






Abstract
We evaluated the effect of monomeric C-reactive protein (CRP) deposition on areas at risk (AAR) of myocardium with ischaemia-reperfusion injury.
Myocardial ischaemia–reperfusion injury model was produced by ligation of the left anterior descending coronary artery for 45 min followed by 45 min of reperfusion using female Sprague-Dawley rats. Tissue from non-ischaemic areas, areas at risk and infarct areas determined by Evans blue and 2,3,5-triphenyltetrazolium chloride staining was obtained from the sham group, the ischaemia–reperfusion injury without C-reactive protein (CRP) injection group (I/R only group), and the ischaemia–reperfusion injury with CRP injection group (I/R + CRP group). We assessed the effect of CRP injection on infarct size, CRP deposition, CRP and IL-6 mRNA expression, the third component of complement (C3) immunodeposition and mitochondrial structural remodelling with apoptosis by quantitative RT-PCR analyses, immunohistochemistry, direct immunofluorescence, electron microscopy and Terminal deoxynucleotide transferase dUTP Nick End Labelling assay, respectively. All images were analysed using an automated morphology tool.
The infarct area significantly increased in the I/R + CRP group compared to the I/R only group. The anti CRP antibody confirmed that CRP deposition occurred in both the infarct and area at risk (AAR) of the I/R + CRP group. The myocardium did not exhibit CRP mRNA expression, and the CRP treatment group showed a tendency for IL-6 to increase without statistical significance. Activated C3, apoptosis and mitochondrial destruction increased on AAR and infarct area in the I/R + CRP group.
These results strongly suggest the active participation of the deposition of CRP on AAR in the progression of myocardial infarction following ischaemia–reperfusion injury, accompanied by complement activation and mitochondrial change.
INTRODUCTION
C-reactive protein (CRP) is an acute phase protein synthesized in the liver and induced by IL-6 when inflammation, infection or cell damage occurs [1–3]. CRP is composed of 5 identical subunits and possesses 2 major forms: pentameric CRP (pCRP) and monomeric CRP (mCRP) [4]. CRP circulates in the serum in the pCRP form. When it comes into contact with the damaged cell membrane, CRP changes into mCRP and is deposited on the activated or apoptotic cell membrane [5, 6]. While pCRP has an anti-inflammatory role, deposited mCRP plays a strong inflammatory role in monocytes and produces oxygen-free radicals in monocytes, aggravating cell injury [5–7].
Elevated serum CRP levels have been found in myocardial infarction and unstable angina [8, 9], and elevated serum CRP levels have been correlated with greater myocardial tissue injury, heart failure progression and mortality rates. CRP was deposited in infarcted myocardium and aggravated myocardial infarction [10–12]. Complement activation and complement-mediated damage play an important role in ischaemia–reperfusion injury and progression of myocardial infarction [13, 14]. CRP is a major activator of complement systems and is deposited (with the complement systems) at tissue injury sites such as myocardial infarction and skin burns [15, 16]. In ischaemia–reperfusion injury, functional and structural changes in mitochondria can induce apoptosis by halting ATP synthesis and structural remodelling, releasing proapoptotic proteins, impairing calcium ionic homeostasis and producing reactive oxygen species [17].
However, no research has been conducted regarding the deposition of CRP on the ischaemic but viable area (area at risk or AAR) in ischaemic reperfusion injury [18]. We hypothesized that CRP also would be deposited on AAR and that deposition would aggravate myocardial infarction in the ischaemia–reperfusion injury through complement activation, mitochondrial remodelling and apoptosis.
MATERIALS AND METHODS
Experimental protocols
We constructed a myocardial ischaemia–reperfusion injury model using female Sprague-Dawley rats weighing between 220 and 260 g. The animals were treated according to the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, Washington, DC, USA). Moreover, we conducted our experimental study in accordance with the ARRIVE guideline. The study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the SMG-SNU Boramae Medical Center Biomedical Research Institute (approval number: 2015-0009). On the sham group (n = 3), we performed left thoracotomy and pericardiotomy without manipulating of the heart to induce myocardial ischaemia. After carrying out the procedures and then waiting for 90 min, we euthanized the rats and then performed autopsies (Fig. 1A). In the ischaemia–reperfusion only group (I/R only group, n = 5), myocardial ischaemia was produced by ligation of the left anterior descending coronary artery approximately 2 mm distal to its origin by snaring it with 6-0 nylon double sutures (buttressed with a small piece of plastic tube for 45 min) and reperfusion for 45 min. After reperfusion, the heart was quickly excised (Fig. 1B). In the ischaemia–reperfusion plus CRP injection group (I/R + CRP group, n = 9), 100 µg bolus of high-purity (>99%) human CRP obtained from human plasma (C4063; Sigma-Aldrich, Saint Louis, MO, USA) were infused via the femoral vein after the release of coronary ligation (Fig. 1C).
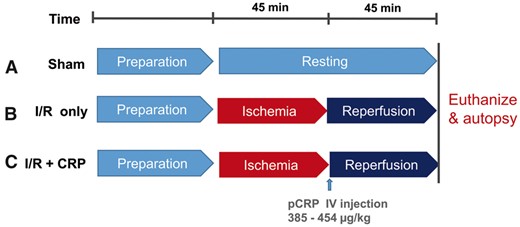
Schematic illustration of experimental protocols.
Histopathologic analysis and immunohistochemistry
The sectioned heart was fixed in 10% buffered formalin and embedded in paraffin. The 4-µm-thick tissue sections were stained with haematoxylin and eosin. The heart sections were immunostained using rabbit polyclonal anti-CRP antibody (ab32412; Abcam, Cambridge, UK; 1:250 dilution) and human monoclonal anti-CRP antibody (C1688; Sigma-Aldrich; 1:400 dilution). Although ab32412 cannot distinguish between pCRP and mCRP, we point out that anti-CRP antibody C1688 recognizes an epitope located on the 24 kD subunit of CRP and that it can detect the monomeric form of CRP.
Immunofluorescence study of complement and electron microscopy
The frozen heart tissue sections were stained with FITC anti-C3, C4, C1q, IgG and IgM (Ventana Medical Systems, Tucson, AZ, USA). Images of tissue sections were captured using a Zeiss LSM 780 confocal microscope (GmBH1997-2015; Carl Zeiss, Oberkochen, Germany) and fluorescence microscopy.
Ultrathin sections were examined with a transmission electron microscope (model GEM-1400; JEOL, Tokyo, Japan). The diameters of 50 mitochondria per sham, I/R only, or I/R + CRP group rat were measured to assess the severity of mitochondrial injury and degree of morphological change.
Analysis of C-reactive protein mRNA levels in rat myocardium
CRP total RNA was isolated from rat myocardium using a miRNeasy Mini Kit (Qiagen, Hilden, Germany). Quantitative RT-PCR analyses of CRP were performed using a TaqMan Gene Expression Assay (Rn000567307_g1; Applied Biosystems, Carlsbad, CA, USA). The ACTB (Rn00667869_m1; Applied Biosystems, Foster City, CA, USA) was used as an endogenous control for normalization purposes.
IL-6 RNA expression assay
RNA was prepared using a miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. The extracted RNA (1 µg) was reverse transcribed using a Reverse Transcription System (Promega, Madison, WI, USA), and cDNAs were amplified using a GeneAmp PCR System 9700 (Applied Biosystems). qRT-PCR analysis of IL-6 was done using TaqMan Gene Expression Assays (Rn01410330_m1; Applied Biosystems) and a 7900HT Fast Real-Time PCR System (Applied Biosystems). The rat ACTB (Rn00667869_m1; Applied Biosystems) endogenous control was used for normalization purposes.
Determination of cardiomyocyte apoptosis with Terminal deoxynucleotide transferase dUTP Nick End Labelling assay
Adjacent serial sections in dewaxed slides were stained using Terminal deoxynucleotide transferase dUTP Nick End Labelling (TUNEL) assay (ApoBrdU-IHC DNA Fragmentation Assay Kit, BV-K403; Biovision, Milpitas, CA, USA) to label DNA breaks and detect apoptotic cells through immunohistochemistry. The nuclei of the apoptotic cells exhibited brown staining, whereas those of non-apoptotic cells and the negative control were stained blue. The number of apoptotic cells in each group was counted, and the apoptotic index (number of apoptotic cells / total number of cells × 100%) of the non-ischaemic areas, the AAR and the infarct in each specimen was calculated [19].
Image analysis
All images of Evans blue and TTC staining and CRP immunohistochemistry were acquired using a Vectra automated imaging system (PerkinElmer, Waltham, MA, USA). The infarct area (white zone after the Evans blue and TTC staining) and the AAR area (red zone after the Evans blue and TTC staining) were automatically calculated by inForm (PerkinElmer) imaging analysis software. The size of the infarct was expressed in terms of the ratio of infarct to the whole ischaemic area [infarct / (AAR + infarct)]. The areas exhibiting CRP immunopositivity were expressed in terms of the ratio of CRP-immunostained area to the whole ischaemic area [CRP-immunostained area / (AAR + infarct)].
Statistical analysis
Data was expressed and plotted as the mean with standard error of the mean. The normality of all parameters was tested using the Kolmogorov-Smirnov test. To compare 2 groups, a 2-tailed t-test was used. For comparison of 3 groups, 1-way analysis of variance and Tukey’s post hoc analysis were used. In cases where normality was excluded, the non-parametric Mann–Whitney U test was used to compare 2 groups. A P-value < 0.05 was considered to be statistically significant. Analysis of data was performed using GraphPad Prism software version 5.0 (GraphPad Software, San Diego, CA, USA) or SPSS V.18.0 (SPSS, Chicago, IL, USA).
An expanded Methods section is available in the Supplementary Data.
RESULTS
Histopathological analysis, C-reactive protein immunohistochemistry and C-reactive protein mRNA expression
After haematoxylin and eosin staining, myocytes in the infarct area showed contraction bands with intensely eosinophilic intracellular stripes, which are representative of ischaemia–reperfusion injury (Fig. 2B). Moreover, cardiomyocytes in the infarct area showed intense cytoplasmic immunostaining with both anti-CRP antibodies (ab32412; Fig. 2B, C1688).
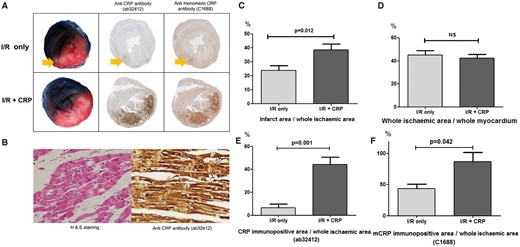
TTC and Evans blue staining and CRP immunohistochemistry. (A) White myocardium represents infarct area, red myocardium represents area at risk and blue myocardium represents non-ischaemic area. In the I/R only group, only infarct area (arrow) was faintly immunostained by both anti-CRP antibody (ab32412) and anti-mCRP antibody (C1688). In contrast, the I/R + CRP group showed that both areas at risk and infarct area (red and white zone) were strongly immunostained by both antibodies. (B) Representative haematoxylin and eosin and CRP immunostaining of damaged myocardium in the I/R + CRP group. Cardiomyocytes showed contractile bands typical of ischaemia–reperfusion injury. The damaged myocardial cells showed diffuse cytoplasmic immunostaining by anti-CRP antibody (magnification ×400). (C) The ratio of infarct area to whole ischaemic area was significantly larger in CRP injection group. (D) However, the ratio (whole ischaemic area to whole myocardium) was not significantly different. (E) CRP-immunostained area (ab32412) increased more in the I/R + CRP group than in the I/R only group. (F) Anti-mCRP antibody-immunostained area (C1688) also increased more in the I/R + CRP group than in the I/R only group.
After Evans blue/TTC staining, the non-ischaemic area was stained blue, the AAR was stained red, and the infarct was stained white (Fig. 2A). The ratio of infarct to the whole ischaemic area was much higher in the I/R + CRP group (38.5 ± 4.2%) than in the I/R only group (23.9 ± 3.4%, P = 0 .012; Fig. 2A and C). On the other hand, the ratio of the whole ischaemic area to the whole myocardium was not statistically different between the 2 groups (I/R only group: 45.2 ± 3.8%, I/R + CRP group: 42.3 ± 3.3%, P = 0.592; Fig. 2D). These results demonstrated that the myocardial infarct size increased in the CRP-deposited myocardium, and that the infused CRP did not affect the perfused area (outside of the left anterior descending coronary artery territory), but affected the ischaemic area.
In the I/R only group, a small amount of CRP immunopositivity was detected in the infarct area. On the other hand, in the I/R + CRP group, CRP was extensively immunostained with the ab32412 and C1688 antibodies (Fig. 2A) not only in the infarct area (white zone) but also in the AAR (red zone). In the whole section of the heart, the anti-CRP antibody positive area (ab32412) was significantly larger after infusion of CRP (I/R only group: 6.4 ± 3.4%, I/R + CRP group: 44.3 ± 6.4%, P = 0.001; Fig. 2E). The anti-mCRP antibody positive area (C1688) was also significantly larger in the I/R + CRP group (I/R only group: 43.8 ± 7.0%, I/R + CRP group: 86.8 ± 14.7%, P = 0.042; Fig. 2F). In sham group, there was no ischaemia, infarction and CRP deposition.
The CRP mRNA was not expressed in rat myocardium using quantitative RT-PCR analyses in any group. These results support the notion that CRP immunostaining was a consequence of deposition rather than CRP production in ischaemic reperfusion injury in rat myocardium.
IL-6 expression increased after C-reactive protein injection
CRP is induced by IL-6, and consequently, the deposited monomeric form of CRP is known to induce proinflammatory reaction. In this study, the IL-6 expression pattern did not show statistically significant difference but it showed tendency to increase after CRP injection (P = 0.076; Fig. 3).
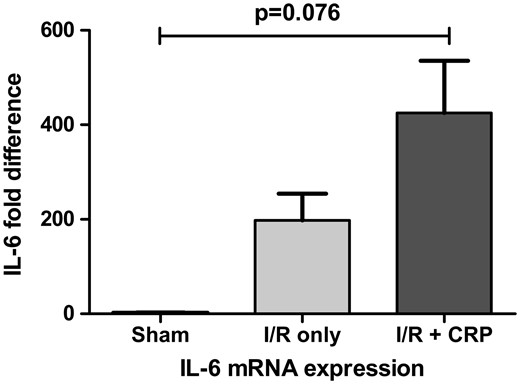
IL-6 mRNA expression showed tendency to increase after CRP injection. However, there was no statistical significance (P = 0.076).
C3 activated in the area at risk and infarct area in the ischaemia–reperfusion injury with C-reactive protein injection group
C3 deposition was mainly deposited in the infarct area in the I/R only group (Fig. 4A), whereas it was deposited more extensively in both the AAR and infarct area in the I/R + CRP group (Fig. 4B). Interestingly, the complement was co-localized in the CRP-immunodeposited area in the AAR (Fig. 4C) in the I/R + CRP group. IgG was deposited mainly in the contraction band of myocardial cells in the infarcted area in both groups [Fig. 4A(d) and B(h)]. C1q and IgM were non-specifically deposited, and C4 was not detected in either group (data not shown).

Relationship among infarct area, area at risk (AAR), C3 and IgG deposition. (A) The I/R only group demonstrated that only infarct (arrow) showed C3 immunofluorescence (b, ×100 tile scan, c, ×200) and only contraction band of infarct showed IgG positivity (d, ×200). (B) The ischaemia–reperfusion (I/R) + C-reactive protein (CRP) group showed more extensive infarction (e), and both infarct area and AAR (arrow) showed diffuse C3 positivity (f, ×100 tile scan, g, ×200). As in the I/R only group, IgG was deposited only in the contraction band of infarct area (h, ×200). (C) Relationship among AAR, CRP (ab32412) immunopositivity, and C3 immunofluorescence in the I/R + CRP group. Arrows indicate AAR. AAR in the CRP infusion group showed intense CRP deposition, and this region correlated with the C3 activation site (k, ×100 tile scan, l, ×400).
Mitochondria swollen and extensive autophages production in the area at risk in the ischaemia–reperfusion injury with C-reactive protein injection group
Electron microscopy showed that the mitochondria of the non-ischaemic area still had normal architecture with multiplicity of cristae in their native configuration and inner/outer mitochondrial membranes (Fig. 5A). In contrast, the mitochondria in the AAR in the I/R + CRP group were swollen and exhibited cristae disorganization. Abundant autophages were produced to a greater extent in the I/R + CRP group than in the I/R only group (Fig. 5B, arrow). The mitochondrial diameters in the AAR were significantly greater in the I/R + CRP group than in the I/R only group (1169.3 ± 31.1 vs 921.9 ± 30.9 nm, P < 0.001; Fig. 5C). In the infarct, both the I/R only group and the I/R + CRP group showed amorphous matrix densities, which are signs of irreversible ischaemic injury (Fig. 5D, arrow). The mitochondria were more shrunken and condensed in the I/R + CRP group than in the I/R only group (1028.0 ± 24.3 vs 1140.2 ± 38.3 nm, P = 0.026; Fig. 5E).
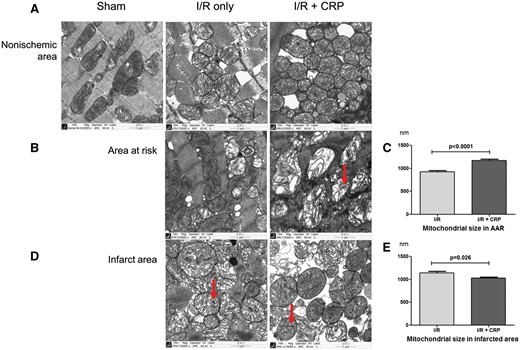
Transmission electron micrographs of non-ischaemic areas, areas at risk and infarct area in myocardium in each group. (A) Non-ischaemic areas in all groups showed normal mitochondria with tightly packed cristae and electron-dense matrices. (B) Mitochondria in areas at risk. In the I/R only group, some mitochondria maintained normal architecture with multiplicity of cristae in their native configuration. In contrast, the CRP infusion group showed that almost all mitochondria had swollen and exhibited disorganization of cristae with abundant autophages formation (arrow) and (C) mitochondrial diameters in AAR were significantly greater after CRP infusion (P < 0.001). (D) Infarct area. Both groups showed amorphous matrix densities that are signs of irreversible ischaemic injury. (E) Mitochondria were more condensed and smaller on AAR in the CRP infusion group (P = 0.026).
Apoptosis increased in the area at risk in the ischaemia–reperfusion injury with C-reactive protein injection group
Representative TUNEL assay results are shown in Fig. 6A–C. In all areas of the sham group and the non-ischaemic area of the I/R only group and the I/R + CRP groups, most myocardial cells were non-apoptotic (Fig. 6A). In the AAR, TUNEL staining showed a significant increase in the cardiomyocyte apoptotic rate in the I/R + CRP group compared to that in the I/R only group (60.1 ± 6.0% vs 31.2 ± 7.7%, P = 0.013; Fig. 6B). In contrast, there was no statistical difference in the cardiomyocyte apoptotic rate in the infarct area in the 2 groups (I/R only group: 75.5 ± 4.6%, I/R + CRP group: 70.2 ± 6.8%, P = 0.373; Fig. 6C).
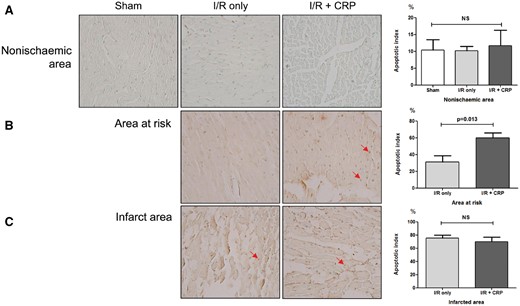
Representative apoptotic cells determined with Terminal deoxynucleotide transferase dUTP Nick End Labelling and quantitative apoptotic indices. Arrows indicate apoptotic cells. Apoptotic nuclei showed brown staining (magnification ×400). (A) In all groups, most myocardial cells in non-ischaemic areas were nonapoptotic. (B) In areas at risk, the CRP infusion group showed increased apoptotic indices (P = 0.013). (C) However, in infarct, there was no significant difference between the 2 groups (P = 0.373).
DISCUSSION
The present study demonstrated 3 main findings. Firstly, myocardial infarct size was increased after CRP injection, and the effect of CRP deposition on ischaemia–reperfusion injury model in rat myocardium occurred in both the infarct area and AAR. Secondly, C3 was also immunodeposited in both the infarct area and AAR after CRP injection, and was co-localized in the CRP deposited area. Thirdly, the region of AAR showed mitochondrial destruction, autophagy formation and increased apoptosis.
Results of the present study are consistent with those of previous studies showing that the presence of serum CRP at the time of myocardial infarction increases the entire myocardial infarct size for human autopsy specimens with acute myocardial infarction [15] and a rat ischaemia–reperfusion model. These studies evaluated CRP deposition only in the irreversibly infarcted area. In our study, however, we reperfused sufficiently after ischaemia to simulate AAR, which is viable but prone to irreversible infarct, and evaluated the effect of CRP deposition in both the infarct area and AAR. To the best of our knowledge, the present study may be the first to show the effect of CRP deposition in the AAR induced by ischaemia–reperfusion injury.
In this study, the CRP treatment group showed a tendency for IL-6 to increase without statistical significance. As IL-6 is an upstream inducer of CRP [20], an increase in IL-6 might enhance endogenous CRP production in the rats’ livers. However, rat CRP does not activate rat complement, whereas human CRP potently activates both rat and human complements [21]. Using human CRP is a good method for controlling complement activation by endogenous CRP [10]. Therefore, we used human CRP as it was done in previous studies [22]. We used anti-human CRP antibodies to detect CRP deposition.
Vilahur et al. [23] showed that short periods of myocardial ischaemia are related to increased mCRP mRNA expression in infiltrated macrophages. In addition, Slevin et al. [24] showed that ischaemic brain tissue and peri-infarcted brain tissue have mCRP expression. We sought to determine whether CRP immune-positivity is a result of myocardial cell-origin mCRP mRNA expression. However, we found no mCRP mRNA expression in the non-ischaemic area, infarct area or AAR of the myocardial tissues in both the I/R only group and I/R + CRP groups. We therefore concluded that mCRP immune-positivity was the result of immunodeposition rather than CRP production in myocardium.
CRP is known as one of the main activators of classical complement pathways. In previous studies, CRP was co-localized with complement, and activation of the complement aggravated myocardial infarction [12, 15]. In addition to the myocardium, ischaemic brain tissue showed co-localization with CRP and complement [24]. After removal of the complement, the CRP failed to aggravate the degree of myocardial infarction [15]. Moreover, the complement activated by CRP deposition has an inflammatory function [12]. While previous studies demonstrated that the complement was activated only in the infarct area, we showed that complement C3 was colocalized to the AAR as well as to the infarct area and that the presence of complement C3 was associated with an increase in myocardial infarct size. Thus, we inferred that CRP immunodeposition on the AAR might also deteriorate ischaemia–reperfusion injury through complement activation. As the classical complement pathway is also activated by IgG or IgM, we studied IgG and IgM. IgG was found only in the infarct area with contraction bands in both groups, and IgM was non-specifically immunostained. The C3 activation in the AAR in the I/R + CRP group therefore appears to be a consequence of CRP rather than IgG or IgM in the classical complement pathway activation.
It has been well recognized that mitochondria are important modulators that decide the fate of cells such as programmed cell death, necrosis, and apoptosis [25]. Structural and functional dysregulation of mitochondria induces irreversible cell damage through the loss of ATP synthesis, the increase in ATP hydrolysis, the impairment of Ca2+ homeostasis, the formation of reactive oxygen species, and the release of proapoptotic proteins in ischaemia–reperfusion injury [26]. Moreover, autophagy driven by mitochondria can be subjected to the extreme stress of ischaemia–reperfusion injury and lead to cell death. Whereas autophagy may be protective under mild ischaemia, it can contribute to damage by causing cell death under severe ischaemia–reperfusion injury [27] and play a significant role in myocyte disappearance and the development of contractile dysfunction in failing hearts [28]. Therefore, mitochondria preservation is important for myocardial protection when dealing with ischaemia–reperfusion injury. We indicated that mitochondria in the AAR with CRP immunodeposition were severely damaged, lost their normal configuration and size, and induced numerous autophages. As we expected, we demonstrated that the apoptotic rate increased after CRP treatment of the AAR. This finding was consistent with those of previous studies such as an in vitro study using stimulated primary neonatal rat cardiomyocytes. In that study, hypoxia-induced cardiomyocytes with co-treatment CRP showed significantly increased apoptosis, which was related to the cytochrome c release by mitochondria into the cytosol. It is concluded that CRP could enhance apoptosis in hypoxia-stimulated cardiomyocytes via the mitochondrial dependent pathway [19]. Therefore, we can speculate that deposited CRP may have a profound effect on mitochondrial damage. As a result of mitochondrial damage, hypoxic myocardium may become more prone to apoptotic death.
Pepys et al. first proposed the inhibition of mCRP deposition for the treatment of cardiovascular diseases [13]. They used 1,6-bis(phosphocholine)-hexane as an inhibitor of CRP conformational change to reduce myocardial infarct size [10, 12]. In a clinical setting, smoking, diabetes, obesity and metabolic syndromes are major risk factors for cardiovascular diseases, and serum CRP is elevated in these clinical settings [29]. As it is almost impossible to salvage the infarcted myocardium, salvaging the AAR might be a new effective cardio-protection strategy. Inhibiting CRP deposition in the AAR may be beneficial during the immediate post-recanalization procedure in patients who have increased serum CRP. Control of CRP conformational change and inhibition of CRP deposition in such sets to undergo coronary artery revascularization could also be good preoperative strategies. In the future, sequestration of pCRP by tailored antibodies (for inhibiting transformation of pCRP into mCRP) or selective inhibition of deposition of mCRP in the injured myocardium will prove to be a promising method for minimizing ischaemia–reperfusion injury in patients with elevated serum CRP.
Limitations
It was reported that sodium azide (contained in human CRP for the preservation of proteins) could make an artefact in the study of vascular endothelial cells [29]. And human-derived CRP could result in alloimmune reaction, and this influence apoptosis and mitochondrial changes. Therefore, the experiment with purified endotoxin-free, azide-free recombinant CRP is being conducted. This study showed that groups having CRP injections had greater apoptotic rates and more CRP immunodeposition. Previous studies suggested that CRP binds to apoptotic cells in skin burns [6] or apoptotic lymphocytes in a Ca2+dependent manner and augmented the classical complement pathway [30]. Therefore, we cannot totally exclude the possibility that the CRP injection increased apoptosis in different, unknown ways (other than mitochondrial damage), and mCRP was deposited in the apoptotic cells.
CONCLUSION
In summary, we demonstrated that CRP was deposited in the infarct area and AAR in ischaemia–reperfusion injury and that monomeric CRP deposition was related to complement activation, mitochondrial injury, apoptosis and aggravated myocardial infarction. Inhibition of CRP deposition in ischaemia–reperfusion damaged myocardium could be a new therapeutic strategy for the post-recanalization of ischaemic myocardium.
SUPPLEMENTARY MATERIAL
Supplementary material is available at ICVTS online.
ACKNOWLEDGMENTS
We thank Kyung Min Park, Gil-Je Lee and Yun Jae Kim for the image analysis, and Eun Jung Jeon and So Ra Kim for the molecular work. We also would like thank So Won Kim for her help with the editing of our manuscript.
Funding
This work was supported by a clinical research grant-in-aid from the Seoul Metropolitan Government Seoul National University (SMG-SNU) Boramae Medical Center (03-2015-0).
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}