research, CNS penetration administered via an intranasal route has been demonstrated for peptides with molecular weights of comparable size to NPY, including insulin (Born et al., 2002).
Despite substantial preclinical data supporting the anxiolytic properties of NPY, no study to date has investigated the safety or efficacy of nose-to-brain delivery of NPY in humans. Previous studies examining the safety of peripheral administration of NPY in nonpsychiatric populations have generally demonstrated good tolerability (Antonijevic et al., 2000; Held et al., 2006). Using a double-blind, randomized, placebo-controlled, dose-ranging study with 5 cohorts, the current study examined the safety and tolerability of NPY via intranasal administration in subjects with PTSD. The tolerability of NPY was examined across a range of doses that included those with potential efficacy at the upper end based on extrapolation from preclinical models (Serova et al., 2013). The secondary goal was to explore the efficacy of NPY on anxiety and PTSD symptoms as a function of dose.
Methods
Study Participants
Study participants were recruited between January 2014 and January 2016 via clinical referral, newspaper, and internet-based advertisement. All study procedures were conducted at the Icahn School of Medicine at Mount Sinai in New York City. Inclusion criteria for eligible participants included being between the ages of 18 and 65, having a primary diagnosis of PTSD as assessed by the Structured Clinical Interview for DSM-IV-TR (First and Pincus, 2002), and a score of at least 50 on the Clinical-Administered PTSD Scale (Weathers et al., 2001). Exclusion criteria included a lifetime history of psychotic or bipolar disorder, current diagnosis of anorexia nervosa or bulimia, alcohol abuse or dependence in the preceding 3 months, any unstable medical condition, active suicidal or homicidal ideation, or current use of any psychotropic medications. All subjects underwent medical clearance, including a physical examination, clinical laboratory screening (including a complete metabolic panel and complete blood count), urine toxicology testing, and an electrocardiogram. Study participants were free of concomitant psychotropic medications for at least 2 weeks prior to randomization and for the duration of the study. The institutional review board at the Icahn School of Medicine at Mount Sinai approved the study and the clinical trial was registered at http://clinicaltrials.gov (NCT01533519). Written informed consent was obtained from all study participants prior to the conduct of any study procedures.
Study Design
Following screening, study participants were randomized to 1 of 2 treatment orders: NPY-placebo or placebo-NPY in a double-blind crossover design (Figure 1). Each patient received intranasal NPY (Bachem) dissolved in saline or intranasal placebo saline on separate treatment days that occurred 1 week apart. The randomization sequence, treatment assignment, and study drug preparation was performed by the investigational drug service at Mount Sinai. Drug or saline placebo was prepared in identical syringes to conceal treatment identity. All study personnel, except the research pharmacist, were blinded to drug identity and treatment order. We utilized a nasal drug delivery device (Kurve Technology) that was designed to maximize drug transport to the CNS and has been used previously in clinical trials involving direct nose-to-brain delivery of peptides (Craft et al., 2012). The device releases a metered dose into a chamber covering the subject’s nose, which is then inhaled by breathing evenly until the prescribed dose is achieved; an electronic atomizer distributes droplets in a vortical pattern to the upper sinuses for optimized access to the nose-to-brain delivery route via the olfactory epithelium.
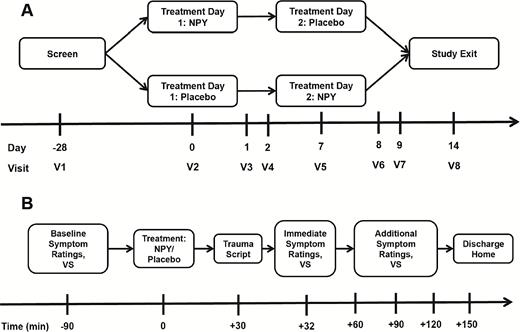
Study flow diagram. (A) Overview of randomized crossover study design. (B) Study procedure timeline occurring on each of 2 treatment days (V2 and V5). NPY, neuropeptide Y; VS, vital signs.
The maximum tolerated dose (MTD) for the current study was operationalized as a protocol-defined dose limiting toxicity (DLT) rate of 20%. DLT was defined as an adverse event or a clinically significant change in vital signs (see supplementary Material for additional details). We employed a combination of a traditional “3+3” dose escalation design (Le Tourneau et al., 2009) and an adaptive Continual Reassessment Method (CRM) (Goodman et al., 1995). Initially, the dose escalation proceeds with cohorts of 3 patients using the 3+3 design; the first cohort is treated at a starting dose that is considered to be safe based on existing literature, and subsequent cohorts are treated at fixed increasing dose levels derived from a modified Fibonacci sequence. Protocol-defined fixed dosing levels were: 1.4 mg, 2.8 mg, 4.6 mg, 6.8 mg, and 9.6 mg. The lower end of the dose range represented an incremental increase from the highest dose previously studied in humans; the number of dose levels and upper end were determined based on study feasibility and designed to include a human dose that may exhibit therapeutic efficacy based on extrapolation from preclinical models (Serova et al., 2013). In this hybrid design, the fixed escalation continues through the dosing levels until the first DLT is observed; subsequent patients are allocated to one of the prespecified dosing levels per a CRM utilizing a Bayesian model (see supplementary Material for additional details). The operating characteristics of our proposed implementation of the CRM were examined by simulation. The simulations, based on 1000 replications of the CRM with 24 patients, produced the probabilities for selecting a given dose as the MTD under different scenarios for dose toxicities. To ensure the robustness of our approach, we assumed starting prior probabilities of toxicity at each respective dose level (1.4 to 9.6 mg) of 5%, 15%, 30%, 50%, and 70%.
Study procedures on each treatment day were as follows. Patients were admitted to a clinical research unit on the morning of each treatment day. Baseline anxiety and PTSD symptom ratings and vital sign measurement were completed on each day (-90 minutes). At time +0, NPY or placebo was administered via nasal delivery device as described above. At +30 minutes, subjects underwent trauma script driven imagery procedures (Pitman et al., 1987) to induce anxiety symptoms. Script driven imagery is an effective method for generating emotional responses, since PTSD patients usually present low symptoms at baseline. The trauma script provocation method (Pitman et al., 1987) was followed immediately by completion of anxiety and PTSD rating scales (+32 minutes). Subsequently, rating scales and vital signs were completed at +60, +90, +120, and +150 minutes. Patients were then discharged home and returned to a research clinic for safety and symptom assessments at +24 hours, +48 hours, and 7 days. See supplementary Material for a detailed description of the trauma script symptom provocation procedure.
Outcome Measures
The primary outcome measure for this study was occurrence of DLTs, and other adverse events, as described above. Rapid anxiolytic efficacy (e.g., immediately following symptom induction subsequent to dosing [+32 minutes]) was secondarily explored using the Beck Anxiety Inventory (BAI) (Steer et al., 1993) and the State-Trait Anxiety Inventory (STAI)–State Form (Spielberger, 1983). PTSD symptoms were explored using the Impact of Event Scale-Revised (IES-R) (Weiss and Marmar, 1996). The IES-R was modified to assess rapid change in PTSD-related symptoms by culling only the 14 items (of total 22) that focus on avoidance, hyper-arousal, and reexperiencing symptoms able to change quickly (IES-R-Rapid). Additional clinician-administered scales were used to explore potential longer term effects of treatment, including the Hamilton Rating Scale for Anxiety and the Montgomery and Asberg Depression Rating Scale (MADRS).
Statistical Analyses
The study sample size was determined via simulations to ensure that at least 3 patients were treated at each dose and that the MTD would be reached with high probability and minimal toxicities, based on a priori estimations of toxicity (see above). The number of patients in early-phase dose-ranging trials is generally small and is not determined by usual statistical considerations. The sample size must be large enough to reliably select an MTD and small enough to be both ethical and feasible. Our sample size of 24 patients was determined by the study statistician (M.P.) to satisfy these constraints. The demographic and clinical characteristics of the study sample, and adverse events are described using summary statistics. We hypothesized greater anxiolytic effects of NPY relative to placebo and that the treatment effect would be larger at higher doses. The influence of treatment on post-provocation anxiety symptoms was examined using a mixed effects model that accounted for the crossover design (Senn, 2002). The model assessed differences in the first post-treatment assessment (+32 minutes) adjusting for period of treatment, treatment order, and baseline value. Variation in treatment effect as a function of dose magnitude was tested using the dose x treatment interaction term, wherein dose was modeled as a continuous variable. We assumed a linear dose effect given the absence of extant knowledge about anxiolytic dose response.
Results
Twenty-six patients with PTSD were randomized and received at least one intranasal administration (either NPY or placebo), representing the analyzed safety set. See supplementary Material for study consort diagram (Figure 1). Two patients elected to discontinue participation, unrelated to adverse events, after the first treatment day and were therefore not crossed over. In one case, the patient discontinued following treatment with NPY; in the other case, the patient discontinued following treatment with placebo. Twenty-four subjects completed the study, representing the analyzed efficacy sample. Table 1 summarizes the clinical and demographic characteristics of the efficacy sample. The mean age of the sample was 39.3 years (SD=13.2) and the proportion of women was 66%. Participants identified themselves predominantly as Caucasian (41%) or African-American (41%).
Demographic and Clinical Characteristics
| Characteristic | |
|---|---|
| Participants treated, n | 24 |
| Age, mean (SD), y | 39.2 (13.0) |
| Female sex, n, % | 16 (66.7) |
| Race, n (%) | |
| African American | 10 (41.7) |
| Caucasian | 10 (41.7) |
| Other | 4 (16.7) |
| Hispanic ethnicity, n (%) | 3 (12.5) |
| Education, n (%) | |
| ≤High school | 2 (8.3) |
| High school graduate | 4 (12.5) |
| Some college or 2-year degree | 7 (29.2) |
| ≥4 years of college or advanced degree | 11 (45.8) |
| Unemployed, n (%) | 11 (45.8) |
| Married or cohabiting, n (%) | 4 (16.7) |
| Primary trauma, n (%) | |
| Domestic violence | 7 (29.2) |
| Unexpected death of a loved one/witnessing violent death | 5 (20.8) |
| Child physical/sexual abuse | 5 (20.8) |
| Physical and/or violent assault as adult | 3 (12.5) |
| Sexual assault/rape as adult | 3 (12.5) |
| Living in war zone | 1 (4.2) |
| Duration of PTSD, mean (SD), y | 12.0 (13.7) |
| CAPS score, mean (SD) | 80.8 (13.7) |
| MADRS score, mean (SD) | 27.8 (7.8) |
| Characteristic | |
|---|---|
| Participants treated, n | 24 |
| Age, mean (SD), y | 39.2 (13.0) |
| Female sex, n, % | 16 (66.7) |
| Race, n (%) | |
| African American | 10 (41.7) |
| Caucasian | 10 (41.7) |
| Other | 4 (16.7) |
| Hispanic ethnicity, n (%) | 3 (12.5) |
| Education, n (%) | |
| ≤High school | 2 (8.3) |
| High school graduate | 4 (12.5) |
| Some college or 2-year degree | 7 (29.2) |
| ≥4 years of college or advanced degree | 11 (45.8) |
| Unemployed, n (%) | 11 (45.8) |
| Married or cohabiting, n (%) | 4 (16.7) |
| Primary trauma, n (%) | |
| Domestic violence | 7 (29.2) |
| Unexpected death of a loved one/witnessing violent death | 5 (20.8) |
| Child physical/sexual abuse | 5 (20.8) |
| Physical and/or violent assault as adult | 3 (12.5) |
| Sexual assault/rape as adult | 3 (12.5) |
| Living in war zone | 1 (4.2) |
| Duration of PTSD, mean (SD), y | 12.0 (13.7) |
| CAPS score, mean (SD) | 80.8 (13.7) |
| MADRS score, mean (SD) | 27.8 (7.8) |
Abbreviations: CAPS, Clinician-Administered PTSD Scale; PTSD, posttraumatic stress disorder; MADRs, Montgomery Asberg Depression Rating Scale.
Demographic and Clinical Characteristics
| Characteristic | |
|---|---|
| Participants treated, n | 24 |
| Age, mean (SD), y | 39.2 (13.0) |
| Female sex, n, % | 16 (66.7) |
| Race, n (%) | |
| African American | 10 (41.7) |
| Caucasian | 10 (41.7) |
| Other | 4 (16.7) |
| Hispanic ethnicity, n (%) | 3 (12.5) |
| Education, n (%) | |
| ≤High school | 2 (8.3) |
| High school graduate | 4 (12.5) |
| Some college or 2-year degree | 7 (29.2) |
| ≥4 years of college or advanced degree | 11 (45.8) |
| Unemployed, n (%) | 11 (45.8) |
| Married or cohabiting, n (%) | 4 (16.7) |
| Primary trauma, n (%) | |
| Domestic violence | 7 (29.2) |
| Unexpected death of a loved one/witnessing violent death | 5 (20.8) |
| Child physical/sexual abuse | 5 (20.8) |
| Physical and/or violent assault as adult | 3 (12.5) |
| Sexual assault/rape as adult | 3 (12.5) |
| Living in war zone | 1 (4.2) |
| Duration of PTSD, mean (SD), y | 12.0 (13.7) |
| CAPS score, mean (SD) | 80.8 (13.7) |
| MADRS score, mean (SD) | 27.8 (7.8) |
| Characteristic | |
|---|---|
| Participants treated, n | 24 |
| Age, mean (SD), y | 39.2 (13.0) |
| Female sex, n, % | 16 (66.7) |
| Race, n (%) | |
| African American | 10 (41.7) |
| Caucasian | 10 (41.7) |
| Other | 4 (16.7) |
| Hispanic ethnicity, n (%) | 3 (12.5) |
| Education, n (%) | |
| ≤High school | 2 (8.3) |
| High school graduate | 4 (12.5) |
| Some college or 2-year degree | 7 (29.2) |
| ≥4 years of college or advanced degree | 11 (45.8) |
| Unemployed, n (%) | 11 (45.8) |
| Married or cohabiting, n (%) | 4 (16.7) |
| Primary trauma, n (%) | |
| Domestic violence | 7 (29.2) |
| Unexpected death of a loved one/witnessing violent death | 5 (20.8) |
| Child physical/sexual abuse | 5 (20.8) |
| Physical and/or violent assault as adult | 3 (12.5) |
| Sexual assault/rape as adult | 3 (12.5) |
| Living in war zone | 1 (4.2) |
| Duration of PTSD, mean (SD), y | 12.0 (13.7) |
| CAPS score, mean (SD) | 80.8 (13.7) |
| MADRS score, mean (SD) | 27.8 (7.8) |
Abbreviations: CAPS, Clinician-Administered PTSD Scale; PTSD, posttraumatic stress disorder; MADRs, Montgomery Asberg Depression Rating Scale.
Safety and Tolerability
The first cohort (n=3) received 1.4 mg of NPY; with no occurrence of a DLT, the study proceeded to the second cohort. The second cohort received 2.8 mg of NPY, wherein one patient experienced a DLT consisting of a decrease in heart rate >20% compared with baseline and an absolute rate of <60 beats/min. Per protocol, this DLT occurrence triggered the CRM, and subjects were allocated to dosing levels sequentially as follows: 2.8 mg (n=6), 4.6 mg (n=5), 6.8 mg (n=6), and 9.6 mg (n=6). Subsequent to the initial DLT, which resolved without sequelae, no other DLTs occurred throughout the study. Table 2 summarizes adverse events experienced by patients.
Adverse Events
| Side Effect | Pooled placebo (n=25) | Pooled active (n=25) | 1.4 mg (n=3) | 2.8 mg (n=5) | 4.6 mg (n=5) | 6.8 mg (n=6) | 9.6 mg (n=6) |
|---|---|---|---|---|---|---|---|
| n(%) | n(%) | n(%) | n(%) | n(%) | n(%) | n(%) | |
| Autonomic | |||||||
| Tachycardia | 1 (4%) | 0 (0%) | – | – | – | – | |
| Bradycardia | 0 (0%) | 1 (4%) | – | 1 (4%) | – | – | – |
| Chest tightness | 2 (8%) | 0 (0%) | – | – | – | – | – |
| CNS/psychiatric | |||||||
| Headache | 2 (8%) | 1 (4%) | – | 1 (4%) | – | – | – |
| Dizziness | 3 (12%) | 0 (0%) | – | – | – | – | – |
| Irritability | 0 (0%) | 1 (4%) | – | – | 1 (4%) | – | – |
| Lightheadedness | 3 (12%) | 0 (0%) | – | – | – | – | – |
| Blurry vision | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Fatigue | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Sleepiness | 2 (8%) | 0 (0%) | – | – | – | – | – |
| Gastrointestinal | |||||||
| Nausea | 0 (0%) | 1 (4%) | 1 (4%) | – | – | – | – |
| Skin disorders | |||||||
| Itchiness | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Other | |||||||
| Flu-like symptoms | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Fever | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Nasal congestion | 0 (0%) | 1 (4%) | – | – | – | – | 1 (4%) |
| Side Effect | Pooled placebo (n=25) | Pooled active (n=25) | 1.4 mg (n=3) | 2.8 mg (n=5) | 4.6 mg (n=5) | 6.8 mg (n=6) | 9.6 mg (n=6) |
|---|---|---|---|---|---|---|---|
| n(%) | n(%) | n(%) | n(%) | n(%) | n(%) | n(%) | |
| Autonomic | |||||||
| Tachycardia | 1 (4%) | 0 (0%) | – | – | – | – | |
| Bradycardia | 0 (0%) | 1 (4%) | – | 1 (4%) | – | – | – |
| Chest tightness | 2 (8%) | 0 (0%) | – | – | – | – | – |
| CNS/psychiatric | |||||||
| Headache | 2 (8%) | 1 (4%) | – | 1 (4%) | – | – | – |
| Dizziness | 3 (12%) | 0 (0%) | – | – | – | – | – |
| Irritability | 0 (0%) | 1 (4%) | – | – | 1 (4%) | – | – |
| Lightheadedness | 3 (12%) | 0 (0%) | – | – | – | – | – |
| Blurry vision | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Fatigue | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Sleepiness | 2 (8%) | 0 (0%) | – | – | – | – | – |
| Gastrointestinal | |||||||
| Nausea | 0 (0%) | 1 (4%) | 1 (4%) | – | – | – | – |
| Skin disorders | |||||||
| Itchiness | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Other | |||||||
| Flu-like symptoms | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Fever | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Nasal congestion | 0 (0%) | 1 (4%) | – | – | – | – | 1 (4%) |
n=24 subjects overlapped in both the pooled placebo and pooled active groups, but one subject dropped out after participating in a placebo condition and did not complete an active condition, and another subject dropped out after participating in an active condition and did not complete a placebo condition.
Number of subjects experiencing an adverse event: only events rated “possibly,” “probably,” or “definitely” related to drug were included.
Adverse Events
| Side Effect | Pooled placebo (n=25) | Pooled active (n=25) | 1.4 mg (n=3) | 2.8 mg (n=5) | 4.6 mg (n=5) | 6.8 mg (n=6) | 9.6 mg (n=6) |
|---|---|---|---|---|---|---|---|
| n(%) | n(%) | n(%) | n(%) | n(%) | n(%) | n(%) | |
| Autonomic | |||||||
| Tachycardia | 1 (4%) | 0 (0%) | – | – | – | – | |
| Bradycardia | 0 (0%) | 1 (4%) | – | 1 (4%) | – | – | – |
| Chest tightness | 2 (8%) | 0 (0%) | – | – | – | – | – |
| CNS/psychiatric | |||||||
| Headache | 2 (8%) | 1 (4%) | – | 1 (4%) | – | – | – |
| Dizziness | 3 (12%) | 0 (0%) | – | – | – | – | – |
| Irritability | 0 (0%) | 1 (4%) | – | – | 1 (4%) | – | – |
| Lightheadedness | 3 (12%) | 0 (0%) | – | – | – | – | – |
| Blurry vision | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Fatigue | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Sleepiness | 2 (8%) | 0 (0%) | – | – | – | – | – |
| Gastrointestinal | |||||||
| Nausea | 0 (0%) | 1 (4%) | 1 (4%) | – | – | – | – |
| Skin disorders | |||||||
| Itchiness | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Other | |||||||
| Flu-like symptoms | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Fever | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Nasal congestion | 0 (0%) | 1 (4%) | – | – | – | – | 1 (4%) |
| Side Effect | Pooled placebo (n=25) | Pooled active (n=25) | 1.4 mg (n=3) | 2.8 mg (n=5) | 4.6 mg (n=5) | 6.8 mg (n=6) | 9.6 mg (n=6) |
|---|---|---|---|---|---|---|---|
| n(%) | n(%) | n(%) | n(%) | n(%) | n(%) | n(%) | |
| Autonomic | |||||||
| Tachycardia | 1 (4%) | 0 (0%) | – | – | – | – | |
| Bradycardia | 0 (0%) | 1 (4%) | – | 1 (4%) | – | – | – |
| Chest tightness | 2 (8%) | 0 (0%) | – | – | – | – | – |
| CNS/psychiatric | |||||||
| Headache | 2 (8%) | 1 (4%) | – | 1 (4%) | – | – | – |
| Dizziness | 3 (12%) | 0 (0%) | – | – | – | – | – |
| Irritability | 0 (0%) | 1 (4%) | – | – | 1 (4%) | – | – |
| Lightheadedness | 3 (12%) | 0 (0%) | – | – | – | – | – |
| Blurry vision | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Fatigue | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Sleepiness | 2 (8%) | 0 (0%) | – | – | – | – | – |
| Gastrointestinal | |||||||
| Nausea | 0 (0%) | 1 (4%) | 1 (4%) | – | – | – | – |
| Skin disorders | |||||||
| Itchiness | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Other | |||||||
| Flu-like symptoms | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Fever | 1 (4%) | 0 (0%) | – | – | – | – | – |
| Nasal congestion | 0 (0%) | 1 (4%) | – | – | – | – | 1 (4%) |
n=24 subjects overlapped in both the pooled placebo and pooled active groups, but one subject dropped out after participating in a placebo condition and did not complete an active condition, and another subject dropped out after participating in an active condition and did not complete a placebo condition.
Number of subjects experiencing an adverse event: only events rated “possibly,” “probably,” or “definitely” related to drug were included.
Therapeutic Efficacy
There was no main effect of treatment on BAI. There was a significant interaction between treatment and dose on BAI score immediately following symptom provocation (+32-minute time point; slope = -1.92, SD=0.86; F1,20=4.95, P=.038), such that at higher dose levels the magnitude of the difference between NPY and placebo was larger (Figure 2). Specifically, for every 1-mg increase in dose, the benefit of NPY over placebo on average was approximately 2 points on the BAI (95%CI: -0.12 – -3.73). Posthoc testing showed no significant effect of treatment at any given dose of NPY (n=3 to n=5 per dosing group). There was no evidence of a carry-over effect of treatment on BAI score (t21=0.20, P=.84). While the pattern of the dose-response relationship was similar for the STAI and the IES-R, the dose-response relationship was not statistically significant (Figure 2). There was no significant effect of treatment or dose on anxiety measures examined following the +32-minute time point on the day of treatment (supplementary Material, supplementary Figure 2).
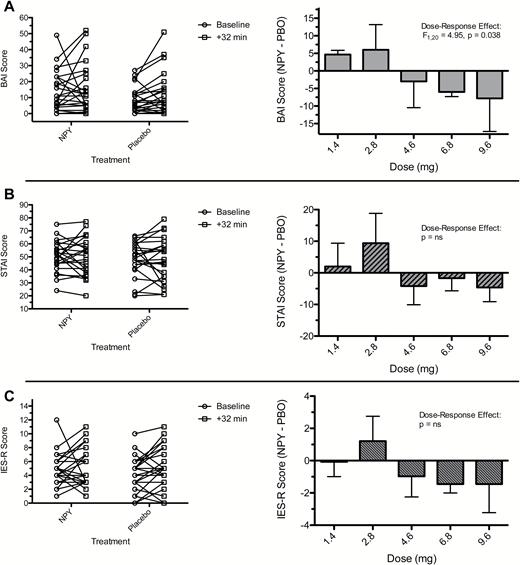
Effect of neuropeptide Y (NPY) treatment and dose on anxiety following symptom provocation in patients with posttraumatic stress disorder (PTSD). Figure displays observed values for anxiety measures before and immediately following a symptom provocation procedure (+32-minute time point) in subjects under NPY and placebo conditions (left column). Mean differences between NPY and placebo in change in symptom measure with associated SEs for each dose of NPY are also displayed (right column). In the case of summarized change scores depicted in the right column, the change in symptom severity (post minus baseline) is compared between NPY and placebo (PBO) as a difference score (NPY minus PBO). Therefore, larger numbers represent greater symptom severity, and negative difference scores indicate less anxiety symptoms under NPY relative to PBO. The dose-response effect was tested using an interaction term from a linear mixed model that adjusted for treatment order and baseline severity. (A) Beck Anxiety Inventory (BAI). (B) State-Trait Anxiety Inventory-State Form (STAI-State). (C) Impact of Events Scale-Revised (IES-R).
Analyses of the longer term outcomes (i.e., beyond the treatment day; +24 hours, +48 hours, and 7 days), including Hamilton Rating Scale for Anxiety and MADRS, showed no difference between the treatment conditions (supplementary Material, supplementary Figure 3).
Discussion
Herein we report the results of a phase Ib dose-ranging study of intranasally administered NPY in patients with PTSD. We observed that NPY was generally well tolerated up to and including the highest studied dose of 9.6 mg. We did not identify the MTD for intranasal NPY, defined in the current study as the dose associated with a toxicity rate of 20%. We found preliminary evidence for a dose-response relationship of NPY on anxiety symptoms, with each 1-mg increase in NPY dose conferring an additional benefit of NPY relative to placebo of 2 points on the BAI. Other measures of anxiety and PTSD symptoms showed a similar but nonsignificant trend. In the case of the STAI, there was on average minimal increase in score following the trauma script, indicating that this measure was not sensitive to the symptom provocation procedure in this study. This first ascending dose study of NPY in patients with PTSD shows that doses up to 9.6 mg are safe and that doses at the upper, but not lower, end of the range examined may be associated with anxiolytic effects.
A large body of preclinical work supports the hypothesis that NPY plays a key role in modulating the stress response, and NPY signaling is associated with resilience to stress and with anxiolytic effects (Thorsell et al., 2002; Heilig, 2004; Wu et al., 2011). NPY counteracts the anxiogenic effect of corticotropin-releasing hormone, which increases in response to stress (Smialowska et al., 2007; Reichmann and Holzer, 2016), and anxiolytic responses are observed following systemic or intraventricular administration of NPY (Kask et al., 2002b), or following viral vector-induced overexpression of NPY in the amygdala (Primeaux et al., 2005). Conversely, NPY knockout mice show a pro-anxiety phenotype (Bannon et al., 2000). Studies involving patients with PTSD show alterations in both peripheral and central NPY levels. For example, NPY levels in CSF were lower in veterans with combat-related PTSD relative to healthy controls (Sah et al., 2009). Another study found lower NPY levels in exposed veterans with PTSD compared with those without PTSD (Sah et al., 2014). NPY levels were higher in special forces soldiers compared with non-special forces soldiers, and these levels negatively associated with dissociation during an interrogation exercise (Morgan et al., 2000).
Most recently, preclinical studies showed that intranasally administered NPY can attenuate or reverse the detrimental effects of stress in the context of the SPS model of PTSD (Serova et al., 2013). Serova et al. initially reported that treatment with NPY via an intranasal route achieved CSF concentrations within a range previously associated with anxiolytic effects; administration either prior to the SPS or immediately following it lessened the subsequent development of anxiety-like behavior (Serova et al., 2014). Intranasal NPY also blocked the stress-induced upregulation of tyrosine hydroxylase in the LC observed under control conditions. In a follow-up study, the same group showed that intranasal NPY could reverse an anxiety phenotype following SPS when given 1 week later, which has important implications for NPY as a potential treatment for PTSD (Serova et al., 2014). Critically, they demonstrated using fluorescent-labeled NPY that intranasally administered NPY travels to multiple brain sites involved in the stress response, including the amygdala, hypothalamus, hippocampus, and LC within 30 minutes of treatment (Sabban et al., 2016).
Despite compelling animal and human data, strategies to enhance NPY signaling as a therapeutic strategy for PTSD or other stress-related disorder (e.g., depression) have not been developed. Due to the connection between the external environment and the CNS by the olfactory neural pathways, direct nose-to-brain transport is a therapeutically viable method of drug delivery (Misra and Kher, 2012). The passage is dependent on molecular weight, polarity, and lipophilicity and includes the advantage of a reduction of effects on other body organs and possibly a reduction in unwanted adverse effects. An initial proof-of-concept study investigated the nose-to-brain delivery of vasopressin (40 and 80 IU), melanocortin (5 and 10 mg), and insulin (40 IU) and found increased concentrations of these peptides in the CSF (Born et al., 2002), adding weight to the feasibility of intranasal delivery of therapeutics. In the current study, we utilized a nasal drug delivery device that was designed specifically for nose-to-brain transport, which had been used previously in clinical trials involving direct nose-to-brain delivery of insulin in Alzheimer’s disease (Craft et al., 2012). Our starting dose was selected based on the highest dose previously administered to human participants; the specific dosing levels and overall dose range reflected standard aspects of dose escalation designs and the feasibility of what could be accomplished within a reasonable timeframe (i.e., no more than 5 dosing levels were used).
Several prior studies have examined peripherally administered NPY in humans. In one study, NPY or saline was administered via 4 bolus i.v. injections (50 or 100 µg) to healthy male volunteers in whom NPY was shown to improve sleep and inhibit adrenocorticotropic hormone (ACTH) and cortisol release (Antonijevic et al., 2000). In a study conducted in both depressed patients and healthy controls, Held et al. (Held et al., 2006) reported that NPY increased prolactin release, decreased sleep onset latency and REM latency, and improved subjective quality and deepness of sleep. A small number of studies have been conducted using intranasal NPY in humans (Baraniuk et al., 1992; Lacroix and Mosimann, 1996; Cervin et al., 1999); only one was concerned with its possible effects on the brain (Hallschmid et al., 2003, 2004). Hallschmid et al. used intranasal NPY (50 nmol) to study brain mechanisms involved in feeding behavior. NPY reduced cortical activity in response to food intake, thereby possibly inhibiting satiation. NPY reduced the decrease in subjective thirst but not hunger following food intake. No side effects were reported (Hallschmid et al., 2003, 2004).
The current study has several limitations. The primary limitation of the study is the sample size. The study does not have sufficient power to provide conclusive information concerning clinical efficacy. In particular, there is a risk of a type I error, since small studies may be biased towards finding large effects. Conversely, there is a risk of not detecting a true effect given the limited sample size. For example, in the current study we found a significant dose-by-treatment interaction for BAI but not for STAI or IES-R, and it is unknown if this lack of significance represents a true effect or a type II error. Also owing to a small sample size, we were not powered to detect an effect of treatment within a given dose cohort, as each cohort included no more than five individuals. The limited sample size also precludes stratifying analyses by trauma type. Because we did not escalate the dose beyond 9.6 mg, the MTD for NPY delivered via intranasal exposure remains unknown. While the current study was not designed to escalate beyond 9.6 mg, future studies will be required to examine the tolerability and efficacy of higher doses. Peripheral NPY levels are not available for analysis, precluding pharmacokinetic or pharmacokinetic-pharmacodynamic modeling. Prior studies show, however, that peripheral levels of NPY are unaffected by intranasal administration (Serova et al., 2013) and that peripheral and central levels may not be correlated (Baker et al., 2013). In most preclinical studies, unless the peptide is radio-tagged, it is difficult to say with certainty whether the intranasal delivery results in direct increases in brain uptake or an increase of endogenous peptides within the CNS. This study did not include any central measure of NPY. Therefore, it is not known if NPY delivered in this study reached its target in the CNS. Future studies that employ neuroimaging or other CNS measures will be critical to establish NPY target engagement. The observed anxiolytic effects of NPY at the higher doses in this study may be taken as an initial efficacy signal and proxy for CNS activity, although the small sample size and inherent exploratory nature of these analyses serve to limit the interpretation.
In conclusion, the current study found that doses of NPY up to 9.6 mg delivered via an intranasal route were safe and well tolerated. There was a significant dose-response effect of NPY on reduction of BAI scores. Additional studies exploring the safety, efficacy, and pharmacodynamics of NPY in humans are warranted. Future studies with chronic dosing of NPY that also employ neuroimaging or other CNS measures will be critical to establish NPY target engagement.
Supplementary Material
Supplementary data are available at International Journal of Neuropsychopharmacology online.
Funding
This work was supported by the Icahn School of Medicine at Mount Sinai, with additional support provided by the National Center for PTSD and the VA CT Healthcare System.
Statement of interest
In the past 3 years, Dr. Murrough has provided consultation services for Novartis, Janssen Research and Development, Genentech, ProPhase, and Impel Neuropharma. Drs. Murrough and Charney are named on a patent pending for NPY as a treatment for mood and anxiety disorders as well as on patents pending for lithium to extend the antidepressant effect of ketamine and ketamine plus lithium as a treatment for suicidal ideation. In the past 5 years, Dr. Iosifescu was a consultant for Avanir, Axsome, CNS Response, INSYS Therapeutics, Lundbeck, Otsuka, Servier, and Sunovion; he has received research support (through the Icahn School of Medicine at Mount Sinai) from Alkermes, Astra Zeneca, Brainsway, Euthymics, Neosync, Roche, and Shire, and he has received speaker honoraria from the Massachusetts General Hospital Psychiatry Academy, Medscape, and Global Medical Education. Dr. Collins has served as a consultant for ProPhase. Dr. Feder is named co-inventor with Dr. Charney (Dean of Icahn School of Medicine at Mount Sinai [ISMMS]) and ISMMS on a use patent application filed by Mount Sinai for the use of ketamine as a treatment for PTSD (patent pending). Dr. Charney also reports issued patents for ketamine for the treatment of depression and ketamine as a treatment for suicidal ideation. Dr. Iacoviello provided consultation services to and reports equity ownership in Click Therapeutics, Inc and is a full-time employee of Click Therapeutics. Drs. Iacoviello and Charney are named inventors on a patent pending for a cognitive-emotional training intervention for major depressive disorder. Dr. Feder’s research is funded by CDC/NIOSH and the Brain and Behavior Research Foundation. All other authors report no conflicts of interest.
Acknowledgments
We thank the Icahn School of Medicine at Mount Sinai research pharmacists, including Ivy Cohen, Alla Khodzhayeva, and Giuseppe Difiore, for their extensive work on this project. We thank Dr. Deborah Ascheim for her assistance in protocol development and adverse event monitoring and Drs. Scott Orr and Roger Pitman for their contribution to the study script driven imagery paradigm. Finally, we wish to thank all of patients who volunteered to participate in the study.
References
Author notes
D.S.C. and J.W.M. contributed equally to this work.

{kind=link}
{kind=link}