





Abstract
In the past few years, several high profiled pharmaceutical companies have decided to shut down major research activities within the central nervous system (CNS) area. For example, in December 2011 Novartis announced that the company is closing its neuroscience facility in Basel, Switzerland, where Novartis is headquartered (Abbott, 2011). It follows similar moves by GlaxoSmithKline and AstraZeneca, both based in the UK, which in 2010 announced the closure of major parts of their neuroscience research divisions globally (Jack, Financial Times, 4 February 2010). Also companies primarily based in the USA, Pfizer and Merck, as well as the French company Sanofi, have pulled back on research into brain disorders. This development is still proceeding, as e.g. AstraZeneca closed their CNS/pain centres (Fiercebiotech, press release, 2 February 2012). Several of the companies have launched smaller new initiatives based on studies of genetics and biomarkers, but as mental disorders such as unipolar depression impose the largest disease burden worldwide, e.g. 6.2% disability-adjusted life year of total (WHO, 2008), and current treatments do not work particularly well for many patients, this has obviously raised a number of concerns related to how the future developments should be carried out, and whether the genetic approach may be sufficient. In June 2012, the International College of Neuropsychopharmacology (http://www.cinp.org) hosted an international workshop in order to discuss and consider the consequences and implications of the withdrawal of these research activities. This paper presents the problem background together with a summary of the viewpoints of the invited speakers and recommendations for future intervention.
Introduction
In the past few years, several high profile pharmaceutical companies, based worldwide, have decided to shut down major research activities within the neuroscience area. The withdrawal of pharmaceutical companies from neuroscience research is a serious concern to the whole community, including researchers, clinicians, patients and scientific organizations, such as the International College of Neuropsychopharmacology (CINP). The major reasons are that the development of new psychotherapeutic drugs has become more and more a high-risk activity with only little success. On the other hand, mental disorders such as, for example, unipolar depression will impose the largest disease burden worldwide in the year 2030 (WHO, 2008) and current treatments do not work particularly well or no really new effective drugs are available, bringing the whole field into a crisis.
The major question is why is the research for psychotherapeutic drugs so unprofitable? At first look this seems not plausible, as each year 38.2% of the EU population suffers from a mental disorder (Wittchen et al., 2011) opening an enormous market for pharmaceuticals. Adjusted for age and co-morbidity, this corresponds to 164.8 million persons affected. The most frequent disorders are anxiety disorders (14.0%), insomnia (7.0%), major depression (6.9%), somatoform disorders (6.3%), alcohol and drug dependence (>4%), attention deficit hyperactivity disorder in the young (5%) and dementia (1–30%, depending on age). Wittchen et al. (2011) concluded that every year over one-third of the total EU population suffers from mental disorders and that the size including neurological disorders is even considerably larger. Disorders of the brain are the largest contributor to the all-cause morbidity burden as measured by disability-adjusted life years in the EU. Obviously, concerted priority action is therefore needed at all levels of basic, clinical and public health research in order to identify better strategies for improved prevention and treatment for brain disorders (Wittchen et al., 2011). Additionally, a new worldwide consortium of researchers, advocates and clinicians supported by the WHO or the NIMH (the grand challenges in global mental health initiative; Collins et al., 2011) announced research priorities for improving the lives of people with mental illness around the world and calls for urgent action and investment. Bearing all this in mind, major concerns arise from the knowledge that leading world pharmaceutical companies are not as much focused on these areas as in previous years, dashing the hopes of millions of patients for new and more effective drugs in the near future.
Hence, the major point is the dramatic increase of costs. As seen in Fig. 1, the cost of drug development during the last decade has been subject to double-digit increases while the rate of production of new drugs remains low and is falling (Munos, 2009). Moreover, with the development and opportunity costs of each new drug, as well as the revenues from highly profitable drugs due to patent expiration, more and more companies reduce their involvement in drug discovery for central nervous system (CNS) diseases (Munos, 2009). CNS development takes time and there is a very short time to recoup expenditure. Of 5000 potential candidate molecules, only 250 reach the pre-clinical stage. Of the 250 reaching the pre-clinical stage, 10 reach clinical development. Of the 10 molecules entering clinical development, only one will be approved by the regulatory authorities and get to the market. The time from first molecule development to drug on the market is typically 12–15 yr. The cost of developing a new drug is typically US$10–15 billion. Moreover, when CNS drugs fail, they tend to do so in late-stage clinical trials, after a significant investment has been made. This is obviously of great concern to the companies for these types of pharmaceuticals as costs for developing new drugs continue to increase. This, of course, puts pressure on healthcare systems. The probability of success is relatively low and the cost of attaining success is disproportionately high.
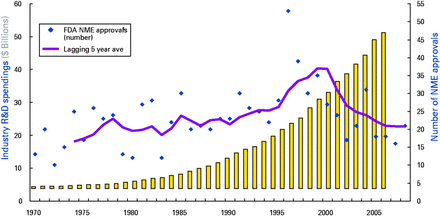
R&D costs and number of registered new molecular entities (NMEs). The costs of R&D (yellow bars) rise in an exponential-like fashion, whereas the number of FDA NME approvals declines. The figure is coloured and reprinted from FDA (2008), under FDA copyright policy. Lagging 5 year average includes new chemical entities (NCEs) and licensed products (BLAs). BLAs included 1986 onward; biologic approvals in prior years assumed negligible. Sources: NME data for 1966–1971 from Peltzman (1973). NME data for 1972–1979 as reported in Hutt (1982). NME data for 1980–2007 from Parexel's Pharma R&D Statistical Sourcebook 2009/2010, FDA and PhRMA. Industry R&D spend data from PhRMA Annual Membership Survey, 2008 and Parexel 2009/2010.
To learn more about the current problems that the companies are dealing with and to discuss and consider the consequences and implications of withdrawal of the research activities, CINP hosted an international workshop in June 2012 (see Appendix 1 for workshop participants). In this paper, we will summarize the discussions that took place and present some recommendations for the field.
Detailed view on the new development
Of course we cannot provide any comprehensive analysis of current drug discovery development as the market is highly complex and several different factors have to be kept in mind. Therefore, we will only raise some main points and discuss them in the concert of opinions from the delegates of our workshop.
The problem of the right target
Before finding a new drug target, e.g. a molecular structure involved in the pathology of interest, it is important to define the pathological phenotype as precisely as possible. Diagnosis was and is a key function and central to develop a plan of treatment for the patients. Psychiatry, however, faces special challenges. The underlying pathology of psychiatric disorders is not known for the majority of conditions. For the most part, the clinician must rely on reports from, and direct observations of, patients to gather the necessary information to determine a diagnosis. Even diagnostic information found in medical records may at some time-points not be useful, since one cannot be certain whether the historical record diagnoses of previous conditions were based on the application of similar diagnostic approaches or even the same system of classification. Obviously, uncertainty and poor reproducibility may affect treatment outcome and response. The current diagnostic system was not designed to facilitate biological differentiation and it does not. Furthermore, due to new technological efforts, such as genome-wide association studies and an exponential increase in new knowledge, current disease classifications become more and more questionable making them not ideal as end-points for target action. Additionally, the DSM-5 classification system, which will be presented in 2013, will rebuild to a certain extent the current nosology of psychiatric diseases (Kupfer and Regier, 2011), although it is unfortunately still not the time for a really new taxonomy or classification of diseases based on molecular fingerprints.
Having more or less unspecific phenotypes has of course a major impact on the success of testing new drugs. Therefore, it is not surprising that pharmaceutical companies are struggling with this as a major problem. What can be done?
Biomarkers
Most of our current clinical trials are based on a descriptive psychiatry of the last decades as well as on, for example, rating scales (Hyman, 2012). This is not satisfactory as objective diagnostic tests and treatment responsive biomarkers are lacking. Although there are several cerebrospinal fluid and/or blood biomarkers that might be used for a more specific target in drug discovery, only some more or less valid markers exist, e.g. in Alzheimer's disease (Hampel et al., 2010). Given the high complexity of the brain – including levels of analysis from molecules to cells, synapses, circuits, and thence to higher cognition, regulation of emotion and executive function – valid biomarkers are highly needed (Hyman, 2012). Science has expanded dramatically during the last decade including the use of new high-throughput methods involving epigenetics, transcriptomics, metabolomics and/or protein × protein interactions, etc. Metabolomics, for instance, can provide valuable information about disease pathogenesis and result in metabolic signatures that could be developed as biomarkers for disease and progression. Comparative studies in plasma could help map peripheral changes in metabolism in patients and enable a more accessible way for biomarker development. The observation that not all patients correct their aberrant metabolic profiles upon treatment suggests that metabolic profiling could be used as an additional tool to complement clinical evaluation in defining drug response phenotypes and variation in response to therapy. Pharmacometabolomics is emerging as a new field that could complement pharmacogenomics by providing precise intermediate phenotypes for drug response. Metabolomics could add significantly to our understanding of both pharmacokinetic and pharmacodynamic properties of drugs. Furthermore, proteomics is a highly innovative tool to measure, for example, hormones, cytokines/chemokines, acute phase reactants, clotting proteins, growth factors, tissue modelling factors and other typical plasma proteins in a high-throughput approach. All these massive screening approaches, which contribute to novel dimensions in disease understanding, make it hard to understand why pharmaceutical companies abandon so many channels.
Endophenotypes
Apart from biomarkers, validated endophenotypes could serve as good starting points for drug research. One intriguing observation derived from genome-wide association studies on inflammatory and metabolic diseases is that there exists a surprisingly high amount of pleiotropy (a gene influences multiple phenotypic traits) and that genes associated with a physiological intermediate phenotype related to the disease (e.g. fasting plasma glucose and insulin response) also are associated with the disease (e.g. diabetes; Lyssenko et al., 2009). Thus, intermediate phenotypes have emerged as a most valuable tool in the search for the genetic underpinning of complex traits and diseases. Clinically defined diseases (e.g. stroke) or behaviours can be regarded as the sum of many risk factors, which can be decomposed into intermediate phenotypes (e.g. lipid levels in the case of stroke and sensory-gating deficits for schizophrenia). There is hope that factors contributing to intermediate phenotypes may be easier to identify because of the improved signal:noise ratio in the fraction of variance explained by any single factor. Studies using a single clinical end-point (e.g. stroke) have one chance of success, whereas studies that also collect intermediate phenotypes are more likely to help us to understand the contribution of factors to characteristic components of disease or behaviour. Therefore, simpler, quantifiable measures of, for example, neuropsychiatric functioning may be of additional usefulness in gene and/or drug discovery. Intermediate phenotypes refer explicitly to core pathophysiological phenomena that bridge the gap between genetic variation and the biologic systems underlying behavioural disturbance. Intermediates can be regarded as syntheses of subsets of proximal risk factors, both environmental and genetic, as shown by Carlson et al. (2004). The rationale for the use of intermediate phenotypes in drug discovery is that the intermediate phenotypes associated with, for example, a behavioural trait are more elementary and less heterogeneous compared to clinical phenotypes. But not all phenotypes that differ between patients and controls are intermediate phenotypes. Several criteria should carefully be achieved to define an intermediate phenotype of a disease (Chan and Gottesman, 2008). For drug research, it is highly important to change study designs incorporating endophenotypes in this manner, to work on approved and specific targets.
The problem of the right samples
Developing new CNS drugs is high risk because there is a relatively poor responder vs. non-responder ratio and a high failure rate, increasing the need for large patient groups when doing clinical trials. Since the definition of diseases is based on symptoms, not on biology, there is a disconnect between the discovery of new drugs and the definition of the patients we treat. Consequently, rarely >30–60% of a patient population responds to a given treatment. Should this area remain attractive for the pharmaceutical industry, the balance needed to drive innovation, i.e. the balance between risk/investment/time and profit, should be redefined and optimized.
Patient stratification may help to decrease both risk and investment, as the responder vs. the non-responder ratio will improve and also contribute to decreased investment with the target of small, specific patient groups. Stratified medicine is working in several areas, as exemplified by several examples from systemic medicine. It does not seem necessary to settle all nosological debates before a pragmatic ‘stratified psychiatry’ can become reality and empirical advances can be made before mechanisms are fully understood. In addition, with the use of molecular well-defined patient groups having a link between drug target and molecular fingerprint, trial outcome may be improved, which may reduce the time needed for drug approval. Smaller clinical trials, based on the above, may potentially shorten the time to registration. This approach will lead to a major cost saving to society and a better and more objective benefit/risk evaluation. Similarly, a new taxonomy/classification of diseases, linking symptoms and biology may provide new targets, gives a better chance for positive efficacy. The overall result is more objective benefit/risk calculations, which may improve profit.
To change the balance between risk/investment/time and profit, a redefinition of the current roles and procedures can be advised. Central in the efforts are novel tools to further stratify the patients (‘personalized medicine’) and also a reshaping of the incentive systems should occur.
Another significant problem is the high and increasing placebo response rates in modern clinical trials. This is problematic not only from a lay-person perspective, with all drugs seemingly useless, impairing the therapeutic alliance between the patient and the psychiatrist, but also from a drug development perspective. The question of why the placebo response rates are so high is of great significance. The problem can be understood within the frame of the diagnostic problems, as highlighted above, but a number of factors may also be relevant when examining the nature of modern trials (Brody et al., 2011). A proportion of the increased placebo effects may be attributable to the broadening of inclusion criteria, i.e. subjects who are less depressed may be more likely to respond to placebos. Clinical trials have become longer in duration, giving room for non-specific factors to arise. The industry-sponsored trials, paying investigators or contract research organizations incrementally by subject, may ‘encourage’ the raters to ‘inflate’ symptoms preferentially at screening visits over symptoms at midtrial visits. Incremental payments also discourage careful screening for exclusion criteria. Finally, the ‘professional patient’ may be incentivized to participate by the cash payments and this may lead the subjects to exaggerate their symptoms. In addition, these symptomatic volunteers may also have participated in other trials, which may potentially conflict with the outcomes.
The problem of translation from animals to humans
A further major point for drug companies to reduce their engagement in neuroscience is the observation that promising effects of novel compounds in animals do not reliably predict efficacious effects in patients. This lack of translation from animal models to clinical efficacy makes it difficult to generate human models of psychiatric disease that provide a good concept for expensive clinical trials in patients. Indeed, a major issue is the use of appropriate animal models. Acute pharmacological models, for example, using PCP, ketamine or similar, are not valid models and they sum up little of the complex development of the disorders. Another major issue is the availability of appropriate screening tools. For example, the ‘forced swim test’, where the time is taken at which time-point the rat stops struggling and gives up, is a standard measurement to detect antidepressant efficacy. But this represents only a single test and the translation from animals to humans is not so easy. It is often observed that molecules work very well in animal models, but when transferring to patients, they do not. Thus, most of the model tests lack the precision and sophistication required to model complex behavioural disorders such as, for example, depression and schizophrenia.
Thus, what is needed is to develop animal models that more closely mimic specific dimensions of psychopathology. This could be developmental interventions (e.g. perinatal/prenatal/early life insults) leading to emergence of symptoms later in the animal's life, which may be more accurate for modelling the disease and its treatment. Specific psychiatric constructs can be assessed using cross-species measures in non-verbal tasks. Such measures are closer to biological and genetic substrates and are more likely to allow better prediction from animal to human experiments. Translational science requires a bidirectional dialogue to develop more quantitative non-verbal measures in humans that are suitable for parallel studies in animals.
The problem of study design
The accepted standard by which the usefulness of a therapeutic treatment is judged has been the double-blind, randomized, controlled trial. The major premise is standardization and blindness, enabling a truly unbiased and objective judgement of the optimal therapy. However, while this is correct in principle, there is a number of issues that are problematic when recommending therapy, such as the right exclusion criteria, recruitment bias, use of additional drugs, etc.
For example, over the past few years, several criticisms occurred regarding standards used by industry to assess the premarket safety of prescription drugs, the industry's conduct of phase IV studies required at the time of product approval or the agency's ability to monitor the use of pharmaceutical products in the marketplace (Kaitin and DiMasi, 2011). The common study design comprises the discovery phase, then the preclinical part and afterwards phases I, II, III before the drug is brought to the market. But this pipeline does not work well for mental disorders as clinical development (phases I–III) accounts for approximately 63% of the costs (53%, phase II), and preclinical drug discovery accounts for only 32% (Paul et al., 2010). Paul et al. have described the need for a new paradigm for drug development based on ‘fast fail’, i.e. focusing on proof-of-concept prior to proceeding to expensive phase III trials. The essence of this approach is validating a target in humans and demonstrating how a new compound engages this target before launching a large-scale clinical trial (Insel, 2012). Therefore, new strategies, including experimental medicine, target validation and testing of involved mechanisms, are necessary. This leads quickly to patients to determine if a lead compound target is effective and success can be defined by ‘fast fail’. Drug repurposing is an even faster way forward by identifying new beneficial effects of approved medications (Insel, 2012).
Pharmaceutical products will be based more and more on a network of innovative stakeholders – including large and small pharmaceutical and biotechnology companies, academic research centres, contract research organizations, public–private partnerships and patient groups (Kaitin and DiMasi, 2011). To do so, the pharmaceutical industry needs to undergo a deep shift in its strategies, such as developing functional outsourcing relationships.
The problem of increasing costs: new ways of collaboration with academia
The most important point for a company is a balance between investment and profit. Every company is struggling to reduce costs and to improve research and development productivity to deal with these challenging times. As costs for drug research increase substantially, it is not surprising that pharmaceutical companies are eliminating or downsizing unprofitable parts. On the other hand, our knowledge of biomedical science has improved substantially over the past decade, but this does not match with real drug development in pharmaceutical companies.
However, there are programmes and ideas to deal with this gap. Precompetitive consortia backed by industry have continued to rise. Most initiatives are focused on developing better disease models and discovering new biomarkers. There are several outsourcing strategies that involve contracting biotech companies and academic researchers to do some of the early drug-discovery work that was previously done in-house. The driving force behind this is that it is too expensive for a single company to solve highly complex issues in neuroscience. Thus, this is a new starting point in collaborations between the pharmaceutical industry and academia. This could bring a new level of shared partnerships in the scientific process, from basic research to clinical testing. Several pharmaceutical companies have started to open new smaller research divisions to study basic mechanisms in collaboration with investigators in academia so as to run research jointly. These are very promising developments especially based on major discoveries in genetics or neuroscience. Given the complexity of neuroscience large-scale, new approaches to data collection and analysis are recommended. They will be necessary to understand heterogeneous, polygenic disorders and will require broad collaboration and sharing of tools and data. Furthermore, some pharmaceutical companies have formed new groups to create an open innovative initiative to reposition drugs and collaborate with anybody and everybody in the outside world. There is a need to overcome research bottlenecks in drug development through collaborative approaches, creating a new culture for accelerating this translation based on standardization, integration and sharing of data through new partnerships (Insel, 2012).
Outlook
In order to meet the demands listed in the WHO (2008) report, a continued effort for CNS drug development is highly needed. This can be done through different sources such as industry, academia, private–public partnerships, government, advocacy groups or private citizens, to mention only a few. In addition, publicity and media issues are especially important to overcome the stigmatization of mental diseases. So how can we handle all these different initiatives, which are ongoing worldwide, and what is the role of large societies of neuropsychopharmacology such as the American, the European or International Colleges of Neuropsychopharmacology, to mention only some of them? They could play an important role in the relationship between industry, academia and the public to develop a real platform to exchange knowledge. Meetings like the workshop organized by the CINP in June 2012 can help to get a better understanding of current developments and to stimulate dialogues between the different partners. CINP will therefore continue with these kinds of workshops, reaching out to all essential parties.
Acknowledgements
G.W. is supported by the Danish Medical Research Council, and Aarhus University Research Foundation.
Statement of Interest
None.
Appendix 1 Names of the workshop participants
Jeff Conn (USA), Jonas Eberhard (Lundbeck), Alan Frazer (USA), Guy Goodwin (UK), Anthony Grace (USA), Hsien-Yuan Lane (Taiwan), Elisabeth Mocaer (Servier), Anthony Phillips (Canada), Dan Rujescu (Germany), Barbara J. Sahakian (UK), Torgny Svensson (Sweden), Gustavo Turecki (Canada), John Waddington (Ireland), Gregers Wegener (Denmark) Shigheto Yamawaki (Japan), Trevor Young (Canada), Joseph Zohar (Israel), Peter Høngaard Andersen (Lundbeck), Brian Dean (Australia), Bjarke Ebert (Lundbeck), Lars Farde (Sweden), Mark Geyer (USA), Shitij Kapur (UK), Siegfried Kasper (Austria), Theo Meert (Janssen), Hans-Jürgen Möller (Germany), Charles Nemeroff (USA), Rainer Rupprecht (Germany), Connie Sanchez (Lundbeck), Armin Szegedi (Merck), Mark Tricklebank (Eli Lilly), Daniel Umbricht (Roche), Eric Wong (USA).
References

{kind=link}