





Abstract
Paliperidone palmitate (PP) is a recently (USA) approved injectable new-generation antipsychotic. This 53-wk, Phase-III double-blind study was designed to assess the non-inferiority of PP to risperidone long-acting injectable (RIS-LAI) in schizophrenia treatment. Acutely symptomatic patients (n=749), with a Positive and Negative Syndrome Scale (PANSS) total score between 60 and 120 were randomly allocated to gluteal injections of either (a) PP: 50 mg eq. on days 1 and 8, and flexible dosing [25–100 mg eq. (i.e. 39–156 mg USA dosing)] once-monthly; or (b) RIS-LAI: bi-weekly injections of 25 mg on days 8 and 22, and flexible dosing (25–50 mg) starting from day 36, with allowed oral supplementation. Patients (n=747) were 59% men, 92% white, mean (s.d.) age of 41 (11.95) yr and 45% (n=339) completed the study. Mean (s.d.) change from baseline to endpoint in PANSS total score was: −11.6 (21.22) PP; and −14.4 (19.76) RIS-LAI (per-protocol analysis set, primary measure); least-squares means difference was −2.6 (95% CI −5.84 to 0.61), with a prespecified 5-point non-inferiority margin. PP's suboptimal dosing regimen (<150 mg eq. initial dose) resulted in lower median plasma levels of the active moiety in PP-treated vs. RIS-LAI-treated patients. Insomnia was the most common treatment-emergent adverse event, with a similar incidence in both groups (15%). PP did not demonstrate comparable efficacy to RIS-LAI, which may be attributable to the initiation dosing strategy employed. Tolerability of both treatments was comparable to previous studies, with no new safety signals detected.
Introduction
Long-acting injectable (LAI) formulations of antipsychotic drugs in the treatment of schizophrenia may alleviate problems of treatment adherence to oral formulations and allow more constant plasma concentrations thereby reducing the occurrence of relapse. Moreover, LAIs provide therapeutic plasma concentrations over several weeks (Rainer, 2008) thereby alleviating the immediate problems associated with treatment non-compliance. Second-generation antipsychotic (SGA) LAIs (‘atypical’ antipsychotic), compared to first-generation (‘conventional’) LAI antipsychotics, are likely to have an improved treatment satisfaction and acceptance in patients with schizophrenia (Fleischhacker 2009; Marinis et al.2007).
The SGA risperidone LAI (RIS-LAI) has been approved for the acute and maintenance treatment of schizophrenia in many countries. Recently, paliperidone palmitate (PP), the palmitate ester of paliperidone (9-hydroxy-risperidone), the major active metabolite of risperidone, has been approved in the USA for the acute and maintenance treatment of schizophrenia. This new LAI is efficacious and tolerable at the recommended doses of 25–150 mg eq. (i.e. 39–195 mg USA dosing) for treatment of schizophrenia (Coppola et al.2009; Hough et al.2009; Kramer et al.2009; Nasrallah et al.2010; Pandina et al.2010a, b). In contrast to RIS-LAI, PP does not require oral supplementation during treatment initiation, as its pharmacokinetic (PK) profile allows both a rapid achievement of therapeutic plasma levels of paliperidone as well as a gradual and continuous release of the drug over the dosing interval (Samtani et al.2009). Additionally, the longer injection interval of 4 wk for PP is advantageous.
Non-inferiority trials are intended to assess whether the efficacy of a new treatment is not worse than that of an active control (Snapinn, 2000). Given the importance of enhancing the long-term treatment armamentarium in schizophrenia, the present study was designed to compare the new LAI PP to RIS-LAI in the long-term treatment of schizophrenia using a non-inferiority design. The study also assessed the safety and tolerability of PP in maintenance therapy of schizophrenia.
Methods
Patients
Consenting men and women (⩾18 yr) with an established DSM-IV diagnosis of schizophrenia for at least 1 yr before screening, a Positive and Negative Syndrome Scale (PANSS) total score between 60 and 120 (inclusive), acutely symptomatic at screening and baseline (day 1), and a body mass index (BMI) ⩾15.0 kg/m2, were enrolled.
Main exclusion criteria included: primary active DSM-IV Axis I diagnosis other than schizophrenia, decrease of ⩾25% in the PANSS total score between screening and baseline, a DSM-IV diagnosis of active substance dependence within 3 months before screening, treatment resistance (failure to respond to two adequate trials at a minimum of 4 wk at a therapeutic dose, of different antipsychotic medications), history of neuroleptic malignant syndrome, or any significant or unstable systemic disease, or suicidal or violent behaviour. Women were excluded if pregnant, nursing, or planning pregnancy.
The Independent Ethics Committee or Institutional Review Board at each study site approved the protocol. The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with Good Clinical Practice (GCP) and applicable regulatory requirements. All patients provided written informed consent before entering the study.
Study medication
Doses of PP can be expressed both in terms of milligram equivalents (mg eq.) of the pharmacologically active fraction, paliperidone, and in milligrams of PP. Thus, the doses expressed as ‘PP 25, 50, 75, or 100 mg eq.’ equate to 39, 78, 117, and 156 mg, respectively, of PP.
In this study, PP was provided as 25, 50, 75, and 100 mg eq. injectable suspensions (supplied as 100 mg eq./ml), or matching placebos [20% Intralipid™ (Fresenius Kabi AB, Sweden) 200 mg/ml]. RIS-LAI was supplied as risperidone depot microspheres in strengths of 25, 37.5, and 50 mg with matching placebo. RIS-LAI required reconstitution before injection, whereas PP was premixed and ready for injection. Oral risperidone was supplied as 1-mg tablets with matching placebo.
Study design, randomization, and blinding
This trial was supported by Johnson & Johnson Pharmaceutical Research & Development (J&JPRD) as part of the Phase III programme for PP. This randomized, double-blind, active-controlled, parallel-group, multicentre, comparative study was conducted from February 2005 to April 2007 in 108 centres in 19 countries in North America, Australia, New Zealand, Western and Eastern Europe. The study consisted of a 7-d screening and oral tolerability testing phase, and a 53-wk double-blind treatment phase. Patients who demonstrated tolerability to oral paliperidone (3 mg/d) for 4 d during screening were randomly assigned (1:1) to flexibly dosed PP+oral placebo (PP group) or flexibly dosed RIS-LAI+oral risperidone (RIS-LAI group) based on a computer-generated randomization scheme (prepared by the sponsor) balanced by using permuted blocks of treatments, stratified by centre and implemented using an interactive voice response system (IVRS). The treatment schedule during the double-blind period is shown in Table 1. The study drug administrator was unblinded to the treatment group but was not allowed to perform any other study-related procedures or communicate patient-related information to study site personnel, including the investigator. The patients and all other study staff were blinded to treatment.
Treatment schedule during the double-blind phase (beginning at baseline, day 1)
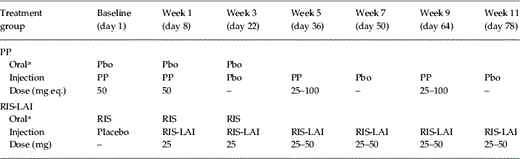
Pbo, Placebo; PP, paliperidone palmitate; RIS-LAI, risperidone long-acting injectable.
The doses expressed as 25, 50, and 75 mg eq. PP equate to 39, 78, and 117 mg PP, respectively.
All injections were administered intramuscularly at the gluteal site.
Patients in PP group continued to receive either flexibly dosed PP or placebo injection every 2 wk from week 11 up to week 51.
Patients in the RIS-LAI group continued to receive RIS-LAI injections every 2 wk from week 11 up to week 51.
Oral supplementation (risperidone 1–6 mg/placebo) was given for the first 4 wk of the double-blind treatment period. Oral supplementation (risperidone 1–4 mg/placebo) was also given at any dose increase from day 36 onwards, continuing for up to 3 wk.
Treatment schedule during the double-blind phase (beginning at baseline, day 1)
Pbo, Placebo; PP, paliperidone palmitate; RIS-LAI, risperidone long-acting injectable.
The doses expressed as 25, 50, and 75 mg eq. PP equate to 39, 78, and 117 mg PP, respectively.
All injections were administered intramuscularly at the gluteal site.
Patients in PP group continued to receive either flexibly dosed PP or placebo injection every 2 wk from week 11 up to week 51.
Patients in the RIS-LAI group continued to receive RIS-LAI injections every 2 wk from week 11 up to week 51.
Oral supplementation (risperidone 1–6 mg/placebo) was given for the first 4 wk of the double-blind treatment period. Oral supplementation (risperidone 1–4 mg/placebo) was also given at any dose increase from day 36 onwards, continuing for up to 3 wk.
All antipsychotics and anti-epileptics were not allowed during the study except for oral risperidone in the RIS-LAI group. Antiparkinsonian medication (at the permitted maximum daily doses) as rescue treatment for extrapyramidal symptoms (EPS), oral lorazepam (2–6 mg/d) or other short-acting benzodiazepines for agitation, anxiety, or sleep difficulties, and oral propranolol for akathisia, were allowed. Antidepressants (except for non-selective or irreversible monoamine oxidase inhibitors) were allowed if they had been used at a stable dose for at least 30 d before screening.
PP was administered as an intramuscular (i.m.) gluteal injection of 50 mg eq. PP at day 1 and day 8 with subsequent gluteal injections of flexibly dosed PP (25, 50, 75, or 100 mg eq.), once every month (Table 1). To preserve the blind, injections of placebo matched to PP were administered starting on day 22 and monthly thereafter in this group. Thus, these patients received an injection of either PP or placebo every 2 wk. RIS-LAI was administered as bi-weekly i.m. (gluteal) injections: 25 mg for the first two injections (days 8 and 22), and flexibly dosed (25, 37.5, or 50 mg) thereafter. To align the injection intervals of the two treatment groups, all patients in the RIS-LAI group received an injection of placebo matched to RIS-LAI on day 1, and the first RIS-LAI injection was delayed until day 8, allowing bi-weekly visits. Doses of PP and RIS-LAI could be changed beginning at day 36 but not more frequently than once a month (Table 1). Patients in the RIS-LAI group received oral risperidone and patients in the PP group received oral placebo tablets.
Efficacy assessments
The primary efficacy variable was the change in PANSS total score from baseline to the last post-randomization assessment in the double-blind period. Secondary efficacy variables included the changes from baseline to endpoint in the Clinical Global Impression – Severity (CGI-S) score and Personal and Social Performance Scale (PSP) score, and responder rate (defined as those patients with a ⩾30% reduction in the PANSS total score from baseline to endpoint).
Safety assessments
Safety evaluations included assessment of treatment-emergent adverse events (TEAEs), clinical laboratory tests, vital sign measurements, physical examination findings, electrocardiograms, EPS rating scales: Simpson and Angus Rating Scale (SAS; Simpson & Angus, 1970), Barnes Akathisia Rating Scale (BARS; Barnes, 1989) and Abnormal Involuntary Movement Scale (AIMS; Guy, 1976), Global Impression of Sexual Function (Guy, 1976), investigator's evaluation of the injection site, and patient's evaluation, using a visual analogue scale (VAS 0–100 mm), of pain both at the injection site and of the injection.
PK assessments
Sparse blood samples were taken for PK analysis at baseline (day 1), on days 64, 204, 260, 267, 274, 288, 302 and at end of the study (day 372) or early withdrawal visits. The samples were collected before study drug administration on the days when study drug was administered.
Sample size determination
Assuming (i) a standard deviation (s.d.) of 20 for the change in PANSS total score, (ii) a true difference between RIS-LAI and PP of 0.1 in favour of RIS-LAI, (iii) a two-sided significance level of 5%, and (iv) 75% of randomly assigned patients could be evaluated for the per-protocol analysis, 350 patients per treatment group were required to demonstrate with 80% power that PP was no worse than RIS-LAI by a margin of 5 points in the change in PANSS total score.
Analysis sets
The per-protocol analysis set (n=570) (for primary efficacy variable) included patients who had received at least four injections (active drug or placebo) during the double-blind phase and for whom the time between any two injections during this period did not exceed 35 d, had a baseline and at least one post-randomization measurement on the primary efficacy variable, and no major protocol violations. The intent-to-treat (ITT) analysis set (n=674) (for secondary efficacy variables) included all randomly assigned patients who received at least one dose of study drug and had at least one efficacy measurement during the double-blind phase. Patients from one site in Hungary (n=2) and one site in Poland (n=53) were excluded from the ITT analysis set due to GCP issues relating to trial conduct, but were included for safety analysis. The efficacy data for these sites was removed due to deficiencies in handling documentation related to trial medication. The safety analysis set (n=747) included all patients who had received at least one dose of double-blind study drug. The PK analysis set (n=598) included all patients randomly assigned to study drug who had evaluable samples at the time of PK assessment.
Statistical analysis
The change from baseline to endpoint in the PANSS total score was analysed using an analysis of covariance (ANCOVA) model with treatment and country as factors and baseline score as the covariate. The point estimate and two-sided 95% confidence interval (CI) based on an ANCOVA model were provided for the difference between RIS-LAI and PP in the change from baseline in PANSS total score at endpoint. Non-inferiority of PP to RIS-LAI was concluded if the lower limit of the two-sided 95% CI of the difference exceeded −5. For the secondary endpoints CGI-S and PSP, the change from baseline to endpoint was analysed using an ANCOVA model similar to that specified for the primary efficacy variable. For responder rate, the point estimate and two-sided 95% CI were provided for the relative risk using a Mantel–Haenszel test controlling for country. The last observation carried forward approach (LOCF) was used to impute missing data. Safety and tolerability measures were evaluated based on descriptive statistics.
During quality control review, a higher than expected overall error rate (1.5%) on safety data was noted when comparing the study database with the data entries in the paper case record forms. The target error rate was 0.05%. Because the observed error rate exceeded the prespecified threshold, the sponsor decided to revalidate and correct all data related to the concomitant medication, exposure, and adverse event fields for the safety analysis set. As the primary efficacy objective of non-inferiority was not achieved, no efficacy data was re-entered. Overall the sponsor found that the final safety conclusions from this study were not changed as a result of this revalidation and data re-entry.
Exploratory analysis
Based on the heterogeneity of treatment effect across baseline BMI categories (normal: <25 kg/m2; overweight: ⩾25 to <30 kg/m2; obese: ⩾30 kg/m2) observed in previous studies (Gopal et al.2010; Nasrallah et al.2010), an additional exploratory analysis was performed to assess the influence of BMI on the change in PANSS total score from baseline to endpoint using an ANCOVA with treatment, country, baseline BMI category, and treatment by BMI category interaction as factors, and baseline PANSS total score as a covariate. The 95% CI for the least-squares mean (LSM) difference between RIS-LAI and PP were derived from a subpopulation model that used all available data to increase the precision of inference. Paliperidone plasma concentrations are presented as box-plots per visit.
Results
Patient characteristics and disposition
Of the 749 enrolled patients, 339 (45%) completed this 53-wk study (Fig. 1). The completion rate in the RIS-LAI group (50%, n=184/370) was better than that in the PP group (41%, n=155/379). The ITT analysis set included 674 patients, and the per-protocol analysis set included 570 patients. Lack of efficacy (PP: 25%, n=95; RIS-LAI: 15%, n=56) and withdrawal of consent (PP: 15%, n=55; RIS-LAI: 17%, n=62) were the most common reasons for discontinuation. Figure 2 shows the Kaplan–Meier estimates of time to withdrawal due to any reason.

Patient disposition (all randomized patients analysis set). LAI, Long-acting injectable; PBO, placebo; RIS, risperidone. A total of l77 patients randomly assigned to RIS-LAI were administered a placebo injection instead of 25 mg RIS-LAI on day 8 because of an interactive voice response system (IVRS) error. The IVRS error was corrected and the trial proceeded as planned. The error was not considered a major protocol violation; therefore, patients were not excluded from the per-protocol analysis set unless they had any other major protocol violation.

Kaplan–Meier estimates of time to withdrawal due to any reason for the safety analysis set. PP, Paliperidone palmitate; RIS-LAI, risperidone long-acting injectable.
The demographic and baseline characteristics were similar between treatment groups (Table 2). Patients had a mean (s.d.) age of 40.7 (11.95) yr, 59% (n=444/747) were men, and 92% (n=688/747) white. Thirty percent were obese. The mean (s.d.) PANSS total score at baseline for the safety analysis set was 81.6 (13.06), and for the per-protocol set was 81.7 (12.67). Before randomization, 20% (n=147/747) of patients in the safety analysis set had received anti-EPS medication; the use was similar in both treatment groups. Supplementary Table S1 (available online) shows the prior psychotropic medications received by patients in the ITT analysis set. Overall, the treatment groups defined for the per-protocol analysis set were similar with respect to demographic and baseline characteristics.
Demographics and baseline characteristics (safety analysis set)

LAI, Long-acting injectable; PANSS, Positive and Negative Syndrome Scale; CGI-S: Clinical Global Impression – Severity.
Demographics and baseline characteristics (safety analysis set)
LAI, Long-acting injectable; PANSS, Positive and Negative Syndrome Scale; CGI-S: Clinical Global Impression – Severity.
In the safety analysis set, 40% (n=151/379) of patients on PP and 47% (n=174/368) of patients on RIS-LAI received all 27 gluteal injections during the double-blind phase. The mean (s.d.) dose received was 63.5 (14.40) mg eq. for PP-treated patients, and 32.4 (7.75) mg for RIS-LAI-treated patients. The mean (s.d.) dose of oral risperidone in the first 4 wk was 3.1 (1.46) mg/d and ranged from 2.1 to 2.9 mg/d in subsequent weeks. Most dose adjustments in both treatment groups were increases and no more than 2% of patients had their dose decreased at any visit. More patients (41%) in the PP group than the RIS-LAI group (36%) needed a dose increase at day 36 (the first time-point when dose adjustment was allowed). Thereafter, there was a progressive decrease in the proportion of patients who required a dose increase.
In the per-protocol analysis set, 49% (n=142/288) of patients on PP and 59% (n=166/282) of patients on RIS-LAI received all 27 gluteal injections during the double-blind phase. The mean (s.d.) dose received in both treatment groups was comparable with the safety analysis set.
Efficacy
For the per-protocol analysis set, the mean (s.d.) change from baseline to endpoint in PANSS total score was −11.6 (21.22) in the PP group, and −14.4 (19.76) in the RIS-LAI group (Supplementary Table S2, online). The LSM difference between PP and RIS-LAI groups for the change in PANSS total score was −2.6 (95% CI −5.84 to 0.61). As the lower limit of the 95% CI was less than −5 (based on the protocol predetermined margin for non-inferiority), PP as dosed in this study was not found non-inferior to RIS-LAI. The results from the ITT analysis set were consistent with these results. A separate analysis including in the ITT set those patients (n=55) from the two sites with GCP issues, indicated that the results were consistent with those excluding the two sites. Figure 3 shows the estimated LSM change in PANSS total score over time.

Least-squares mean (LSM) (±s.e.) change from baseline in total PANSS score over time – LOCF (per-protocol analysis set). PP, Paliperidone palmitate; RIS-LAI, risperidone long-acting injectable.
The ITT analysis set was used for analysis of secondary efficacy measures. The reduction in the severity of schizophrenia symptoms as assessed by CGI-S score was higher in the RIS-LAI group than in the PP group, whereas PSP scores showed similar changes in both treatment groups (Supplementary Table S2). In the ITT analysis set at endpoint, 43% (n=138/322) of patients in the PP group and 46% (n=148/323) of patients in the RIS-LAI group had improved PSP scores compared to baseline.
The percentage of ITT responders (⩾30% improvement in the PANSS total score compared to baseline) was 44% (n=152/343) in the PP group and 54% (n=179/329) for the RIS-LAI group. The point estimate of the relative risk of PP vs. RIS-LAI treatment for responder rate at endpoint was 0.8 (95% CI 0.70–0.95).
The exploratory post-hoc analysis of treatment by baseline BMI category interaction approached statistical significance (p=0.108) at the 10% level for the analysis of the change in PANSS total score at endpoint. The mean change in PANSS total score at endpoint in the PP group was lower (indicating less clinical improvement) in obese patients than in normal-weight or overweight patients. The mean change in PANSS total score was similar in the RIS-LAI group regardless of baseline BMI category. The point estimate for the difference in LSMs between PP and RIS-LAI indicated non-inferiority for the normal and overweight subgroup (−0.5, 95% CI −4.01 to 3.08), but not for the obese subgroup (−7.5, 95% CI −12.1 to −2.82).
Safety
Overall, the rates of TEAEs were similar in the PP (76%, n=289/379), and RIS-LAI (79%, n=289/368) groups. The most common (>10%) TEAEs noted in either treatment group were insomnia (15% in each treatment group), psychotic disorder (14% PP, 12% RIS-LAI), worsening or relapse of schizophrenia (12% PP, 9% RIS-LAI), anxiety (10% PP, 15% RIS-LAI) and headache (9% PP, 11% RIS-LAI) (Fig. 4).

Treatment-emergent adverse events experienced by at least 5% of patients in any treatment group (safety analysis set). PP, Paliperidone palmitate; RIS-LAI, risperidone long-acting injectable.
Four deaths were reported during the study: three in the PP group [one acute myocardial infarction, one unknown cause (parents refused to provide any information about the cause or circumstances to the investigator), one food aspiration] and one in the RIS-LAI group (pulmonary carcinoma). Three of the deaths were considered to be not related to study drug, and the one due to food aspiration was considered as doubtfully related. The proportion of serious TEAEs was slightly greater in the PP group [29% (n=111/379) of patients] compared to RIS-LAI group [22% (n=80/368) of patients]. Across both treatment groups, serious TEAEs were mostly related to psychiatric disorders [20–25% (n=72–96)]; most common were psychotic disorders (8–9%) and schizophrenia (6–8%). Serious TEAEs related to relapse were noted in 25% (95/379) of patients in the PP group and 18% (67/368) of patients in the RIS-LAI group. The percentage of patients who discontinued due to TEAEs was similar in both treatment groups (7% in PP group, 6% in RIS-LAI group); psychiatric disorders were the most frequent reason for discontinuation in both groups (5% PP group, 3% RIS-LAI group).
The incidence of treatment-emergent EPS-related adverse events was low and similar for both treatment groups, with the exception of hyperkinesia (6% PP vs. 10% RIS-LAI). Akathisia as a serious TEAE occurred in two patients in the PP group, and did not result in study discontinuation. Neuroleptic malignant syndrome as a serious TEAE was reported in one patient on PP and resulted in study discontinuation. No occurrence of tardive dyskinesia was reported.
Anti-EPS medication was administered to 67 (18%) patients in the PP group and to 76 (21%) patients in the RIS-LAI group during the double-blind phase. The use of anti-EPS medication decreased from baseline to endpoint in both groups [PP 21% (n=78) to 9% (n=36); RIS-LAI 19% (n=69) to 12% (n=44)]. There were the following changes in EPS rating scales during the double-blind phase: elevated SAS indicative of parkinsonism [PP 47 (12%) patients, RIS-LAI 53 (14%) patients]; elevated BARS indicative of akathisia [PP 24 (6%) patients, RIS-LAI 24 (7%) patients], and elevated AIMS indicative of dyskinesia [PP 15 (4%) patients, RIS-LAI 12 (3%) patients]. Overall, these results indicated that treatment with either PP or RIS-LAI was associated with a low incidence of EPS-related adverse events in this study.
Treatment-emergent glucose-related AEs occurred in eight patients in the PP group and 14 patients in the RIS-LAI group as follows: PP (number of patients) – blood glucose increased (n=4), hyperglycaemia (n=3), diabetes mellitus non-insulin-dependent (n=1), glycosuria (n=1), ketonuria (n=1), urine ketone body present (n=1); RIS-LAI (number of patients) – blood glucose increased (n=6), hyperglycaemia (n=4), diabetes mellitus (n=4), and hypoglycaemia (n=1). One event of diabetes mellitus in a patient in the RIS-LAI group was considered serious and was attributed to the underlying diabetes (onset unknown) and non-compliance with the diabetes medication.
There were three reports of ischaemic events, one each of myocardial ischaemia, myocardial infarction (resulting in death), and transient ischaemic attack (serious). There was also one ischaemia-related event of mild angina pectoris. The myocardial ischaemia occurred in a RIS-LAI group patient; the other three events occurred in PP patients: The angina pectoris and transient ischaemic attack were considered as possibly related to the study drug while the myocardial infarction and myocardial ischaemia were not considered as related to the drug. Tachycardia occurred in 12 (3%) patients in the PP group and four (1%) in the RIS-LAI group: none were serious or resulted in discontinuation. There were no reports of ventricular tachycardia or fibrillation, torsade de pointes, or syndromes of inappropriate antidiuretic hormone secretion. No patient had a linear-derived corrected QT interval (QTcLD) value ⩾480 ms during the treatment or an increase of >60 ms from baseline.
The proportion of patients with abnormally high prolactin levels during the double-blind period was larger in the RIS-LAI group [men 53% (n=115/219), women 51% (n=68/133)] than in the PP group [men 31% (n=64/205), women 42% (n=68/161)]. Mean (s.d.) increases in prolactin levels from baseline to endpoint were comparatively higher in women [PP 22.5 (45.89) ng/ml, RIS-LAI 22.4 (68.65) ng/ml] than in men [PP 6.9 (16.73) ng/ml, RIS-LAI 9.1 (14.46) ng/ml]. Potentially prolactin-related TEAEs were low and similar in both groups (<4%). There was no statistically significant difference between the two treatment groups in Global Impression of Sexual Function at endpoint for either sex.
The proportion of patients with a ⩾7% increase in body weight from baseline to endpoint was similar in both groups [PP 14% (n=50/346), RIS-LAI 15% (n=52/338)]. Overall, the mean (s.d.) change from baseline to endpoint in body weight was −0.2 (6.01) kg for the PP group and 0.8 (5.65) kg for the RIS-LAI group. There were no clinically relevant changes in vital signs or notable changes in most chemistry, haematology, or urinalysis laboratory parameters.
Treatment-emergent injection-site pain was reported with similar frequency (2–3%) across both groups. Investigators' evaluations showed the number of patients with moderate or severe redness, pain, or swelling at the injection site was lower at endpoint than at baseline in both treatment groups. The mean (s.d.) patient-assessed injection-site pain VAS (0–100 mm) scores decreased from baseline [PP 7.8 (14.12), RIS-LAI 9.6 (14.3)] to endpoint [PP 3.4 (7.22), RIS-LAI 3.4 (7.64)] in both treatment groups. No one discontinued from the study due to injection-site pain.
PK findings
The median plasma concentrations of paliperidone in the PP treatment group were lower on day 64 (first time-point) compared to later time-points (Fig. 5). They were also considerably lower than those found for the active moiety (i.e. paliperidone+risperidone) in the RIS-LAI group, which reached apparent steady-state plasma concentrations at the first measured post-baseline time-point (day 64). The concomitant intake of drugs, inhibiting or inducing the CYP450 enzyme system was comparable across groups and is therefore unlikely to have had an influence on median plasma level comparisons, but, may have contributed to the observed inter-subject variability in drug plasma concentrations presented in Fig. 5.

Plasma concentrations over time of paliperidone and active moiety by treatment group. Paliperidone palmitate (PP) treatment group represents paliperidone concentration and risperidone long-acting injectable (RIS-LAI) treatment group represents active moiety (sum of risperidone and paliperidone) concentrations. Plasma levels were analysed only at the nominal time-points shown. Up (↑) arrows indicate days when PP injections were given and down (↓) arrows indicate days when RIS-LAI injections were given. The plasma concentrations of paliperidone were determined by means of a validated liquid chromatography coupled to mass spectrometry/mass spectrometry method with a target limit of quantification of 0.1 ng/ml, at the Bioanalytical Department, Johnson & Johnson Pharmaceutical Research & Development, Beerse, Belgium. The solid line in the box is the median of individual paliperidone (PP treatment) or active moiety (Risperdal Consta treatment) plasma concentrations. The lower boundary of the box represents the 25th percentile, the higher boundary of the box represents the 75th percentile. The whiskers are the nearest values within 1.5 times the interquartile range below and above the 25th and 75th percentile, respectively. PBO, Placebo.
Discussion
Based on the predetermined margin of 5 points in the change in PANSS total score, statistical non-inferiority of flexibly dosed PP to flexibly dosed RIS-LAI was not achieved in this study. PP was not as efficacious as RIS-LAI in this trial. Consistent with this, a larger reduction of CGI-S scores in the RIS-LAI group compared to the PP group was also noted. This randomized, double-blind study was the first to compare the efficacy, safety and tolerability of PP with RIS-LAI. The results from this study helped in the design of subsequent studies for PP, including the development of an improved dosing regimen.
Previous studies have shown that antipsychotic efficacy is associated with a central D2-receptor occupancy of 60–80% (Kapur, 1996; Nyberg et al.1995). For paliperidone, a plasma concentration of 7.5 ng/ml led to a central D2-receptor occupancy of approximately 60% (Karlsson et al.2006). However, in the present study, overall median paliperidone plasma concentrations had not reached 7.5 ng/ml by day 64, suggesting that the initiation dosing regimen was not adequate to rapidly and consistently achieve therapeutically effective plasma levels of paliperidone (the active moiety) after initiating therapy with PP.
Rapid attainment of therapeutic plasma concentration is important when initiating treatment with a LAI (Samtani et al.2009). Moreover, the timing of systemic drug release of a LAI depends on the distribution of muscle and adipose tissue at the injection site. The hypovascularity of adipose tissue in the gluteal muscle may result in slow uptake of the drug in the systemic circulation. At the deltoid site, the likelihood of a true i.m. injection is higher (Samtani et al.2009). Thus the initial gluteal injections in the current study may have contributed to the initial lower paliperidone plasma concentrations as a rate-limiting factor (Samtani et al.2009). Further, the initial two low initiation doses of 50 mg eq. PP appeared to be suboptimal as paliperidone concentrations (active moiety) in PP-treated patients were lower during most of the study than the levels of the risperidone+paliperidone concentrations (active moiety) in RIS-LAI-treated patients, at equivalent doses of risperidone. Consistent with these observations, the PP group showed a higher rate of discontinuation due to lack of efficacy, a lower responder rate, and less improvement in symptom severity. More patients in the PP group required a dose increase when dose adjustment was allowed (day 36).
More recent studies demonstrated that: (a) use of weight-adjusted needle length with a 1-inch needle for patients weighing <90 kg and with a 1.5-inch needle for patients weighing ⩾90 kg for deltoid initiation (Pandina et al.2010a); and (b) use of a 150 mg eq. initiation dose (Pandina et al.2010a) improved the quick and consistent attainment of plasma concentrations. In fact, to rapidly achieve therapeutic plasma concentrations of PP, the currently recommended initiation dosing regimen is a high-dose injection administered on day 1 (150 mg eq.) and day 8 (100 mg eq.) into the deltoid muscle using weight-adjusted needle length, followed by flexibly dosed once-monthly injections (25–150 mg eq.) into the deltoid or gluteus (Citrome, 2010). No oral supplementation is required. The use of the higher PP dose in the modified initiation dosing did not result in increased incidence of TEAEs and no new safety signals were detected in studies that used higher initiation doses (Gopal et al.2010; Pandina et al.2010a, b).
The magnitude of change in PANSS total score from baseline to endpoint in patients receiving PP was consistent with that observed in other clinical studies that demonstrated significant improvement of PP over placebo (Nasrallah et al.2010; Pandina et al.2010a, b). PSP score changes were similar in both treatment groups.
The overall incidence of TEAEs was similar in the two groups. The overall use of anti-EPS medication decreased from baseline and was low at endpoint for both PP and RIS-LAI, consistent with the shorter duration studies of PP (Gopal et al.2010; Pandina et al.2010a, b) as well as RIS-LAI (Kane et al.2003; Pandina et al.2010b). High prolactin levels were observed in both treatment groups, although the incidence of potentially prolactin-related TEAEs was low in each, consistent with other studies for PP and risperidone (Hough et al.2009; Knegtering et al.2005; Kramer et al.2009; Melkersson, 2006; Nasrallah et al.2010; Spina & Cavallaro, 2007). Systematic evaluations of sexual function in this study, which used male and female versions of a sexual functioning scale, showed a low incidence of sexual side-effects and no difference between the treatment groups. Other groups have reported higher ratings of sexual adverse events when using different scales for evaluating effect of antipsychotics on sexual function (Fujii et al.2009; Knegtering et al.2004, 2006; Konarzewska et al.2009; Nagaraj et al.2009). Local injection-site tolerability was good and comparable for both treatments.
In this study, exclusion of the two sites with GCP issues could be considered a study limitation, although the results of the analysis including these two sites were consistent with those of the ITT analysis set excluding these two sites. Another study limitation is the high number of dropouts observed throughout the trial in both the treatment groups, which is not unexpected in long-term open-label and double-blind studies (Lieberman et al.2005). An additional limitation was that the oral doses cannot be directly compared to the long-acting formulation doses. Furthermore, oral formulations of the two drugs are different as risperidone is an immediate release (IR) formulation, whereas paliperidone is an extended release (ER) formulation for which only 30% of actual dose is absorbed.
Both PP and RIS-LAI improved schizophrenia symptom severity and reduced severity of illness. However, PP did not demonstrate comparable efficacy to RIS-LAI, which may be attributable to the initiation dosing strategy employed, which used a low PP dose injected in the gluteus. Tolerability and safety results, however, were comparable between PP and RIS-LAI.
Acknowledgements
This work was funded by Johnson & Johnson Pharmaceutical Research & Development, L.L.C., Raritan, New Jersey, USA. Dr Madhavi Patil (SIRO Clinpharm Pvt Ltd) provided writing assistance and Dr Wendy P. Battisti (Johnson & Johnson Pharmaceutical Research & Development, L.L.C.) provided additional editorial support for this manuscript. The authors thank the study participants, without whom this study could not have been accomplished.
[This study is registered at www.clinicaltrials.gov (NCT00210717).]
Appendix. Investigators and sites
Australia: Hustig, Harry, MD; Kulkarni, Jayashri, MBBS, MPM, FRANZCP, PhD. Austria: Geretsegger, Christian, MD; Hofer, Alex, MD; Rittmannsberger, Hans, MD; Sandner, Wolfgang, MD; Schoenbeck, Georg, MD; Wrobel, Margit, MD. Belgium-Luxemburg: Hellebuyck, Hans; Janssen, Firmin; Kint, Jean-Pascal; Kusters, Johan; Peuskens, Joseph. Canada: Bawa, Hussam; Beauclair, Linda; Chuang, Henry; Delva, Nick; Fallu, Angelo; Johnson, Sunny; Koczerginski, David; Labelle, Allain; Reiss, Jeffrey; Tibbo, Phillip. Czech Republic: Kalusova, Zuzana, MD; Lochmannova, Milena, MD; Marholdova, Kamila, MD; Papezova, Simona, MD; Pulkrabkova, Lucie, MD; Slavomir, Pietrucha, MD; Tuma, Ivan, MD; Zdenek, Solle, MD. Denmark: Aagaard, Jorgen; Dahl, Erik; Dam, Jesper; Kristan, Oellegaard, Anderson; Kristensen, Karsten, Haderup; Moller-Andersen, Peter; Olstrup, Leif. Estonia: Aadamsoo, Kaire, MD; Karindi, Margit, MD; Kiens, Tuuli, MD; Puusild, Ants, MD. France: Bougerol, Thierry, MD; Dassa, Daniel, MD; Favre, Pascal, MD; Gorwood, Philip, MD; Louvrier, Jacques, MD; Palazzolo, Jerome, MD; Raymondet, Phillippe, MD. Germany: Backhaus, Dolores, MD; Boehringer, Johannes, MD; Deuschle, Michael, MD; Freyberger, Harald, MD; Gunther, Rolf, MD; Heres, Stephan, MD; Lambert, Martin, MD; Richter, Rainer A, MD; Schmauss, Max, MD; Schumann, Gunther, MD; Uebelhack, Ralf, MD. Hungary: Bitter, Istvan, MD; Faludi, Gabor, MD; Frecska, Ede, MD; Hal, Viktor, MD; Kassai-Farkas, Akos, MD; Kurimay, Tamas, MD; Varga, Csilla, MD; Vincze, Gabor, MD. Lithuania: Baliuniene, Jolanta-Daiva; Dembinskiene, Laisve, MD; Matoniene, Valdone; Rudzianskiene, Sonata; Siurkute, Aldona. Netherlands: De Veen, Rene, MD; Dries, Pieter, MD; Moctezuma, Jorge, MD; Rijnders, Cees, MD; van Kooten, Marcel. New Zealand: Miles, Waynes, MBChB, MD, FRANZCP, MRANZCP. Poland: Jarema, Marek, MD, PhD; Matysiakiewicz, Jerzy, MD; Prewlocka, Anna, MD; Ryebakowski, Janusz, MD, PhD; Rzewuska, Malgorzata, MD, PhD; Wozniak, Kryzsztof, MD. Spain: Arias, Francisco; Baeza, Jose, Vincente; Bobes-Garcia, Julio; Gonzalez, Miguel, Angel; Olivera, Angustias; Pons, Alexandre. Sweden: Bjorner, Annika, MD; Brain, Cecila, MD; Briksson, Lars, MD, MA; Gidlund, Lars, MD; Helldin, Lars, MD; Lindstrom, Leif, MD, PhD. Switzerland: Strik, Werner, MD. USA: Bortnick, Brian, MD; Brams, Mathew, MD; Brar, Saroj, MD, Goenjian, Armen, MD; Logue, H Edward, MD; Sendi, Ismail, MD; Walling, David, PhD.
Note
Supplementary material accompanies this paper on the Journal's website.
Statement of Interest
This study was sponsored by Johnson & Johnson Pharmaceutical Research and Development, L.L.C. The sponsor provided a formal review of the manuscript. Dr Fleischhacker was a coordinating investigator on this study and has received consultancy and advisory board fees from AstraZeneca, Eli Lilly, BMS, Otsuka, Johnson & Johnson, Pfizer, Merck, Targacept, NeuroSearch and UBC; Grant/Research support from Alkermes, Johnson & Johnson, Otsuka and Pfizer. Drs Gopal, Gassmann-Mayer, Lim, Hough, and Ms. Lane are employees of Johnson & Johnson Pharmaceutical Research & Development, L.L.C. Mr Remmerie is an employee of Johnson & Johnson Pharmaceutical Research & Development, Division of Janssen Pharmaceutica, N.V., Beerse, Belgium. Dr Eerdekens was employed by Johnson & Johnson Pharmaceutical Research & Development, Division of Janssen Pharmaceutica, N.V., Beerse, Belgium at the time of this study and is currently employed by Grünenthal GmbH, Aachen, Germany. Drs Lim, Gassmann-Mayer, and Eerdekens, and Ms. Lane hold Johnson & Johnson stock and stock options.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}