





Abstract
Studies examining the epigenetic effects of nicotine are limited, but indicate that nicotine can promote a transcriptionally permissive chromatin environment by increasing acetylation of histone H3 and H4. To further explore nicotine-induced histone modifications, we measured histone methyltransferase (HMT) mRNA expression as well as total and promoter-specific H3K9me2 levels. Following administration of nicotine, HMT mRNA and H3K9me2 levels were examined in mouse primary cortical neuronal culture and cortex extracted from mice injected intraperitoneally, as well as in human lymphocyte culture. Furthermore, Bdnf/BDNF mRNA levels were examined as an epigenetically regulated read-out of gene expression. There was a significant decrease of the HMT GLP, G9a and Setdb1 mRNA expression in the nicotine-treated tissue examined, with significant decreases seen in both total and promoter-specific H3K9me2 levels. Increasing doses of nicotine resulted in significant decreases in Bdnf/BDNF promoter specific H3K9me2 binding, leading to enhanced Bdnf/BDNF transcription. Taken together, our data suggest that nicotine reduces markers of a restrictive epigenomic state, thereby leading to a more permissive epigenomic environment.
Introduction
Epigenetic modifications result in protein assemblies that are commonly described as ‘permissive’ or ‘restrictive’. ‘Permissive’ assemblies, or euchromatin, (acetylation of histones) leave exposed the naked DNA sequences, permitting interaction with DNA-binding proteins as a prelude to gene regulation. Transcriptionally repressive assemblies, or heterochromatin, (methylation of specific histones, DNA methylation) effectively seal the gene promoter from interaction with transcription factors and transcriptional machinery (Oberdoerffer & Sinclair, 2007). Di-methylation of the ninth lysine on the n-terminus tail of histone H3 (H3K9me2) is a signature modification of repressed chromatin and is catalysed by the actions of highly conserved and dedicated histone methyltransferases (HMT) such as GLP, G9a and Setdb1 (Volkel & Angrand, 2007). The interdependent G9a/GLP dimer is responsible for the bulk of H3K9me2 modifications across the genome (Shinkai & Tachibana, 2011). In mammals, loss of GLP, G9a or Setdb1 protein can result in embryonic lethality, reduced H3K9 di-methylation and inappropriate gene transcription (Tachibana et al.2005).
Nicotine is the primary psychoactive component of tobacco and acts as an agonist at ligand-gated ionotropic nicotinic acetylcholine receptors (nAChR). nAChR have a pentameric structure, which includes a heteromeric stoichiometric arrangement of five different subunits (α2-α7, α9, α10 and β2-β4) or homomeric receptors predominantly composed of the α7 subunit. Data from binding studies reveal the existence of a high-affinity nicotine-binding site in brain, predominantly composed of α4 and β2 subunits, and a low-affinity binding site that is mostly the homomeric α7 subunit (Barik & Wonnacott, 2009). Stimulation of nAChR induce Ca2+, K+ and Na+ influx, and in the nervous system has been shown to facilitate GABAergic, glutamatergic, serotonergic, cholinergic and dopaminergic transmission (Dani & de Biasi, 2001; Wonnacott et al.2005). Three different tissues were examined in this paper, two neuronal models and one marker of the periphery, the lymphocyte. Neuronal cultured cells are primarily GABAergic (Noh et al.2005), thus express α7 nAChR (Burli et al.2010). Additionally, studying the effect of nicotine on epigenetic regulation in the lymphocyte is possible through identified nAChR on the cell surface (Fujii et al.2008; Hiemke et al.1996) specifically the homomeric α7 nAChR, which is activated by nicotine (de Rosa et al.2005; Sato et al.1999) and α4β2 (Benhammou et al.2000), therefore serving as a viable model to study the epigenetic effects of circulating nicotine levels.
Although the studies examining the epigenetic effects of nicotine are limited, all data suggest its role in contributing to a more permissive epigenomic state. In HL-60 cells, acute nicotine treatment results in increases of histone H3 acetylation, a transcriptionally permissive chromatin modification. This effect is inhibited by mecamylamine, a non-competitive antagonist of nAChR. Through this blocking effect of pretreatment with mecamylamine, it was shown that the epigenomic-modifying effects of nicotine are through direct action on nAChR (Landais et al.2005). Furthermore, in mice, chronic 7-d treatment of nicotine increases levels of acetylated histone H3 and H4 in total striatum extracts and at the promoter regions of FosB, an immediate early gene (Levine et al.2011). Chronic nicotine treatment also results in down-regulation of DNA methyltransferase (DNMT) expression, thereby decreasing DNA methylation and increasing cortical and hippocampal mRNA expression in mice. Again, these effects are inhibited by mecamylamine (Satta et al.2008). Additionally, nicotine administration results in up-regulation of genes that have been shown to be epigenetically modified, specifically brain-derived neurotrophic factor (Bdnf/BDNF; Kenny et al.2000; Son & Winzer-Serhan, 2009).
Previous literature indicates a chromatin-relaxing role of nicotine through increases in histone acetylation and decreases in DNA methylation. Whether or not nicotine also alters the expression of restrictive histone modifications, such as H3K9me2, remains to be addressed. We hypothesize that nicotine will reduce histone methyltransferase mRNA, enzymes associated with restrictive chromatin modifications. By decreasing these enzymes, levels of the restrictive modification, H3K9me2 will also decrease both globally as well as locally at Bdnf/BDNF promoter regions, resulting in consequent increases in Bdnf/BDNF mRNA gene expression.
Method
Mouse primary cortical neuronal culture
Primary cortical neurons were dissected from E13 Harlan ICR outbred mice. Cells were plated at an average of one foetus per pre-coated poly-d-lysine six-well plate in minimal growth medium (Gibco 21103-049) with B27 supplement (Gibco 17504-044), l-glutamine and gentamycin. Proliferation of non-neuronal cells was halted by adding cytosine β-d-arabinofuranoside (Sigma C-1768) at 3 d in vitro (Noh & Gwag, 1997).
Mouse treatment
Adult male Swiss albino mice were given a single acute dose of 3 mg/kg (−)-nicotine hydrogen tartrate salt (Sigma N-5260, Sigma, USA), via i.p. injections 6 h before killing (Schreiber et al.2002; Stolerman et al.1973). Nicotine was dissolved in a volume of saline solution corresponding to 0.1 ml/10 g body weight. Vehicle mice were injected with a saline solution. All animals were anaesthetized via CO2 followed by cervical dislocation. All animal use was approved by the Animal Care and Use Committee of the University of Illinois at Chicago, College of Medicine.
Lymphocyte cell culture
Lymphocyte extraction was performed using techniques as previously described (Gavin et al.2009a; Jayaraman et al.1999). Briefly, 80 ml blood was collected in tubes rinsed with 0.5 m EDTA, pH 8.0 and extracted using the Ficoll–Paque gradient method (GE 17-1440-02). Cells were cultured at a concentration of 1 × 106 cells/ml in complete media consisting of RPMI 1640, penicillin/streptomycin, l-glutamine, sodium pyruvate, non-essential amino acids, and foetal bovine serum, and incubated in 5% CO2 at 37 °C. The study was approved by the Institutional Review Board of the University of Illinois at Chicago, College of Medicine. Lymphocytes were collected from non-smoking healthy male subjects between the ages of 18–65 yr.
Cell culture treatment
Both in-vitro models (primary cortical neurons and lymphocytes) were incubated with vehicle (DMSO) or increasing concentrations of (−)-nicotine hydrogen tartrate salt (Sigma N-5260) added to culture media at a concentration of 1:1000, 6 h before harvest. All doses are expressed in terms of the salt, not nicotine base. Nicotine base is roughly equivalent to one-third of the nicotine tartrate salt weight (Schreiber et al.2002). Furthermore, concentrations examined in this paper are within the range noted in other published work (Calabresi et al.1989; Fang et al.1991; Misbahuddin et al.1985). In human lymphocytes, cells were pretreated with 10 µm mecamylamine, a non-selective and non-competitive antagonist of nicotinic acetylcholine receptors, at a concentration of 1:1000, for 7 h before harvest (Bose et al.2005; Cattaneo et al.1993).
Cell viability was measured both at treatment and harvest via cell counts obtained from a representative well, per condition, following 5 min incubation in a media/Trypan blue mixture, which stains non-viable cells. Conditions that exhibited a significant amount of cell loss were excluded from data analysis (Bach & Bach, 1972; Choi et al.1987; Dawson et al.1991). A significant amount of cell loss was deemed >90% decrease of viable cells between pretreatment and before harvest (Gavin et al.1974).
mRNA extraction
Total RNA from all tissue examined was isolated using the TRIzol reagent method (Life Technologies 15596-026; Life Techologies Corporation, USA; Mannhalter et al.2000).
Real-time polymerase chain reaction (PCR) quantification
Total RNA was converted to cDNA using the Applied Biosystems (USA) High Capacity Archive Kit (4368813). For detection and measurement of expression, Fermentas Maxima SYBR Green/ROX qPCR Master Mix (K0222; Fermentas International Inc., Canada) was used. PCR mixtures were run on a Stratagene (USA) Mx3005P™ QPCR System. Primers were designed to cross over one intron to amplify only cDNA and yielding an amplicon of between 75–200 base pairs. Dissociation curves were conducted to establish the presence of a single amplicon at the predicted melting temperature and a lack of primer–dimer formation. A comparative threshold cycle (CT) validation experiment was done to determine target and reference primer efficiency. CT value was used for relative quantification of target gene expression and normalized to GAPDH. Fold-changes relative to vehicle condition were calculated as ΔCT (Livak & Schmittgen, 2001; Schmittgen & Livak, 2008). Primer sequences are listed in Table 1, with mouse primers (mm) used with primary neuronal culture and mouse cortex extracts and human primers (h) used on lymphocyte cultures. Additionally, mRNA primers and Chromatin Immunoprecipitation primers are listed separately.
Primers used in polymerase chain reactions
| Primer name/location | Sequence |
|---|---|
| mRNA expression primers | |
| hGLP 5′ + 1801 | 5′ TCCTGGCTGTGGCTACTTCT 3′ |
| hGLP 3′ + 1875 | 5′ AAACGGTGAGAGATGCTGCT 3′ |
| hG9a 5′ + 610 | 5′ TTCCGCATGAGTGATGATGT 3′ |
| hG9a 3′ + 745 | 5′ TCGTCAGGGTCACTTCTCCT 3′ |
| hSetdb1 5′ + 463 | 5′ AAGACCAGAAGCTCCGTGAA 3′ |
| hSetdb1 3′ + 561 | 5′ CCTGGGAACTGCTCTTCTTG 3′ |
| hBDNFIXabcd 5′ | 5′ AACCTTGACCCTGCAGAATG 3′ |
| hBDNFIXabcd 3′ | 5′ TGGTCATCACTCTTCTCACCTG 3′ |
| hGAPDH 3′ + 340 | 5′ CGAGATCCCTCCAAAATCAA 3′ |
| hGAPDH 5′ + 509 | 5′ TTCACACCCATGACGAACAT 3′ |
| mmGLP 5′ + 3569 | 5′ ATTGACGCTCGGTTCTATGG 3′ |
| mmGLP 3′ + 3799 | 5′ ACACTTGGAAGACCCACACC 3′ |
| mmG9a 5′ + 3112 | 5′ TGCCTATGTGGTCAGCTCAG 3′ |
| mmG9a 3′ + 3247 | 5′ GGTTCTTGCAGCTTCTCCAG 3′ |
| mmSetdb1 5′ + 794 | 5′ GATTCTGGGCAAGAAGAGGA 3′ |
| mmSetdb1 3′ + 989 | 5′ GTACTTGGCCACCACTCGAC 3′ |
| mmBdnfI 5′ | 5′ GTCTTCTGTAGTCGCCAAGGTGG 3′ |
| mmBdnfI 3′ | 5′ GCACACCTGGGTAGGCCAAG 3′ |
| mmBdnfIV 5′ | 5′ GTCTTCTGTAGTCGCCAAGGTGG 3′ |
| mmBdnfIV 3′ | 5′ TCGCTGAAGGCGTGCGAGTA 3′ |
| mmBdnfIXA 5′ | 5′ GCAGCTGGAGTGGATCAGTAA 3′ |
| mmBdnfIXA 3′ | 5′ TGGTCATCACTCTTCTCACCTG 3′ |
| mmGapdh 3′ + 13 | 5′ ACGGCCGCATCTTCTTGTGCAGTG 3′ |
| mmGapdh 5′ + 238 | 5′ GGCCTTGACTGTGCCGTTGAATTT 3′ |
| Chromatin immunoprecipitation primers | |
| hBDNFIXabcd 5′ | 5′ AAACATCCGAGGACAAGGTG 3′ |
| hBDNFIXabcd 3′ | 5′ TTCGAAAGTGTCAGCCAATG 3′ |
| mmBdnfI 5′ | 5′ CAAAATAGGGCAGCGACTCT 3′ |
| mmBdnfI 3′ | 5′ CTGAGCGAAAAGGTGTAGGC 3′ |
| mmBdnfIV 5′ | 5′ CCCTGGAACGGAATTCTTCT 3′ |
| mmBdnfIV 3′ | 5′ AGTCCTCTCCTCGGTGAATG 3′ |
| mmBdnfIXA 5′ | 5′ CATGAGACCGGGCAAGTC 3′ |
| mmBdnfIXA 3′ | 5′ CCTTGGGAGGAATGTGTGAT 3′ |
| Primer name/location | Sequence |
|---|---|
| mRNA expression primers | |
| hGLP 5′ + 1801 | 5′ TCCTGGCTGTGGCTACTTCT 3′ |
| hGLP 3′ + 1875 | 5′ AAACGGTGAGAGATGCTGCT 3′ |
| hG9a 5′ + 610 | 5′ TTCCGCATGAGTGATGATGT 3′ |
| hG9a 3′ + 745 | 5′ TCGTCAGGGTCACTTCTCCT 3′ |
| hSetdb1 5′ + 463 | 5′ AAGACCAGAAGCTCCGTGAA 3′ |
| hSetdb1 3′ + 561 | 5′ CCTGGGAACTGCTCTTCTTG 3′ |
| hBDNFIXabcd 5′ | 5′ AACCTTGACCCTGCAGAATG 3′ |
| hBDNFIXabcd 3′ | 5′ TGGTCATCACTCTTCTCACCTG 3′ |
| hGAPDH 3′ + 340 | 5′ CGAGATCCCTCCAAAATCAA 3′ |
| hGAPDH 5′ + 509 | 5′ TTCACACCCATGACGAACAT 3′ |
| mmGLP 5′ + 3569 | 5′ ATTGACGCTCGGTTCTATGG 3′ |
| mmGLP 3′ + 3799 | 5′ ACACTTGGAAGACCCACACC 3′ |
| mmG9a 5′ + 3112 | 5′ TGCCTATGTGGTCAGCTCAG 3′ |
| mmG9a 3′ + 3247 | 5′ GGTTCTTGCAGCTTCTCCAG 3′ |
| mmSetdb1 5′ + 794 | 5′ GATTCTGGGCAAGAAGAGGA 3′ |
| mmSetdb1 3′ + 989 | 5′ GTACTTGGCCACCACTCGAC 3′ |
| mmBdnfI 5′ | 5′ GTCTTCTGTAGTCGCCAAGGTGG 3′ |
| mmBdnfI 3′ | 5′ GCACACCTGGGTAGGCCAAG 3′ |
| mmBdnfIV 5′ | 5′ GTCTTCTGTAGTCGCCAAGGTGG 3′ |
| mmBdnfIV 3′ | 5′ TCGCTGAAGGCGTGCGAGTA 3′ |
| mmBdnfIXA 5′ | 5′ GCAGCTGGAGTGGATCAGTAA 3′ |
| mmBdnfIXA 3′ | 5′ TGGTCATCACTCTTCTCACCTG 3′ |
| mmGapdh 3′ + 13 | 5′ ACGGCCGCATCTTCTTGTGCAGTG 3′ |
| mmGapdh 5′ + 238 | 5′ GGCCTTGACTGTGCCGTTGAATTT 3′ |
| Chromatin immunoprecipitation primers | |
| hBDNFIXabcd 5′ | 5′ AAACATCCGAGGACAAGGTG 3′ |
| hBDNFIXabcd 3′ | 5′ TTCGAAAGTGTCAGCCAATG 3′ |
| mmBdnfI 5′ | 5′ CAAAATAGGGCAGCGACTCT 3′ |
| mmBdnfI 3′ | 5′ CTGAGCGAAAAGGTGTAGGC 3′ |
| mmBdnfIV 5′ | 5′ CCCTGGAACGGAATTCTTCT 3′ |
| mmBdnfIV 3′ | 5′ AGTCCTCTCCTCGGTGAATG 3′ |
| mmBdnfIXA 5′ | 5′ CATGAGACCGGGCAAGTC 3′ |
| mmBdnfIXA 3′ | 5′ CCTTGGGAGGAATGTGTGAT 3′ |
Bdnf/BDNF, Brain-derived neurotrophic factor.
Mouse primers used for post-mitotic primary cortical neuronal cell culture and mouse cortex extracts are indicated by a preceding ‘mm’, while human primers used for lymphocyte are indicated by an ‘h’.
Primers used in polymerase chain reactions
| Primer name/location | Sequence |
|---|---|
| mRNA expression primers | |
| hGLP 5′ + 1801 | 5′ TCCTGGCTGTGGCTACTTCT 3′ |
| hGLP 3′ + 1875 | 5′ AAACGGTGAGAGATGCTGCT 3′ |
| hG9a 5′ + 610 | 5′ TTCCGCATGAGTGATGATGT 3′ |
| hG9a 3′ + 745 | 5′ TCGTCAGGGTCACTTCTCCT 3′ |
| hSetdb1 5′ + 463 | 5′ AAGACCAGAAGCTCCGTGAA 3′ |
| hSetdb1 3′ + 561 | 5′ CCTGGGAACTGCTCTTCTTG 3′ |
| hBDNFIXabcd 5′ | 5′ AACCTTGACCCTGCAGAATG 3′ |
| hBDNFIXabcd 3′ | 5′ TGGTCATCACTCTTCTCACCTG 3′ |
| hGAPDH 3′ + 340 | 5′ CGAGATCCCTCCAAAATCAA 3′ |
| hGAPDH 5′ + 509 | 5′ TTCACACCCATGACGAACAT 3′ |
| mmGLP 5′ + 3569 | 5′ ATTGACGCTCGGTTCTATGG 3′ |
| mmGLP 3′ + 3799 | 5′ ACACTTGGAAGACCCACACC 3′ |
| mmG9a 5′ + 3112 | 5′ TGCCTATGTGGTCAGCTCAG 3′ |
| mmG9a 3′ + 3247 | 5′ GGTTCTTGCAGCTTCTCCAG 3′ |
| mmSetdb1 5′ + 794 | 5′ GATTCTGGGCAAGAAGAGGA 3′ |
| mmSetdb1 3′ + 989 | 5′ GTACTTGGCCACCACTCGAC 3′ |
| mmBdnfI 5′ | 5′ GTCTTCTGTAGTCGCCAAGGTGG 3′ |
| mmBdnfI 3′ | 5′ GCACACCTGGGTAGGCCAAG 3′ |
| mmBdnfIV 5′ | 5′ GTCTTCTGTAGTCGCCAAGGTGG 3′ |
| mmBdnfIV 3′ | 5′ TCGCTGAAGGCGTGCGAGTA 3′ |
| mmBdnfIXA 5′ | 5′ GCAGCTGGAGTGGATCAGTAA 3′ |
| mmBdnfIXA 3′ | 5′ TGGTCATCACTCTTCTCACCTG 3′ |
| mmGapdh 3′ + 13 | 5′ ACGGCCGCATCTTCTTGTGCAGTG 3′ |
| mmGapdh 5′ + 238 | 5′ GGCCTTGACTGTGCCGTTGAATTT 3′ |
| Chromatin immunoprecipitation primers | |
| hBDNFIXabcd 5′ | 5′ AAACATCCGAGGACAAGGTG 3′ |
| hBDNFIXabcd 3′ | 5′ TTCGAAAGTGTCAGCCAATG 3′ |
| mmBdnfI 5′ | 5′ CAAAATAGGGCAGCGACTCT 3′ |
| mmBdnfI 3′ | 5′ CTGAGCGAAAAGGTGTAGGC 3′ |
| mmBdnfIV 5′ | 5′ CCCTGGAACGGAATTCTTCT 3′ |
| mmBdnfIV 3′ | 5′ AGTCCTCTCCTCGGTGAATG 3′ |
| mmBdnfIXA 5′ | 5′ CATGAGACCGGGCAAGTC 3′ |
| mmBdnfIXA 3′ | 5′ CCTTGGGAGGAATGTGTGAT 3′ |
| Primer name/location | Sequence |
|---|---|
| mRNA expression primers | |
| hGLP 5′ + 1801 | 5′ TCCTGGCTGTGGCTACTTCT 3′ |
| hGLP 3′ + 1875 | 5′ AAACGGTGAGAGATGCTGCT 3′ |
| hG9a 5′ + 610 | 5′ TTCCGCATGAGTGATGATGT 3′ |
| hG9a 3′ + 745 | 5′ TCGTCAGGGTCACTTCTCCT 3′ |
| hSetdb1 5′ + 463 | 5′ AAGACCAGAAGCTCCGTGAA 3′ |
| hSetdb1 3′ + 561 | 5′ CCTGGGAACTGCTCTTCTTG 3′ |
| hBDNFIXabcd 5′ | 5′ AACCTTGACCCTGCAGAATG 3′ |
| hBDNFIXabcd 3′ | 5′ TGGTCATCACTCTTCTCACCTG 3′ |
| hGAPDH 3′ + 340 | 5′ CGAGATCCCTCCAAAATCAA 3′ |
| hGAPDH 5′ + 509 | 5′ TTCACACCCATGACGAACAT 3′ |
| mmGLP 5′ + 3569 | 5′ ATTGACGCTCGGTTCTATGG 3′ |
| mmGLP 3′ + 3799 | 5′ ACACTTGGAAGACCCACACC 3′ |
| mmG9a 5′ + 3112 | 5′ TGCCTATGTGGTCAGCTCAG 3′ |
| mmG9a 3′ + 3247 | 5′ GGTTCTTGCAGCTTCTCCAG 3′ |
| mmSetdb1 5′ + 794 | 5′ GATTCTGGGCAAGAAGAGGA 3′ |
| mmSetdb1 3′ + 989 | 5′ GTACTTGGCCACCACTCGAC 3′ |
| mmBdnfI 5′ | 5′ GTCTTCTGTAGTCGCCAAGGTGG 3′ |
| mmBdnfI 3′ | 5′ GCACACCTGGGTAGGCCAAG 3′ |
| mmBdnfIV 5′ | 5′ GTCTTCTGTAGTCGCCAAGGTGG 3′ |
| mmBdnfIV 3′ | 5′ TCGCTGAAGGCGTGCGAGTA 3′ |
| mmBdnfIXA 5′ | 5′ GCAGCTGGAGTGGATCAGTAA 3′ |
| mmBdnfIXA 3′ | 5′ TGGTCATCACTCTTCTCACCTG 3′ |
| mmGapdh 3′ + 13 | 5′ ACGGCCGCATCTTCTTGTGCAGTG 3′ |
| mmGapdh 5′ + 238 | 5′ GGCCTTGACTGTGCCGTTGAATTT 3′ |
| Chromatin immunoprecipitation primers | |
| hBDNFIXabcd 5′ | 5′ AAACATCCGAGGACAAGGTG 3′ |
| hBDNFIXabcd 3′ | 5′ TTCGAAAGTGTCAGCCAATG 3′ |
| mmBdnfI 5′ | 5′ CAAAATAGGGCAGCGACTCT 3′ |
| mmBdnfI 3′ | 5′ CTGAGCGAAAAGGTGTAGGC 3′ |
| mmBdnfIV 5′ | 5′ CCCTGGAACGGAATTCTTCT 3′ |
| mmBdnfIV 3′ | 5′ AGTCCTCTCCTCGGTGAATG 3′ |
| mmBdnfIXA 5′ | 5′ CATGAGACCGGGCAAGTC 3′ |
| mmBdnfIXA 3′ | 5′ CCTTGGGAGGAATGTGTGAT 3′ |
Bdnf/BDNF, Brain-derived neurotrophic factor.
Mouse primers used for post-mitotic primary cortical neuronal cell culture and mouse cortex extracts are indicated by a preceding ‘mm’, while human primers used for lymphocyte are indicated by an ‘h’.
Acid histone extraction and western blot
Nuclear histone extractions were performed as previously described (Simonini et al.2006). Briefly, the cultured pellet was homogenized in a solution of 0.25 m sucrose, 3.3 mm calcium acetate and 1 mm PMSF. To precipitate the histones, 0.4 N H2SO4 was added, followed by TCA. The pellet was washed with acetone/HCl, then 100% acetone. Pellets were re-suspended in double-distilled H2O. The extract yields a relatively pure sample of basic histone proteins. Protein levels were measured using the Bradford Method with bovine serum albumin (BSA) as the standard.
Equal concentrations of protein from each sample were boiled in Laemmli buffer for 10 min and loaded into a 10–20% Tris-glycine gel (EC61355BOX; Invitrogen, USA) in 1 × running buffer. Proteins were then transferred to a 0.45 µm pore size nitrocellulose membrane (Invitrogen LC2001), which was then treated with anti-H3K9me2 rabbit polyclonal antibody (05-768; Millipore, USA) for human samples and anti-H3K9me2 mouse monoclonal antibody (1220; Abcam plc, UK) for mouse samples at a dilution of 1:3000 overnight. All membranes were developed with chemiluminescence methodology using ECL Plus (W319851Amersham Biosciences, UK) after incubation with the species-appropriate secondary antibody and developed by a Storm 840 processor using ImageQuant 5.0 software. After the H3K9me2 assay, membranes were further processed for histone 1 protein (H1; Millipore antibody 05-457), at a dilution of 1:1000, for normalization across samples (Gavin et al.2009b; Terranova et al.2006). Gel loading and membrane transfer was monitored using Coomassie Blue staining and unsatisfactory transfers were discarded.
Chromatin immunoprecipitation (ChIP)
The fast chromatin immunoprecipitation (ChIP) method was performed as previously published (Nelson et al.2006). Cells were cross-linked with 1.5% formaldehyde and quenched with 125 mm glycine. Cells were lysed in immunoprecipitation buffer [150 mm NaCl, 50 mm Tris-HCl (pH 7.5), 5 mm EDTA, IGEPAL 100% (0.5% vol/vol), and Triton X-100 (1.0% vol/vol)] and sonicated for 15 min on the ‘high’ setting (Diagenode Bioruptor Sonicator; Diagenode s.a., Belgium). Anti-H3K9me2 mouse monoclonal antibody (Abcam ab1220) was used to precipitate H3K9me2 associated DNA. A ‘no antibody’ control was used to normalize for non-specific background interference (Gupta et al.2010; Miao et al.2008, 2012). The protein/DNA complexes were pulled down using agarose beads (Protein A/G PLUS-Agarose, Santa Cruz SC-2003). DNA isolation was performed using 10% Chelex100 and 20 ug/µl proteinase K. Real time PCR was used to amplify Bdnf/BDNF promoter sequences, as listed in Table 1. Three Bdnf transcripts were examined in the mouse samples (post-mitotic neuronal culture and cortex extracts) and one BDNF transcript was studied in human lymphocytes.
Statistical analysis
SPSS statistical package (version 15.0 for Windows) was used for all statistical analyses. Analysis of all mRNA and ChIP data was conducted on ΔCT (experimental gene CT-normalizing gene CT) values and was presented as mean ΔΔCT values ±s.e.m. Western blot analysis was conducted on optical density levels and was presented as percent change from vehicle ±s.e.m. A probability level of p < 0.05 was the criterion to achieve statistical significance.
In post-mitotic neuronal culture, a biological replicate was defined as surgery/culture from a single pregnant mouse dam. In mouse cortex, a biological replicate was defined as a single mouse injected i.p. In lymphocyte culture, a biological replicate was defined as a blood draw from a single participant.
Results
Post-mitotic neuronal culture
In primary cortical neuronal cultures treated with nicotine for 6 h (six biological replicates), analysis of variance (ANOVA) revealed an effect of nicotine on mRNA expression for GLP (F3,20 = 12.8, p < 0.001), G9a (F3,20 = 46.1, p < 0.001) and Setdb1 (F3,20 = 18.4, p < 0.001) relative to vehicle. Tukey post hoc comparisons indicated that there was a significant nicotine-induced decrease in mRNA expression for GLP, G9a and Setdb1 (Fig. 1a).
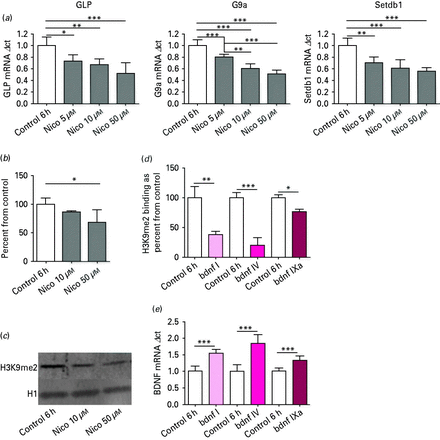
(a) Histone methyltransferases GLP, G9a and Setdb1 mRNA expression in post-mitotic primary cortical neuronal cell culture. After nicotine (Nico) treatment for 6 h, mRNA levels showed a significant decrease, as determined by Tukey post hoc. n = 6; * p < 0.05, ** p < 0.01, *** p < 0.001. (b) Global histone post-translational modification H3K9me2 expression in post-mitotic primary cortical neuronal cell culture after Nico treatment for 6 h. Protein levels showed a significant decrease. n = 7; * p < 0.05, as determined by Tukey post hoc. (c) Representative Western blot for post-mitotic primary cortical neuronal culture treated with various concentrations of Nico. (d) Chromatin immunoprecipitation: H3K9me2 binding at brain-derived neurotrophic factor (Bdnf) promoter regions I, V and IXa in post-mitotic primary cortical neuronal cell culture after 6 h of 50 µm Nico treatment. * p < 0.05, ** p < 0.01, *** p < 0.001, n = 3, as determined by two-tailed t test. (e) BDNF mRNA: increases in Bdnf transcript I, V and IXa mRNA expression in post-mitotic primary cortical neuronal cell culture after Nico treatment for 6 h. n = 6; *** p < 0.001, as determined by two-tailed t test.
For Western blot H3K9me2 analysis, seven biological replicates were used. Optical density levels of H3K9me2 protein levels between vehicle and nicotine-treated cells were analysed using one-way ANOVA, and demonstrated a significant decrease (F2,18 = 3.31, p < 0.05). All data are presented as percent change from vehicle ±s.e.m (Fig. 1b). Figure 1c is a representative Western blot of H3K9me2 in post-mitotic neuronal cell culture treated with nicotine.
Changes of H3K9me2 binding to the Bdnf/BDNF promoter were examined using the ChIP technique, and isolated DNA was measured using real-time PCR (Nelson et al.2006). PCR data was normalized for differences in amount of total chromatin (input) and background noise (no antibody), using both the ‘percent of input’ and ‘background subtraction’ methods (Haring et al.2007). For ChIP analysis, two-tailed t tests were performed for each individual transcript between nicotine and vehicle treated cell culture in three biological replicates. In primary cortical neurons exposed to 50 µm nicotine for 6 h, there was a significant decrease in H3K9me2 binding at promoter I of Bdnf (t4 = 5.38, p < 0.01), Bdnf promoter IV (t4 = 8.9, p < 0.001), and Bdnf promoter IXa (t4 = 3.9, p < 0.05; Fig. 1d).
We next confirmed that decreased promoter occupancy by a restrictive H3K9me2 resulted in increased Bdnf transcript mRNA. As expected, increases in Bdnf transcript (I, IV, IXa) mRNA were also seen between vehicle and nicotine treated neurons, in six biological replicates, using a two-tailed t test [Bdnf transcript I mRNA (t10 = 7.01, p < 0.001), Bdnf transcript IV (t10 = 6.5, p < 0.001) and Bdnf transcript IXa (t10 = 5.0, p < 0.001; Fig. 1e)].
Mouse cortex
In mouse cortical extracts (five biological replicates), 3 mg/kg i.p. injections of nicotine significantly decreased mRNA expression for GLP (t8 = 5.823, p < 0.001), G9a (t8 = 4.23, p < 0.01) and Setdb1 (t8 = 8.06, p < 0.001) relative to vehicle-treated mice (Fig. 2a). For Western blot H3K9me2 analysis, a two-tailed t test demonstrated a significant decrease in H3K9me2 levels between vehicle and nicotine-treated mice (t8 = 2.83, p < 0.05; Fig. 2b). Figure 2c is a representative Western blot of H3K9me2 in mouse cortex treated with 3 mg/kg nicotine. In nicotine-treated mice, there was a significant decrease in H3K9me2 binding at Bdnf promoter I (t8 = 5.89 p < 0.001), Bdnf promoter IV (t8 = 9.36 p < 0.001) and Bdnf promoter IXa (t8 = 8.49 p < 0.001; Fig. 2d). Increases in Bdnf transcript (I, IV, IXa) mRNA were also seen between vehicle and nicotine-treated mice using a two-tailed t test [Bdnf transcript I (t8 = 5.04 p < 0.01), Bdnf IV (t8 = 13.48 p < 0.001), Bdnf IXa (t8 = 3.91 p < 0.001); Fig. 2e].
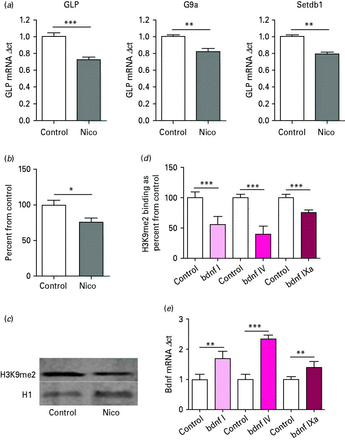
(a) Histone methyltransferases GLP, G9a and Setdb1 mRNA expression in mouse cortex extracts after nicotine (Nico) treatment. mRNA levels showed a significant decrease, as determined by two-tailed t test. n = 5; ** p < 0.01, *** p < 0.001. (b) Global histone post-translational modification H3K9me2 expression in mouse cortex extracts after Nico treatment. Protein levels showed a significant decrease. * p < 0.05, as determined by t test. (c) Representative Western blot for mouse cortex extracts treated with Nico. (d) Chromatin immunoprecipitation: H3K9me2 binding at brain-derived neurotrophic factor (Bdnf) promoter regions in mouse cortex extracts after Nico treatment. *** p < 0.001, as determined by two-tailed t test. (e) BDNF mRNA: increases in Bdnf transcript mRNA expression in mouse cortex extracts after Nico treatment. ** p < 0.01, *** p < 0.001, as determined by two-tailed t test.
Lymphocyte culture
In lymphocyte cultures treated with nicotine for 6 h (nine biological replicates), one-way ANOVA revealed an effect of nicotine on mRNA expression for GLP (F3,32 = 16.40, p < 0.001), G9a (F3,32 = 7.61, p < 0.001) and SETDB1 (F3,32 = 5.87, p < 0.01) relative to vehicle treatment. Additionally, selected cultures were pretreated with mecamylamine, a non-competitive antagonist of nAChR, in an attempt to inhibit the changes shown. Graphs are shown together for efficiency. Similar to unpublished data in NIH-3T3 cells in our laboratory, pretreatment with mecamylamine significantly abolished the nicotine-induced decreases in HMT mRNA, as demonstrated by ANOVA: GLP (F3,12 = 24.9, p < 0.001), G9a (F3,12 = 7.89, p < 0.01) and SETDB1 (F3,12 = 6.37, p < 0.01) (Fig. 3a). In Western blot analysis in lymphocyte culture (four biological replicates), demonstrated a significant decrease in overall H3K9me2 levels (F3,12 = 63.25, p < 0.001; Fig. 3b). Figure 3c is a representative western blot of H3K9me2 in lymphocyte culture treated with nicotine. There was a significant decrease in H3K9me2 binding at promoter IXabcd of BDNF in lymphocytes exposed to 50 µm nicotine for 6 h (t10 = 25.03 p < 0.001; six biological replicates; Fig. 3d). As demonstrated in the other tissue examined in this paper, there was also a significant increase in mRNA of BDNF transcript IXabcd upon nicotine treatment in lymphocytes (t6 = 11.81 p < 0.001; four biological replicates; Fig. 3e). There were no significant correlations between age of participant and HMT mRNA or H3K9me2 levels.
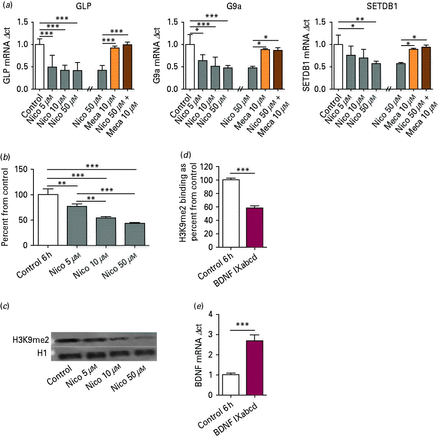
(a) Histone methyltransferases (HMT) GLP, G9a and SETDB1 mRNA expression in lymphocyte cell culture. After nicotine (Nico) treatment for 6 h, mRNA levels showed a significant decrease, as determined by Tukey post hoc. Nicotine-induced decreases on HMT mRNA were abolished with mecamylamine pretreatment. n = 9; * p < 0.05, ** p < 0.01, *** p < 0.001. (b) Global histone post-translational modification H3K9me2 expression in lymphocyte cell culture after Nico treatment for 6 h. Protein levels showed a significant decrease. n = 4; **p < 0.01, ***p < 0.001 as determined by t test. (c) Representative Western blot for lymphocyte culture treated with various concentrations of Nico. (d) Chromatin immunoprecipitation: H3K9me2 binding at BDNF promoter regions IXabcd in lymphocyte cell culture after 6 h of 50 µm Nico treatment. n = 6; *** p < 0.001, as determined by two-tailed t test. (e) BDNF mRNA: increases in BDNF transcript IXabcd mRNA expression in lymphocyte cell culture after Nico treatment for 6 h. n = 4; *** p < 0.001, as determined by two-tailed t test.
Collectively we conclude that nicotine is decreasing HMT mRNA levels, resulting in total and promoter specific decrease in protein levels of the restrictive chromatin mark H3K9me2. Through this mechanism, nicotine promotes a more transcriptionally permissive chromatin state, allowing for increased gene transcription, as demonstrated through increases in Bdnf/BDNF mRNA levels.
Discussion
From the results demonstrated in this paper, the nAChR agonist nicotine acts as a chromatin modifier, working to encourage a more transcriptionally permissive chromatin environment. At the concentrations examined, nicotine significantly decreased HMT mRNA and both total and promoter specific H3K9me2 levels, resulting in increases in Bdnf/BDNF mRNA levels. These results were demonstrated in two mouse models, one in vitro (post-mitotic neuronal culture) and one in vivo (mice injected intraperitoneally), as well as in a human in vitro model (lymphocyte culture from normal non-smoking participants).
Previous literature has demonstrated the role of nicotine as a chromatin-modifying drug, leading to a more transcriptionally permissive epigenomic state (Landais et al.2005; Levine et al.2011; Satta et al.2008). But the effect of nicotine administration on the restrictive histone modification H3K9me2 was unknown. We examined the effect of nicotine on H3K9me2 and its catalysing enzymes, and chose this specific restrictive chromatin marker for a number of reasons. First, the ninth lysine of histone H3 can receive either an acetyl (permissive) or methyl (restrictive) post-translational modification (Sims et al.2003), thus acting as a ‘switch’ between both restrictive and permissive chromatin states. Furthermore, histone methylation is a stable and durable modification (Jenuwein, 2001) and thus can aggregate and propagate a repressive message. Although it is durable, this modification is influenced by external stimuli, including drugs of abuse (Maze et al.2010), and is necessary in exploratory behaviours, motivation and environmental adaption such as contextual fear conditioning (Gupta et al.2010; Schaefer et al.2009; Shinkai & Tachibana, 2011; Tachibana et al.2005; Tzeng et al.2007). The enzymes that catalyse the addition of the methyl groups to H3K9 are part of a family of HMT and are critically important to development and normal functioning. In mice, GLP and G9a conditional knockouts produce a significant decrease in total H3K9me2 levels, which results in behavioural deficits in learning, motivation and adaption (Kramer et al.2011; Schaefer et al.2009). HMT have been implicated in brain disorders: genetic alterations of GLP results in Kleefstra Syndrome, a genetic disorder that results in learning disability and a developmental delay (Nillesen et al.2011) and over-expression of SETDB1 is implicated in Huntington's disease (Fox et al.2004; Ryu et al.2006). Given the critical role histone methylation plays in development and learning and memory, the effect nicotine may have on this epigenetic mechanism could not be ignored. We found that nicotine significantly decreases mRNA levels of all three HMT examined, GLP, G9a and Setdb1/SETDB1. These changes in mRNA levels lead to decreases in H3K9me2 levels. These findings complement the previous literature, all indicating nicotine acts as a chromatin-relaxer.
We selected Bdnf/BDNF as an epigenetically regulated ‘readout’ of nicotine-induced chromatin remodelling, with previous studies indicating an up-regulation upon nicotine exposure (Kenny et al.2000; Son & Winzer-Serhan, 2009). To examine the implication of decreases in histone methylation on Bdnf/BDNF expression, we used ChIP. We selected several mouse promoter regions, Bdnf exon I, IV and IXa, due to their previously demonstrated ability to be epigenetically modified, through both in vivo and in vitro depolarization models. Human BDNF IXabcd was selected as this transcript is well expressed widely throughout the body and the brain, including cortical pyramidal neurons. Additionally, it demonstrates similar patterns of activity as mouse Bdnf IXa, including induction by neuronal activity (Aid et al.2007; Chen et al.2003; Gavin et al.2012; Koppel et al.2009, Pruunsild et al.2007, 2011; Timmusk et al.1993; Tsankova et al.2004). Through our finding that H3K9me2 levels were reduced at Bdnf/BDNF promoter regions, and Bdnf/BDNF mRNA levels were up-regulated, this paper is able to delineate the important role of histone methylation upon the chromatin-relaxing effects of nicotine administration, which coincides with previous literature. Other epigenetic modifications can also affect BDNF levels, including DNA methylation and DNA methylation binding proteins, both of which are important to histone modifications. Given the results of this paper, these other epigenetic events are worthy of further investigation.
Our laboratory has published some of the early studies on the abnormal regulation of epigenetic modifications in patients with schizophrenia (Gavin et al.2008, 2009a, b; Sharma et al.2008), suggesting that a hyper-restrictive epigenetic environment is inherent to the disease (Issidorides et al.1975; Sharma et al.2012; Stefanis & Issidorides, 1976). Concurrently, patients with schizophrenia have higher rates of smoking than the general population or any other mental illness (Bobes et al.2010; Lohr & Flynn, 1992; McClave et al.2010; Sagud et al.2009). This paper utilized lymphocytes as a cell model as a basis for future clinical studies in patients with schizophrenia. Lymphocytes can be obtained from living subjects, acting as a real-time measurement of epigenetic responses to stimuli (Gavin & Sharma, 2009). Limited literature suggests that nicotine increases permissive chromatin environments, which could be beneficial to overly repressive conditions, as we have proposed in schizophrenia.
In conclusion, nicotine induced epigenetic modifications lead to a more permissive epigenomic environment, through significant down-regulation of HMT mRNA (GLP, G9a and Setdb1/SETDB1), and H3K9me2 total and promoter-specific levels. Furthermore, in lymphocytes, pretreatment with mecamylamine, a non-selective and non-competitive antagonist of nicotinic acetylcholine receptors effectively abolishes the nicotine-induced significant decreases in HMT mRNA. Additionally, increases in Bdnf/BDNF mRNA were a further indicator of a transcriptionally permissive chromatin state.
These findings were replicated in two different cross species in vitro models and one in vivo model, thus demonstrating a general effect of nicotine on intracellular processes related to the control of gene expression.
Acknowledgements
We thank Drs Auta, Gavin, Grayson and Guidotti for their comments and support.
Statement of Interest
None.
References

{kind=link}
{kind=link}
{kind=link}