






Abstract
Currently available cardioplegic solutions provide excellent protection in patients with normal surgical risk; in high-risk patients, however, such as in emergency coronary artery bypass surgery, there is still room for improvement. As most of the cardioplegic solutions primarily protect myocytes, the addition of substances for protection of the endothelium might improve their protective potential. The nitric oxide donor, S-nitroso human serum albumin (S-NO-HSA), which has been shown to prevent endothelial nitric oxide synthase uncoupling, was added to the newly developed histidine-tryptophan-ketoglutarat (HTK-N) cardioplegia in an isolated heart perfusion system after subjecting rats to acute myocardial infarction (MI) and reperfusion.
In male Sprague-Dawley rats, acute MI was induced by ligation for 1 h of the anterior descending coronary artery. After 2 h of in vivo reperfusion hearts were evaluated on an isolated erythrocyte-perfused working heart model. Cold ischaemia (4°C) for 60 min was followed by 45 min of reperfusion. Cardiac arrest was induced either with HTK (n = 10), HTK-N (n = 10) or HTK-N + S-NO-HSA (n = 10). In one group (HTK-N + S-NO-HSA plus in vivo S-NO-HSA; n = 9) an additional in vivo infusion of S-NO-HSA was performed.
Post-ischaemic recovery of cardiac output (HTK: 77 ± 4%, HTK-N: 86 ± 7%, HTK-N + S-NO-HSA: 101 ± 5%, in vivo S-NO-HSA: 93 ± 8%), external heart work (HTK: 79 ± 5%, HTK-N: 83 ± 3%, HTK-N + S-NO-HSA: 101 ± 8%, in vivo S-NO-HSA: 109 ± 13%), coronary flow (HTK: 77 ± 4%, HTK-N: 94 ± 6%, HTK-N + S-NO-HSA: 118 ± 15%, in vivo S-NO-HSA: 113 ± 3.17%) [HTK-N + S-NO-HSA vs HTK P < 0.001; HTK-N + S-NO-HSA vs HTK-N P < 0.05] and left atrial diastolic pressure (HTK: 122 ± 31%, HTK-N: 159 ± 43%, HTK-N + S-NO-HSA: 88 ± 30, in vivo S-NO-HSA: 62 ± 10%) [HTK-N + S-NO-HSA vs HTK P < 0.05; in vivo S-NO-HSA vs HTK-N P < 0.05] were significantly improved in both S-NO-HSA-treated groups compared with HTK and HTK-N, respectively. This was accompanied by better preservation of high-energy phosphates (adenosine triphosphate; energy charge) and ultrastructural integrity on transmission electron microscopy. However, no additional benefit of in vivo S-NO-HSA infusion was observed.
Addition of the NO donor, S-NO-HSA refines the concept of HTK-N cardioplegia in improving post-ischaemic myocardial perfusion. HTK-N with S-NO-HSA is a possible therapeutic option for patients who have to be operated on for acute MI.
INTRODUCTION
Currently postoperative outcome and mortality of routine coronary artery bypass grafting (CABG) in patients with stable angina are excellent. However, in patients, who have to be operated with acute coronary syndrome, the mortality rate ranges from 1.6% up to 46.7%, depending on the patient’s preoperative haemodynamic condition [1, 2]. Avoiding further damage to the myocardium, preservation of coronary perfusion and minimizing the ‘no-reflow phenomenon’ seem to be of great importance in the case of ongoing ischaemia [3].
Major determinants of endothelial injury leading to the ‘no-reflow phenomenon’ are the generation of reactive oxygen species as a consequence of ischaemia/reperfusion and reduced nitric oxide (NO) bioavailability [4]. During ischaemia the constitutive endothelial NO synthase (eNOS) is activated leading to a burst of NO production which causes the depletion of l-arginine and tetrahydrobiopterin [5, 6]. As a consequence of substrate and co-factor deficiency, eNOS may become uncoupled leading to high production of superoxide after reperfusion, and consequently high production of peroxynitrite [7, 8]. Thereby NO bioavailability is further decreased and oxidative stress increased. The supplementation of exogenous NO, on the one hand, can prevent eNOS-uncoupling due to its negative feedback on the production of NO by eNOS (product inhibition of the enzyme) and, on the other hand, restores NO levels acting as vasodilator and radical scavenger [9, 10].
The direct NO donor, S-nitroso human serum albumin (S-NO-HSA) is a high-molecular-weight S-nitrosothiol with a high nitrosograde and a prolonged half-life compared with low-molecular-weight S-nitrosothiols [7]. In the setting of 15 min of unprotected warm ischaemia in the pig heart S-NO-HSA stabilized the basal production of NO, prevented the uncoupling of the constitutive eNOS and reduced the production of superoxide (O2−) and peroxynitrite (ONOO−), thereby improving coronary flow and haemodynamic performance [9].
In the present series of experiments S-NO-HSA was added to the histidine-tryptophan-ketoglutarat (HTK-N) cardioplegic solution in a rat model of acute MI subjected to 1 h of cold ischaemia on an isolated organ perfusion model. By this experimental setting we wanted to test S-NO-HSA in ischaemically damaged hearts before undergoing global ischaemia due to cardioplegic arrest such as in the case of emergency CABG in patients with unstable angina or previous MI.
We tested two hypotheses depending on the time point of S-NO-HSA supplementation:
Does the addition of S-NO-HSA to the new cardioplegic solution HTK-N further improve post-ischaemic recovery of endothelial and myocardial function?
Is there additional benefit if S-NO-HSA therapy is started at the time point of reperfusion of the ischaemic left anterior descending coronary artery (LAD) territory in evolving infarction; i.e. at the time point when the decision for emergency CABG is made after incomplete revascularization due to failed percutaneous coronary intervention (PCI) in the clinical setting?
MATERIALS AND METHODS
Animal preparation
All experiments were approved by the Committee for Animal Research, Vienna Medical University and were in compliance with the European Convention on Animal Care. In total 55 adult male Sprague-Dawley rats were anaesthetized by intraperitoneal administration of Xylazin (4 mg/kg; Bayer) and Ketamin (100 mg/kg; Dr E. Gräub AG, Bern), intubated and ventilated (Ugo Basile 7025 rodent ventilator, 4.5–5 ml/stroke, 70 strokes/min). To maintain anaesthesia, an intravenous infusion of 1.5 mL/h saline with 0.5 mg/h Ketamin (Ketasol, Dr E. Gräub AG) and 0.018 mg/h Xylazin (Rompun, Bayer) was applied via a perfusor.
Model of myocardial infarction
MI was induced by ligature of the LAD using a 7-0 Prolene suture which was tied by a tourniquet [11]. To simulate patients with ongoing infarction, the tourniquet was reopened after 60 min of ischaemia and followed by 2 h of in vivo reperfusion. Immediately after LAD occlusion the animals were randomly assigned into two different groups. In one group (n = 16) intravenous infusion of S-NO-HSA started 15 min after LAD occlusion and was continued for 75′ (0.3 µmol/kg/h); i.e. for the last 45 min during in vivo occlusion and for 30′ during in vivo reperfusion. In the other animals (n = 39) an equal volume of saline was infused. In total 49 animals completed the in vivo part of the experiment; 2 animals receiving S-NO-HSA and 4 animals with saline infusion did not complete the study protocol due to haemodynamic instability; i.e. 14 animals receiving S-NO-HSA, and 35 animals receiving saline infusion. At the end of the reperfusion period five animals receiving S-NO-HSA infusion and five animals receiving saline infusion were randomly taken to assess infarct size by Evans Blue and 2,3,5-triphenyl tetrazolium chloride (TTC) staining.
Experimental protocol and perfusion system
The remaining 30 animals having received saline during reperfusion were randomly assigned into three groups for the experiments on the isolated working heart: In Group 1 (HTK, n = 10) cardioplegic arrest was induced by HTK cardioplegia (2.48 ml/g heart weight), in Group 2 (HTK-N, n = 10) HTK-N cardioplegia was used (2.48 ml/g heart weight) and in Group 3 (HTK-N + S-NO-HSA, n = 10) the NO donor, S-NO-HSA was added to cardioplegia (2.48 ml/g heart weight HTK-N + 0.3 µmol S-NO-HSA). Group 4 (in vivo S-NO-HSA) consisted of the remaining 9 animals with S-NO-HSA infusion (0.3 µmol/kg/h) during in vivo reperfusion and cardioplegic arrest in vitro induced by HTK-N + S-NO-HSA (2.48 ml/g heart weight HTK-N + 0.3 µmol S-NO-HSA).
In Fig. 1A, study groups are summarized. Figure 1B shows the time schedule of the entire experiment.
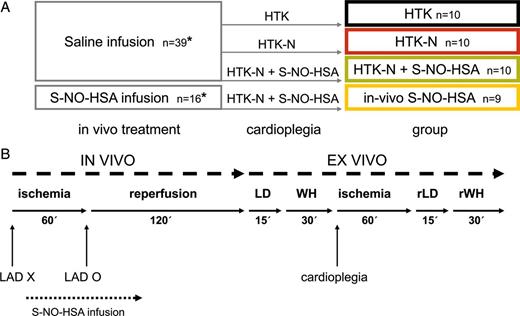
(A) Treatment scheme for the four groups (1) HTK, (2) HTK-N, (3) HTK-N + S-NO-HSA, (4) in vivo S-NO-HSA; *at the end of the in vivo part of the experiment five animals receiving S-NO-HSA infusion and five animals receiving saline infusion were randomly taken to assess infarct size by Evans Blue and 2,3,5-triphenyl tetrazolium chloride (TTC) staining. (B) Time schedule of the experimental protocol. LAD X: occlusion of LAD; LAD O: reopening of LAD; LD: langendorff mode; WH: working heart mode; rLD: langendorff mode during ex vivo reperfusion; rWH: working heart mode during ex vivo reperfusion.
After completion of 2 h of in vivo reperfusion rats were heparinized with 200 IU/kg intravenously, the hearts were excised and weighed, and haemodynamic performance was evaluated on an isolated erythrocyte perfused working heart system (Hugo Sachs Elektronik, Freiburg, Germany), as described previously [12]. As perfusate, a suspension of purified bovine red blood cells in a modified Krebs–Henseleit buffer was used with a haematocrit of ∼30%. The perfusate was oxygenated with a gas mixture of 20% oxygen, 5% carbon dioxide and 75% nitrogen by a hollow fibre dialysator.
In the Langendorff mode (LD before ischaemia, rLD during reperfusion) coronary arteries were perfused at a constant pressure of 70 mmHg while the left ventricle was beating without volume load. After cannulation of the left atrium the perfusion system was converted to the working heart mode, where the left ventricle was ejecting against a predefined afterload corresponding to a mean aortic pressure of 70 mmHg. The mean arterial pressure is set to 70 mmHg by pressurizing the Windkessel on top of the aortic cannula at the beginning of the experiment. For induction of cold cardioplegic arrest HTK and HTK-N cardioplegia, respectively, were used at a temperature of 4–6°C.
Drugs
HTK and HTK-N both are intracellular, crystalloid cardioplegic solutions. The major modifications of HTK-N compared with HTK are the enrichment with the amino acids aspartate, l-arginine, l-glycine and l-alanine, with the two iron chelators desferal (deferoxamine) and LK-614, and the partial substitution of l-histidine by N-α-acetyl-l-histidine. The exact composition of HTK and HTK-N is given in Table 1 [13]. S-NO-HSA is a high-molecular-weight S-nitrosothiol with a high S-nitrosograde and exact equimolar nitrosation (S-NO- in position Cys-34 of HSA: ≈0.8 mol/mol protein). It has a prolonged half-life compared with low-molecular-weight S-nitrosothiols. S-NO-HSA preparation is described in detail in Hallström et al. [9].
Composition of HTK and HTK-N
| HTK-N46b | HTK (Custodiol®) | |
|---|---|---|
| mmol/l | mmol/l | |
| Ions | ||
| Na+ | 16 | 15 |
| K+ | 10 | 9 |
| Ca2+ | 0.02 | 0.015 |
| Mg2+ | 8 | 4 |
| Cl− | 30.04 | 50.03 |
| Amino acids | ||
| l-histidine | 124 | 180 + l-histidine HCl |
| N-alpha-acetyl-l-histidine | 57 | – |
| l-tryptophan | 2 | 2 |
| l-aspartate | 5 | – |
| l-arginine | 3 | – |
| l-glycine | 10 | – |
| l-alanine | 5 | – |
| Iron chelators | ||
| Desferal (deferoxamine) | 0.025 | – |
| LK-614 | 0.0075 | – |
| Others | ||
| Ketoglutarate | 2 | 1 |
| Saccharose | 33 | – |
| Mannitol | – | 30 |
| HTK-N46b | HTK (Custodiol®) | |
|---|---|---|
| mmol/l | mmol/l | |
| Ions | ||
| Na+ | 16 | 15 |
| K+ | 10 | 9 |
| Ca2+ | 0.02 | 0.015 |
| Mg2+ | 8 | 4 |
| Cl− | 30.04 | 50.03 |
| Amino acids | ||
| l-histidine | 124 | 180 + l-histidine HCl |
| N-alpha-acetyl-l-histidine | 57 | – |
| l-tryptophan | 2 | 2 |
| l-aspartate | 5 | – |
| l-arginine | 3 | – |
| l-glycine | 10 | – |
| l-alanine | 5 | – |
| Iron chelators | ||
| Desferal (deferoxamine) | 0.025 | – |
| LK-614 | 0.0075 | – |
| Others | ||
| Ketoglutarate | 2 | 1 |
| Saccharose | 33 | – |
| Mannitol | – | 30 |
Composition of HTK and HTK-N
| HTK-N46b | HTK (Custodiol®) | |
|---|---|---|
| mmol/l | mmol/l | |
| Ions | ||
| Na+ | 16 | 15 |
| K+ | 10 | 9 |
| Ca2+ | 0.02 | 0.015 |
| Mg2+ | 8 | 4 |
| Cl− | 30.04 | 50.03 |
| Amino acids | ||
| l-histidine | 124 | 180 + l-histidine HCl |
| N-alpha-acetyl-l-histidine | 57 | – |
| l-tryptophan | 2 | 2 |
| l-aspartate | 5 | – |
| l-arginine | 3 | – |
| l-glycine | 10 | – |
| l-alanine | 5 | – |
| Iron chelators | ||
| Desferal (deferoxamine) | 0.025 | – |
| LK-614 | 0.0075 | – |
| Others | ||
| Ketoglutarate | 2 | 1 |
| Saccharose | 33 | – |
| Mannitol | – | 30 |
| HTK-N46b | HTK (Custodiol®) | |
|---|---|---|
| mmol/l | mmol/l | |
| Ions | ||
| Na+ | 16 | 15 |
| K+ | 10 | 9 |
| Ca2+ | 0.02 | 0.015 |
| Mg2+ | 8 | 4 |
| Cl− | 30.04 | 50.03 |
| Amino acids | ||
| l-histidine | 124 | 180 + l-histidine HCl |
| N-alpha-acetyl-l-histidine | 57 | – |
| l-tryptophan | 2 | 2 |
| l-aspartate | 5 | – |
| l-arginine | 3 | – |
| l-glycine | 10 | – |
| l-alanine | 5 | – |
| Iron chelators | ||
| Desferal (deferoxamine) | 0.025 | – |
| LK-614 | 0.0075 | – |
| Others | ||
| Ketoglutarate | 2 | 1 |
| Saccharose | 33 | – |
| Mannitol | – | 30 |
Ex vivo haemodynamic data acquisition
Haemodynamic data were recorded at predefined time points: WH30 (baseline), which means the time point measured in minutes after conversion from the Langendorff to the working heart mode before cardioplegic arrest. rWH5, rWH10, rWH15, rWH20, rWH25 and rWH30 indicate the respective time points during reperfusion.
All haemodynamic data were registered as mean values derived from the respective pressure and flow tracings. The following parameters were recorded: cardiac output (CO, ml/min) identical to left atrial flow (Flowmeter Narcomatic RT-%00, Narco Biosystems) in a closed-circuit perfusion system, and coronary flow (CF, ml/min). Coronary flow was measured as the difference between left atrial flow and aortic flow after the accuracy of the system had been checked by timed collection of blood solution at the beginning of the experiment. As the mean aortic pressure is set at 70 mmHg, which remains constant throughout the experiment, alterations in coronary flow primarily reflect alterations in coronary resistance, allowing evaluation of microvasculature function. LV systolic pressure was measured by a high-fidelity MicroTip catheter (Millar SP-407; Millar Instruments, Inc., Houston, TX, USA) inserted into the left ventricle. To assess the pressure–volume work performed per minute, external heart work (EHW) was calculated according to the following formula: CO × LV systolic pressure (g × m/min). Diastolic compliance was measured as the diastolic left atrial pressure. Haemodynamic data of the reperfusion period are presented as recovery (mean percentage of baseline values) to adjust for differences in baseline between different groups.
Histological evaluation
At the end of the in vivo part of the experiment five animals per group having received saline and S-NO-HSA during reperfusion, respectively, were randomly taken to assess infarct size by Evans Blue and TTC staining. Staining was performed based on the method of Vivaldi et al. [14]. In each heart in four different slices at the midpapillary level infarcted area (which remained white), area at risk (AAR) (which stained red additionally) and vital myocardium (which stained blue) were measured using the ImageJ software. The mean value of these four measurements (infarcted area/total area of the left ventricle) was taken as the infarct size.
At the end of the entire experiment heart tissue samples (HTK: n = 10, HTK-N: n = 10, HTK-N + S-NO-HSA: n = 10, in vivo S-NO-HSA: n = 9) were taken to enable both histological as well as biochemical analysis of the hearts. Electron microscopy was performed according to a standardized protocol, as described elsewhere [15]. Ischaemic damage was graded according to the following scheme based on mitochondrial damage [15]: no visible damage, normal matrix granula; slight damage: loss of matrix granula, light clearing of matrix; moderate damage: moderate clearing of matrix, moderate swelling and partial fragmentation of cristae; severe damage: severe matrix clearing, severe swelling and loss of cristae and irreversible damage: amorphous dense granula.
Biochemical evaluation
Blood gas analysis
Samples from arterial and coronary sinus blood were taken at time points WH5 before ischaemia and rWH5 and rWH25 during reperfusion. Blood gas analysis was performed and myocardial oxygen consumption (MVO2) and myocardial oxygen delivery (MDO2) were calculated according to the formula by Dworschak et al. [16].
Measurement of high-energy phosphates
Freeze-clamp biopsies were taken for the determination of the high-energy phosphates adenosine monophosphate (AMP), adenosine diphosphate (ADP), adenosine triphosphate (ATP) and phosphocreatine (PCr) by high-performance liquid chromatography.
The sample preparation and high-performance liquid chromatography (HPLC) measurement of ATP, ADP, AMP and PCr were performed as previously described [7, 17]. A piece of frozen tissue (50–100 mg) was homogenized with 500 µl of 0.4 mol/l perchloric acid in a ball mill (mikro-dismembrator, Braun, Melsungen, Germany) pre-cooled with liquid nitrogen. After centrifugation (12 000 g), 150 µl of the acid extract was neutralized with 15–20 µl of 2 mol/l potassium carbonate (4°C). The supernatant (20 µl injection volume), obtained after centrifugation, was used for HPLC analysis. Separation was performed on a Hypersil octadecylsilane column (5 µm; 250 × 4 mm I.D.) using an L-2200 autosampler, two L-2130 HTA pumps and an L-2450 diode array detector (all: VWR Hitachi, VWR, Vienna, Austria). Detector signals (absorbance at 214 and 254 nm) were recorded, and the programme EZchrom Elite (VWR) was used for data acquisition and analysis.
Energy charge (EC) was calculated according to the following formula: EC = (ATP + ½ ADP)/(ATP + ADP + AMP).
The pellets of the acid extract were dissolved in 1 ml of 0.1 mol/l sodium hydroxide and further diluted 1:10 with physiologic saline for protein determination (BCA [bicinchoninic acid] Protein Assay, Pierce, Thermo Fischer Scientific, Inc., Rockford, IL, USA).
Statistical analysis
Data are expressed as mean ± SEM. Comparison between groups was performed by analysis of variance using the GraphPad Prism (Version 4.03 for Windows) statistical software. Whenever significance was indicated, Bonferroni multiple comparison analysis was performed. A value of P < 0.05 was considered statistically significant.
RESULTS
Animal characteristics
The first and second part of Table 2 summarize baseline haemodynamic parameters and blood gas measurements after 30 min on the working heart apparatus. Immediately prior to ischaemia there was no significant difference between the four treatment groups.
Baseline data
| HTK (n = 10) | HTK-N (n = 10) | HTK-N + S-NO-HSA (n = 10) | In vivo S-NO-HSA (n = 9) | |
|---|---|---|---|---|
| CF (ml min−1 g−1 HW) | 2.21 ± 0.11 | 1.87 ± 0.15 | 1.74 ± 0.09 | 1.80 ± 0.16 |
| CO (ml min−1 g−1 HW) | 25.23 ± 2.23 | 25.86 ± 1.49 | 26.99 ± 1.24 | 27.23 ± 2.51 |
| EHW (g × m/min) | 4269 ± 301 | 5019 ± 318 | 4805 ± 158 | 4625 ± 472 |
| HR (beats/min) | 236 ± 8 | 237 ± 4 | 239 ± 7 | 230 ± 6 |
| mDO2 (ml min−1 g−1) | 0.33 ± 0.03 | 0.31 ± 0.03 | 0.31 ± 0.03 | 0.27 ± 0.02 |
| mVO2 (ml min−1 g−1) | 0.07 ± 0.00 | 0.06 ± 0.00 | 0.07 ± 0.00 | 0.06 ± 0.01 |
| BDW (g) | 464.6 ± 17.6 | 417.1 ± 16.9 | 455.8 ± 4.7 | 446.3 ± 12.1 |
| Total HW (g) | 1.99 ± 0.08 | 2.02 ± 0.08 | 2.01 ± 0.09 | 2.03 ± 0.05 |
| HW/BDW ratio | 0.0044 ± 0.0006 | 0.0047 ± 0.0004 | 0.0045 ± 0.0005 | 0.0046 ± 0.0003 |
| HTK (n = 10) | HTK-N (n = 10) | HTK-N + S-NO-HSA (n = 10) | In vivo S-NO-HSA (n = 9) | |
|---|---|---|---|---|
| CF (ml min−1 g−1 HW) | 2.21 ± 0.11 | 1.87 ± 0.15 | 1.74 ± 0.09 | 1.80 ± 0.16 |
| CO (ml min−1 g−1 HW) | 25.23 ± 2.23 | 25.86 ± 1.49 | 26.99 ± 1.24 | 27.23 ± 2.51 |
| EHW (g × m/min) | 4269 ± 301 | 5019 ± 318 | 4805 ± 158 | 4625 ± 472 |
| HR (beats/min) | 236 ± 8 | 237 ± 4 | 239 ± 7 | 230 ± 6 |
| mDO2 (ml min−1 g−1) | 0.33 ± 0.03 | 0.31 ± 0.03 | 0.31 ± 0.03 | 0.27 ± 0.02 |
| mVO2 (ml min−1 g−1) | 0.07 ± 0.00 | 0.06 ± 0.00 | 0.07 ± 0.00 | 0.06 ± 0.01 |
| BDW (g) | 464.6 ± 17.6 | 417.1 ± 16.9 | 455.8 ± 4.7 | 446.3 ± 12.1 |
| Total HW (g) | 1.99 ± 0.08 | 2.02 ± 0.08 | 2.01 ± 0.09 | 2.03 ± 0.05 |
| HW/BDW ratio | 0.0044 ± 0.0006 | 0.0047 ± 0.0004 | 0.0045 ± 0.0005 | 0.0046 ± 0.0003 |
Data are given as mean ± SEM.
CF: coronary flow; CO: cardiac output; EHW: external heart work (calculated as CO × LV systolic pressure); HR: heart rate; mDO2: myocardial oxygen delivery; mVO2: myocardial oxygen consumption; BDW: body weight; HW: heart weight.
Baseline data
| HTK (n = 10) | HTK-N (n = 10) | HTK-N + S-NO-HSA (n = 10) | In vivo S-NO-HSA (n = 9) | |
|---|---|---|---|---|
| CF (ml min−1 g−1 HW) | 2.21 ± 0.11 | 1.87 ± 0.15 | 1.74 ± 0.09 | 1.80 ± 0.16 |
| CO (ml min−1 g−1 HW) | 25.23 ± 2.23 | 25.86 ± 1.49 | 26.99 ± 1.24 | 27.23 ± 2.51 |
| EHW (g × m/min) | 4269 ± 301 | 5019 ± 318 | 4805 ± 158 | 4625 ± 472 |
| HR (beats/min) | 236 ± 8 | 237 ± 4 | 239 ± 7 | 230 ± 6 |
| mDO2 (ml min−1 g−1) | 0.33 ± 0.03 | 0.31 ± 0.03 | 0.31 ± 0.03 | 0.27 ± 0.02 |
| mVO2 (ml min−1 g−1) | 0.07 ± 0.00 | 0.06 ± 0.00 | 0.07 ± 0.00 | 0.06 ± 0.01 |
| BDW (g) | 464.6 ± 17.6 | 417.1 ± 16.9 | 455.8 ± 4.7 | 446.3 ± 12.1 |
| Total HW (g) | 1.99 ± 0.08 | 2.02 ± 0.08 | 2.01 ± 0.09 | 2.03 ± 0.05 |
| HW/BDW ratio | 0.0044 ± 0.0006 | 0.0047 ± 0.0004 | 0.0045 ± 0.0005 | 0.0046 ± 0.0003 |
| HTK (n = 10) | HTK-N (n = 10) | HTK-N + S-NO-HSA (n = 10) | In vivo S-NO-HSA (n = 9) | |
|---|---|---|---|---|
| CF (ml min−1 g−1 HW) | 2.21 ± 0.11 | 1.87 ± 0.15 | 1.74 ± 0.09 | 1.80 ± 0.16 |
| CO (ml min−1 g−1 HW) | 25.23 ± 2.23 | 25.86 ± 1.49 | 26.99 ± 1.24 | 27.23 ± 2.51 |
| EHW (g × m/min) | 4269 ± 301 | 5019 ± 318 | 4805 ± 158 | 4625 ± 472 |
| HR (beats/min) | 236 ± 8 | 237 ± 4 | 239 ± 7 | 230 ± 6 |
| mDO2 (ml min−1 g−1) | 0.33 ± 0.03 | 0.31 ± 0.03 | 0.31 ± 0.03 | 0.27 ± 0.02 |
| mVO2 (ml min−1 g−1) | 0.07 ± 0.00 | 0.06 ± 0.00 | 0.07 ± 0.00 | 0.06 ± 0.01 |
| BDW (g) | 464.6 ± 17.6 | 417.1 ± 16.9 | 455.8 ± 4.7 | 446.3 ± 12.1 |
| Total HW (g) | 1.99 ± 0.08 | 2.02 ± 0.08 | 2.01 ± 0.09 | 2.03 ± 0.05 |
| HW/BDW ratio | 0.0044 ± 0.0006 | 0.0047 ± 0.0004 | 0.0045 ± 0.0005 | 0.0046 ± 0.0003 |
Data are given as mean ± SEM.
CF: coronary flow; CO: cardiac output; EHW: external heart work (calculated as CO × LV systolic pressure); HR: heart rate; mDO2: myocardial oxygen delivery; mVO2: myocardial oxygen consumption; BDW: body weight; HW: heart weight.
The third part of Table 2 shows morphometric data. There was no significant difference in body weight, heart weight and heart weight to body weight ratio between groups. Infarct size at the end of the in vivo part of the experiment was 34.6 ± 1.5% in the animals receiving saline infusion and 36.5 ± 2% in the animals receiving S-NO-HSA infusion (P = 0.15); i.e. there was no significant difference in infarct size between treatment groups at the beginning of evaluation on the working heart apparatus.
Ex vivo haemodynamic data
During reperfusion on the working heart apparatus post-ischaemic recovery of haemodynamic values showed the following results (Fig. 2): Recovery of left atrial flow showed significantly higher values in HTK-N + S-NO-HSA compared with both HTK and HTK-N; in vivo S-NO-HSA showed significantly higher values compared with HTK (Fig. 2A). Recovery of CF was significantly higher in both HTK-N + S-NO-HSA and in vivo S-NO-HSA compared with HTK-N and HTK, respectively. There was neither a significant difference between HTK and HTK-N nor between HTK-N + S-NO-HSA and in vivo S-NO-HSA (Fig. 2B). Recovery of external heart work showed a similar picture with higher values in HTK-N + S-NO-HSA and in vivo S-NO-HSA compared with HTK-N and HTK, respectively; there was no difference between HTK-N + S-NO-HSA and in vivo S-NO-HSA. Between HTK and HTK-N, there was again no difference (Fig. 2C). Diastolic left atrial pressure during reperfusion, which is an indicator for left ventricular diastolic compliance, was reduced in HTK-N + S-NO-HSA and in vivo S-NO-HSA compared with both HTK and HTK-N, while there was no difference between HTK and HTK-N (Fig. 2D).
![Ex vivo haemodynamic measurements. Data of the reperfusion period are represented as recovery values (percentage of baseline). (A) Recovery of cardiac output [left atrial flow (LAF)]. †HTK-N + S-NO-HSA vs HTK P < 0.001, *HTK-N + S-NO-HSA vs HTK-N P < 0.05, #in vivo S-NO-HSA vs HTK P < 0.05, in vivo S-NO-HSA vs HTK-N n.s., HTK vs HTK-N n.s. (B) Recovery of coronary flow (CF). †HTK-N + S-NO-HSA vs HTK P < 0.001, *HTK-N + S-NO-HSA vs HTK-N P < 0.05, #in vivo S-NO-HSA vs HTK P < 0.001, **in vivo S-NO-HSA vs HTK-N P < 0.01, HTK vs HTK-N n.s. (C) Recovery of external heart work (EHW). †HTK-N + S-NO-HSA vs HTK P < 0.001, *HTK-N + S-NO-HSA vs HTK-N P < 0.05, #in vivo S-NO-HSA vs HTK P < 0.001, **in vivo S-NO-HSA vs HTK-N P < 0.001, HTK vs HTK-N n.s. (D) Recovery of left atrial pressure (LAP). †HTK-N + S-NO-HSA vs HTK P < 0.05, *HTK-N + S-NO-HSA vs HTK-N P < 0.05, #in vivo S-NO-HSA vs HTK P < 0.05, **in vivo S-NO-HSA vs HTK-N P < 0.05, HTK vs HTK-N n.s.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/icvts/20/3/10.1093_icvts_ivu383/5/m_ivu38302.jpeg?Expires=1750227648&Signature=bBE~~e7OHyjCQmdqku2zimBVjVXa5J0spGR22RWLMTs8j6ZF5RMZWzV7jVKMgz7GlArxFsU5h~JeNW2kyQu4CUIGi~rnGFO-LfJaMs97FplgOcgy0FbImIJi1R1B124fLCUtKDtOfhjyf7OB26M-PgVtKlDO34yXZU~jcEZ~YYauXiMGqk2hCSkh5Woec3ioxmMUN0lBAGol86g8f9~17-U7cH34wXpUwl8MKeijwfirApe-2XTqWx~ASImCViNAnx7uO9Mna0zwN46w~UqUEAVFuQQCemngz7k3H9LcOVnjiuwe35PYwhaEatYnOZymzt3jaXjrwB4nj8ZwOnr~BA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Ex vivo haemodynamic measurements. Data of the reperfusion period are represented as recovery values (percentage of baseline). (A) Recovery of cardiac output [left atrial flow (LAF)]. †HTK-N + S-NO-HSA vs HTK P < 0.001, *HTK-N + S-NO-HSA vs HTK-N P < 0.05, #in vivo S-NO-HSA vs HTK P < 0.05, in vivo S-NO-HSA vs HTK-N n.s., HTK vs HTK-N n.s. (B) Recovery of coronary flow (CF). †HTK-N + S-NO-HSA vs HTK P < 0.001, *HTK-N + S-NO-HSA vs HTK-N P < 0.05, #in vivo S-NO-HSA vs HTK P < 0.001, **in vivo S-NO-HSA vs HTK-N P < 0.01, HTK vs HTK-N n.s. (C) Recovery of external heart work (EHW). †HTK-N + S-NO-HSA vs HTK P < 0.001, *HTK-N + S-NO-HSA vs HTK-N P < 0.05, #in vivo S-NO-HSA vs HTK P < 0.001, **in vivo S-NO-HSA vs HTK-N P < 0.001, HTK vs HTK-N n.s. (D) Recovery of left atrial pressure (LAP). †HTK-N + S-NO-HSA vs HTK P < 0.05, *HTK-N + S-NO-HSA vs HTK-N P < 0.05, #in vivo S-NO-HSA vs HTK P < 0.05, **in vivo S-NO-HSA vs HTK-N P < 0.05, HTK vs HTK-N n.s.
Histological evaluation
Ultrastructural integrity in transmission electron microscopy was evaluated in 10 specimens (HTK, HTK-N, HTK-N + S-NO-HSA) and 9 specimens (in vivo S-NO-HSA), respectively, in 3 randomly selected fields in a blinded manner. In the HTK group 4 of 10 specimens showed severe to irreversible damage, 5 showed moderate damage and 1 was graded as showing slight to moderate damage. In HTK-N 2 of 10 samples were graded as severe to irreversible damage, and 8 samples showed moderate to severe damage. The HTK-N + S-NO-HSA group was graded as moderately damaged in five samples, and five samples showed slight damage. None of the hearts showed severe to irreversible damage. In the in vivo S-NO-HSA group all nine hearts showed moderate damage; none was graded as severely to irreversibly damaged. Representative micrographs are depicted in Fig. 3.
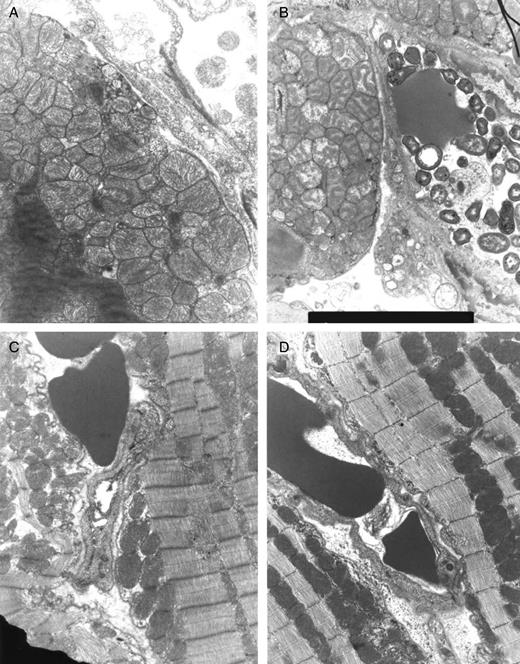
Representative micrographs of transmission electron microscopy at the end of ex vivo reperfusion. (A) HTK, (B) HTK-N, (C) HTK-N + S-NO-HSA, (D) in vivo S-NO-HSA.
Biochemical evaluation
At baseline there were no significant differences in mDO2 and mVO2 between groups (Table 2). As shown in Fig. 4A, during reperfusion at time point rWH5′ the recovery of mDO2 was significantly higher in HTK-N (P < 0.01), HTK-N + S-NO-HSA (P < 0.01) and in vivo S-NO-HSA (P < 0.001) compared with HTK; at time point rWH25′ there was no difference between HTK and HTK-N, whereas both HTK-N + S-NO-HSA as well as in vivo S-NO-HSA showed an improvement of mDO2 compared with HTK (P < 0.01 HTK vs HTK-N + S-NO-HSA; P < 0.001 HTK vs in vivo S-NO-HSA) and HTK-N (P < 0.05 HTK-N vs HTK-N + S-NO-HSA; P < 0.001 HTK-N vs in vivo S-NO-HSA).
![Biochemical data and high-energy phosphates in LV biopsy tissue. (A) Recovery of myocardial oxygen delivery, mDO2 [%]; (B) Recovery of myocardial oxygen consumption, mVO2 [%]; (C) phosphocreatine and adenine nucleotide levels; (D) energy charge, *P < 0.05; **P < 0.01 and ‘P < 0.001.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/icvts/20/3/10.1093_icvts_ivu383/5/m_ivu38304.jpeg?Expires=1750227648&Signature=Gh3gJYl~~DXjJ-jWWABK93zRIFZIEwybByaRF6TgHA59QDd2ncgPe74JqiGFdQ2XBpDgXOBumyYrfrc80LdbRM3W2Ji3EBy0cQxdGbq-sfxmsnpiuBfpS9bHEn95iqsHBgQGttazSDJ6NRO4r~SuIYdWPTvq0YRsSaxCTdrUkRzso5MIr5zM9gA2pCJ3k5cWmbIxlvPt8Px5-j00nGlHRo5wyXAk9VSGRk08LiBx0stY-wC71dpNlC-WeW-tC6VUnaMhTmsHUZ5VFLBDJJYlEsOB~y8PvznJKJHV8x~qUen-21I1ojdQs4xwtGZCQl2UhhYIvHNyNJ2xp7mQ2rpMSg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Biochemical data and high-energy phosphates in LV biopsy tissue. (A) Recovery of myocardial oxygen delivery, mDO2 [%]; (B) Recovery of myocardial oxygen consumption, mVO2 [%]; (C) phosphocreatine and adenine nucleotide levels; (D) energy charge, *P < 0.05; **P < 0.01 and ‘P < 0.001.
The recovery of mVO2 showed no difference between groups at time point rWH5′ ; after 25′ of reperfusion, recovery of mVO2 was significantly higher in HTK-N (P < 0.01), HTK-N + S-NO-HSA (P < 0.01) and in vivo S-NO-HSA (P < 0.01) compared with HTK (Fig. 4B).
Evaluation of high-energy phosphates at the end of reperfusion showed superior preservation in both S-NO-HSA treated groups compared with HTK, whereas there was only a slight improvement in HTK-N protected hearts compared with HTK without reaching statistical significance with the exception of ATP (Figure 4C). In detail, ATP content was significantly higher in HTK-N compared with HTK (P < 0.05); this improvement was even more pronounced in both S-NO-HSA treatment groups (HTK-N + S-NO-HSA vs HTK P < 0.001; in vivo S-NO-HSA vs HTK P < 0.001). In parallel, PCr content, the buffering source of ATP during energy demand, was significantly elevated in HTK-N + S-NO-HSA (P < 0.05) and in vivo S-NO-HSA (P < 0.05) compared with HTK; HTK-N showed only a trend towards better recovery with no statistical significance. In addition, AMP content was significantly higher in HTK-N + S-NO-HSA (P < 0.01) and in vivo S-NO-HSA (P < 0.05) compared with HTK. Similar results were seen with ADP. EC showed a significant improvement in HTK-N (P < 0.05), HTK-N + S-NO-HSA (P < 0.05) as well as in vivo S-NO-HSA (P < 0.05) compared with HTK (Fig. 4D).
DISCUSSION
In our model of acute myocardial infarction the addition of the direct NO donor, S-NO-HSA to HTK-N led to a significant improvement of post-ischaemic recovery of coronary flow, cardiac output, LV systolic pressure, external heart work and a reduction of left atrial diastolic pressure compared with both HTK and HTK-N. Haemodynamic improvement was accompanied by a better preservation of energy state and ultrastructural integrity.
The new HTK-N solution was developed with four major modifications compared with its predecessor HTK: (i) the addition of the amino acids aspartate, l-arginine, l-alanine and l-glycine; (ii) the use of two iron chelators LK-614 and deferoxamine; (iii) the use of N-α-acetyl-l-histidine instead of l-histidine alone; and (iv) the reduction of its chloride concentration [18–21]. In a model of heart transplantation in the rat, Loganathan et al. [22] observed an improvement of cardiac function, and coronary flow with HTK-N accompanied by a reduction of apoptosis compared with HTK. Similarly, in chronic failing hearts 6 weeks post-myocardial infarction (MI), we saw an improvement of postischaemic recovery of cardiac function after 1 h of ischaemia but we did not see an improvement of coronary flow in HTK-N protected hearts compared with HTK [13]. We speculated that this might be due to the fact that the aforementioned modifications might not be sufficient to maintain NO production in a diseased endothelium. Therefore, we refined the concept of HTK-N by addition of the direct NO donor, S-NO-HSA.
It is known that one of the key factors of endothelial dysfunction in ischaemia/reperfusion injury is the deficiency and/or reduced bioavailability of NO leading to the ‘no- or low-reflow’ phenomenon after blood flow has been restored. During ischaemia an increase of intracellular calcium activates eNOS leading to an initially high NO production followed by the release of reactive oxygen species and cytotoxic substances that cause endothelial injury, oedema formation and vascular constriction [4]. As a consequence of the burst of NO during ischaemia and subsequent reperfusion, local l-arginine and/or tetrahydrobiopterin concentrations are depleted, leading to the so-called eNOS uncoupling’ [5, 6]; i.e. under the condition of substrate or cofactor deficiency, one or two subunits of eNOS divert electrons to oxygen forming superoxide which can react with NO to form peroxynitrite [7, 8]. It could be shown that the production of oxygen-derived free radicals by uncoupled eNOS is a major contributor of endothelial dysfunction [8]. The strategy for substituting NO by S-NO-HSA is based upon three deliberations: (i) the production of NO by eNOS is reduced; (ii) uncoupling of eNOS is prevented or at least attenuated as the turnover rate of l-arginine and tetrahydrobiopterin is decreased; and (iii) consequently the release of superoxide and peroxynitrite is reduced [9]. This concept proves to be effective in the setting of warm, unprotected ischaemia in the pig heart where S-NO-HSA supplementation preserved the function of eNOS, maintained the basal production of NO and decreased the release of superoxide and peroxynitrite [9]. Similar results were obtained in the isolated rabbit heart after 6 h of cardioplegic arrest [10]. In the present study we tested the supplementation of S-NO-HSA to HTK-N in a model of acute heart failure due to a reperfused MI. By this experimental setting, we wanted to simulate the clinical situation in which a patient with acute MI cannot be stabilized by PCI but has to be operated on in the acute phase of infarction. These patients often suffer from severe haemodynamic instability after weaning from cardiopulmonary bypass, which can partially be mitigated by beating-heart strategies most likely due to avoiding a no- or low-reflow phenomenon as a consequence of cardioplegic arrest [3]. Whereas HTK-N was able to maintain endothelial function in a healthy heart and vessels in acute failing hearts due to ischaemia/reperfusion injury, only the supplementation of exogenous NO via S-NO-HSA improved myocardial perfusion and energy metabolism, and attenuated ultrastructural damage [22].
However, starting S-NO-HSA infusion immediately before reopening the occluded LAD did not show an additional positive effect on haemodynamic recovery after cardioplegic arrest. It is known that after reopening of a previously occluded coronary vessel, tissue perfusion might be impaired for several hours due to the so-called ‘no-reflow’ phenomenon [4]. As the impairment of myocardial reperfusion after PCI negatively affects early and late clinical outcome, several vasoactive substances have been tested both experimentally and clinically to reduce endothelial dysfunction [23, 24]. If a patient has to be operated on in the acute phase of MI after partial or total reperfusion of ischaemic areas, endothelial and myocardial dysfunction are already present before the insult of ischaemia/reperfusion injury by cardioplegic arrest. Therefore, we hypothesized that starting the NO donor, S-NO-HSA before reopening the LAD might attenuate endothelial and myocardial dysfunction during early reperfusion and improve the basis for subsequent cardioplegic arrest. Postischaemic recovery on the isolated heart perfusion system, however, was similar to those hearts that received S-NO-HSA only as an adjunct to cardioplegia. One may speculate that, on the one hand, the damage caused by global ischaemia during cardioplegic arrest overlies local ischaemia/reperfusion injury in the LAD territory and that, on the other hand, the effect of S-NO-HSA supplementation with cardioplegia overlies the effect of in vivo supplementation. Our study protocol was not suited to differentiate these effects. Therefore, according to the existing data we can only conclude that in vivo S-NO-HSA supplementation has no additional benefit on postischaemic recovery after subsequent cardioplegic arrest. Whether S-NO-HSA supplementation after reperfusion of an occluded coronary artery is a possible therapeutic option to improve infarct development, functional recovery and long-term outcome cannot be answered by our study protocol and needs further investigation.
In conclusion, after MI in rats S-NO-HSA as an adjunct to the new HTK-N revealed improved myocardial protection during cardioplegic arrest compared with HTK-N alone. HTK-N with S-NO-HSA is therefore a possible therapeutic option for patients operated on for acute MI due to failed or impossible percutaneous coronary intervention.
Acknowledgements
We thank Friederike Neumann for statistical support, and Azra Saric and Gerd Kager for technical assistance. HTK and HTK-N46b were kindly provided by Gernot Köhler (Dr Köhler Chemie, Bensheim, Germany).
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}