





Abstract
Initially identified as suppressors of metastasis in various types of cancer, kisspeptins are a family of neuropeptides that are key regulators of the mammalian reproductive axis. Accumulating evidence has shown that kisspeptin is able to control both the pulsatile and surge GnRH release, playing fundamental roles in female reproduction, which include the secretion of gonadotropins, puberty onset, brain sex differentiation, ovulation and the metabolic regulation of fertility. Furthermore, recent studies have demonstrated the involvement of the kisspeptin system in the processes of implantation and placentation. This review summarizes the current knowledge of the pathophysiological role and utility of these local placental regulatory factors as potential biomarkers during the early human gestation.
A successful pregnancy, from the initiation of embryo implantation to parturition, is a complex process that requires the orchestration of a series of events. This review aims to concisely summarize what is known about the role of the kisspeptin system in implantation, placentation, early human pregnancy and pregnancy-related disorders, and to develop strategies for predicting, diagnosing and treating these abnormalities.
Using the PubMed and Google Scholar databases, we performed comprehensive literature searches in the English language describing the advancement of kisspeptins and the kisspeptin receptor (KISS1R) in implantation, placentation and early pregnancy in humans, since its initial identification in 1996 and ending in July 2018.
Recent studies have shown the coordinated spatial and temporal expression patterns of kisspeptins and KISS1R during human pregnancy. The experimental data gathered recently suggest putative roles of kisspeptin signaling in the regulation of trophoblast invasion, embryo implantation, placentation and early pregnancy. Dysregulation of the kisspeptin system may negatively affect the processes of implantation as well as placentation. Clinical studies indicate that the circulating levels of kisspeptins or the expression levels of kisspeptin/KISS1R in the placental tissues may be used as potential diagnostic markers for women with miscarriage and gestational trophoblastic neoplasia.
Comprehensive research on the pathophysiological role of the kisspeptin/KISS1R system in implantation and placentation will provide a dynamic and powerful approach to understanding the processes of early pregnancy, with potential applications in observational and analytic screening as well as the diagnosis, prognosis and treatment of implantation failure and early pregnancy-related disorders.
Introduction
The hypothalamic-pituitary-gonadal (HPG) axis plays a critical role in maintaining the normal function of the reproductive system in adolescents (Knobil et al., 1980). This axis is governed by the pulsatile secretion of gonadotropin-releasing hormone (GnRH) which, in turn, stimulates the secretion of gonadotropins (FSH and LH). The pulsatile secretion of GnRH/gonadotropins is modulated by feedback inhibition from the target gonadal hormones (Herbison, 1998). During the periovulatory stage, the surge secretion of GnRH/gonadotropins is induced by the positive feedback of serum estrogen (Goodman, 1978), which consequently triggers ovulation in female mammals (Moenter et al., 1992). Being the upstream regulator of pulsatile and surge GnRH release, kisspeptin has been much heralded in the past decade, with its fundamental roles in regulating female reproduction in several aspects, which include gonadotropin secretion, puberty onset, brain sex differentiation and ovulation (Roa et al., 2008; Pinilla et al., 2012; Hu et al., 2017). Kisspeptin neurons are located within two regions in the hypothalamus, the anteroventral periventricular nucleus (AVPV) and arcuate nucleus (ARC), which are responsible for mediating the positive and negative feedback effects of estrogen on GnRH/gonadotropin secretion, respectively (Smith et al., 2005; Tomikawa et al., 2012). Specifically, estrogen binds to ERα on kisspeptin neurons in the ARC or infundibular nucleus, which further inhibit kisspeptin and subsequently the GnRH release (Smith et al., 2005). Additionally, estrogen exerts positive feedback (also mediated via ERα) on kisspeptin neurons in the AVPV nucleus, which accounts for the LH surge in the menstrual cycle (Tomikawa et al., 2012).
In many mammals, kisspeptins function to stimulate the release of GnRH (and subsequently the secretion of FSH and LH) by binding to the kisspeptin receptor (KISS1R), which is a G-protein-coupled receptor. Notably, inactivating mutations of either KISS1 or KISS1R lead to the phenotype of hypogonadotropic hypogonadism in humans and mice (Tena-Sempere, 2006; Tenenbaum-Rakover et al., 2007; Roseweir and Millar, 2009; Nimri et al., 2011; Topaloglu et al., 2012). Additionally, recent studies have indicated the potential pathophysiological roles of extra-hypothalamic kisspeptins in the central and peripheral regulation of the activity of diverse systems (Terao et al., 2004; Pinto et al., 2012; Laoharatchatathanin et al., 2015; Comninos et al., 2016; Uenoyama et al., 2016). Outside the hypothalamic area, kisspeptins and their putative receptor KISS1R have been identified across different types of tissues, particularly those involved in reproductive and metabolic functions, including the hippocampus, forebrain, ovary, uterus, placenta, testis, adipose tissue, pancreas and liver (Ohtaki et al., 2001; Arai et al., 2005; Castellano et al., 2006; Cejudo Roman et al., 2012; Pinto et al., 2012; Xu et al., 2012; Zhang et al., 2014; Dudek et al., 2016). Indeed, kisspeptins can exert several different physiological actions on various types of tissues in an autocrine/paracrine manner. During the processes of implantation and placentation, many studies have shown that the functional form of the kisspeptin/KISSR system is expressed at the maternal-fetal interface, including the endometrial and placental tissues of various species, including humans (Shahed and Young, 2009; Cejudo Roman et al., 2012; Zhang et al., 2014). Furthermore, recent studies have indicated that locally expressed kisspeptin/KISS1R directly participates in various physiological and pathophysiological activities at the maternal-fetal interface.
This review will mainly focus on the available literature related to the pathophysiological roles of the kisspeptin/KISS1R system in regulating the processes of implantation and placentation. We summarize our current knowledge regarding the underlying mechanisms by which kisspeptin exerts its cellular activities in the processes of implantation, embryo development and placentation. We will also mention the potential implications of kisspeptins in the prediction and diagnosis of early pregnancy-related disorders.
Methods
Literature searches using either PubMed or Google Scholar were performed to review articles written in English focusing on the advancement of kisspeptins and KISS1R in embryo implantation, placentation and early pregnancy-related complications. The search included human and other mammalian studies starting from the initial identification of kisspeptin in 1996 until July 2018. Search terms included ‘kisspeptin’, ‘metastin’, ‘GPR54’ and ‘KISS1R’. We also combined the findings that best aligned with the themes of ‘implantation’, ‘uterus’, ‘pregnancy’, ‘placenta’, ‘miscarriage’ and ‘gestational trophoblastic neoplasia’. The search strategy and study selection for the references included in this review are presented in Supplemental Figure S1.
A short review of the KISS1 gene and peptide and its receptor
Kisspeptin (a 145-amino acid polypeptide) and its encoding gene KISS1 were originally identified as a human metastasis suppressor (and a gene) of malignant melanoma in Hershey, Pennsylvania, the USA, the hometown of the famous chocolate Hershey’s kisses (Lee et al., 1996). The name of KISS1 was derived from these sweets, with the terminology ‘SS’ in KISS1 indicating the ‘suppressor sequence’. In humans, the KISS1 gene that encodes kisspeptins is located on the long (q) arm of chromosome 1 at q32 (West et al., 1998). Initially, this gene encodes an intermediate 145-amino-acid prepropeptide, which is unstable and biologically inactive. The prepropeptide is then cleaved into four biologically active peptides distinguished by their number of amino acids: kisspeptin-10, 13, 14 and 54 (52 in rodent animals) (Figure 1). Sharing a common characteristic of the RF-amide signal peptide, all of these peptides have a C-terminal region that contains an Arg-Phe-NH2 motif, which allows these peptides to bind to and fully activate KISS1R. Because of the structural similarities and their common origin derived from the prepropeptide, the term ‘kisspeptins’ is now globally used to describe this family of peptides (Kotani et al., 2001; Gottsch et al., 2009; Pinilla et al., 2012).
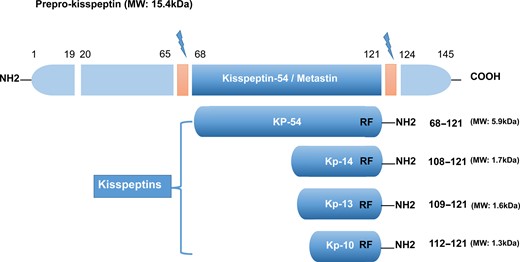
The major structure of kisspeptins in humans, the peptides encoded by theKISS1gene. All kisspeptins are the products generated by the cleavage of a common precursor named prepro-kisspeptin. The prepro-kisspeptin is a 145-amino acid peptide, which is further cleaved into the following four peptides with lower molecular weights: KP-54, KP-14, KP-13 and KP-10. All kisspeptins contain the RF-amide motif that can bind and activate KISS1R. This figure is modified from Pinilla et al. (2012); Hu et al. (2017).
To acknowledge its metastasis inhibitory properties, kisspeptin-54 was initially termed ‘metastin’. This peptide was considered the major product of the human KISS1 gene, and it could be further cleaved into 14, 13 and 10 amino acid peptides (Ohtaki et al., 2001) (Figure 1). However, the largest proteolytic product of the kisspeptin precursor comprises 52 amino acids, and the terminal RF-amide signature is substituted by an Arg-Tyr-NH2 motif in rats and mice (Pinilla et al., 2012). In both humans and rats, the kisspeptin-54/52, -14, -13 and a shorter peptide kisspeptin-10 have the same affinity and binding efficacy onto their receptor, indicating that the C-terminal region of these peptides contributes to the high-affinity binding and the activation of KISS1R (Kotani et al., 2001).
KISS1R belongs to the G-protein-coupled receptor family that was first identified as an orphan receptor in the rat brain. This seven-transmembrane receptor is structurally similar to the transmembrane region of galanin receptors, with ~40% sequence identity (Lee et al., 1999). Subsequently, the orthologue gene of KISS1R in humans was cloned and recognized as a putative receptor for KISS1 gene-derived peptides (Muir et al., 2001; Ohtaki et al., 2001). In terms of nomenclature, KISS1R has different names, including KISS1R, AXOR12, GPR54, CPPB1, hOT7T175 and HH8 because various groups of researchers have independently identified its presence or studied its physiological roles (Lee et al., 1999; Kotani et al., 2001; Muir et al., 2001; Ohtaki et al., 2001; Gottsch et al., 2009).
Kisspeptin signaling pathways
KISS1R is a G-protein-coupled receptor that belongs to the subgroup of typical Gq/11 protein-associated receptors. Following the stimulation of KISS1R by kisspeptin peptides, the phosphorylated Gq/11 protein activates phospholipase C (PLC)-β, leading to the activation or formation of various second messengers, including those involved in the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2), the accumulation of inositol-(1,4,5)-triphosphate (IP3), diacylglycerol and protein kinase C (PKC) and the mobilization and release of intracellular Ca2+ into the cytoplasm (Ohtaki et al., 2001; Stafford et al., 2002; Jiang et al., 2005) (Figure 2).
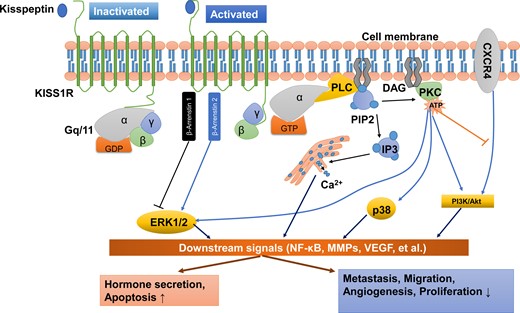
Kisspeptin/KISS1R signaling and potential functions at a glance. KISS1R is a seven-transmembrane domain, Gq/11-coupled receptor. Upon binding of kisspeptin, the intracellular portion of KISS1R phosphorylates Gq/11. The α-subunit of Gq/11 activates PLC, which subsequently cleaves PIP2 into IP3 and DAG. IP3 promotes intracellular Ca2+ release from the endoplasmic reticulum, while DAG activates a signaling cascade by phosphorylating PKC. PKC activation induces the phosphorylation of ERK1/2 and p38. In addition, activation of KISS1R recruits arrestin-1 and -2, which down-regulates and up-regulates phosphorylated ERK1/2 levels, respectively. The activation of KISS1R can also stimulate the phosphorylation of PI3K/Akt, and suppress the phosphorylation of PI3K/Akt by blocking the prometastatic chemokine receptor CXCR4 (the receptor for the chemokine stromal cell-derived factor 1) signaling. These signals act on downstream pathways, including NF-κB, MMPs and VEGF, to promote hormone secretion and apoptosis and inhibit metastasis, migration, angiogenesis and proliferation. DAG, diacylglycerol; ERK1/2, extracellular signal-regulated kinase; IP3, inositol 1,4,5-triphosphate; PI3K, phosphatidylinositol-3-kinase; MMPs, matrix metalloproteinases; NF-κB, nuclear factor κB; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; PLC, phospholipase C; VEGF, vascular endothelial growth factor.
Apart from the PLC–PKC–Ca2+ pathway, other intracellular transduction pathways are also activated in response to kisspeptin stimulation. In many cell types, the phosphorylation of ERK1/2 is thought to be the most conserved kinase signal. However, some cell types that show the activation of ERK1/2 do not have stable p38 MAPK and PI3K/Akt activation in response to kisspeptins (Ringel et al., 2002; Castano et al., 2009; Peng et al., 2013). For instance, treatment with kisspeptin-10 induces strong and sustained phosphorylation of ERK1/2 but very weak phosphorylation of p38 and no phosphorylation of stress-activated protein kinase/c-Jun NH2-terminal kinase in Chinese hamster ovary (CHO)-K1 cells (Kotani et al., 2001). Another example is that kisspeptin activates ERK1/2 signaling without significant effects on P38 phosphorylation in rat luteal cells (Peng et al., 2013). In many mammalian cells, the kisspeptin-induced activation of Kiss1r can recruit β-arrestin 1 and β-arrestin 2 to the plasma membrane, which further increases the intracellular level of phosphorylated ERK1/2. In particular, β-arrestin 2 potentiates Kiss1r signaling to ERK, but β-arrestin 1 inhibits it (Pampillo et al., 2009; Szereszewski et al., 2010; Goertzen et al., 2016). Additionally, activation of PKC is required to promote the ERK1/2 signaling cascade (Woods and Johnson, 2007). Taking these data together, kisspeptin may activate the ERK1/2 signaling via the β-arrestin 1, β-arrestin 2 and PLC–PKC-ERK1/2 signaling pathways (Figure 2). The experimental results obtained from functional studies showed that kisspeptin activates the PI3K/Akt signaling pathway in a cell type-specific manner. Kisspeptin can induce the phosphorylation of PI3K/Akt in stably KISS1R-overexpressed thyroid cancer cells (Stathatos et al., 2005), but not in rat luteal cells (Peng et al., 2013). Similarly, previous studies demonstrated that kisspeptin-10 promotes the phosphorylation of PI3K/Akt in preoptic neurons (Hanchate et al., 2012), while kisspeptin-10 suppresses the phosphorylation of PI3K/Akt in tumor cells (Navenot et al., 2005, 2009; Chen et al., 2016). In fact, kisspeptin-10 does not directly inhibit the activation of PI3K/Akt signaling pathway in tumor cells. The suppressive effect of kisspeptin-10 on PI3K/Akt signaling is mediated by suppressing the capacity of the prometastatic chemokine receptor CXCR4 (the receptor for the chemokine stromal cell-derived factor 1) in response to stromal cell-derived factor 1, which subsequently abolishes the activation of PI3K/Akt signaling by CXCR4 in some tumor cells (Navenot et al., 2005, 2009).
In tumor cells, numerous studies have documented that kisspeptin inhibits the activity of matrix metalloproteinase-9 (MMP-9) and MMP-2 by blocking nuclear translocation of the nuclear factor κB (NF-κB), which subsequently reduces the capacity of NF-κB to bind to the MMP-9 promoter (Yan et al., 2001; Gao et al., 2007, 2008; Yoshioka et al., 2008; Cho et al., 2009a; Takeda et al., 2012). Studies have shown that the phosphorylation of ERK and PI3K/Akt is the upstream of NF-κB that regulates the NF-κB DNA-binding activity in tumor cells (Dhawan and Richmond, 2002; Bai et al., 2009; Lee et al., 2012). Interestingly, the enhanced expression of MMP-9 and MMP-2 via the PI3K/Akt/NF-κB (Cheng et al., 2006) or ERK/NF-κB signaling pathways (Birkenkamp et al., 2000; Kang et al., 2011) has also been identified in different cell types. Collectively, these findings suggest that ERK1/2 or PI3K/Akt signaling is involved in kisspeptin-induced regulation of NF-κB/MMP-9 or NF-κB/MMP-2 (Figure 2). Previous studies have also demonstrated that the administration of kisspeptin suppresses the expression of vascular endothelial growth factor (VEGF) via ERK1/2 signaling, to inhibit angiogenesis in tumors (Cho et al., 2009b) and in trophoblast cells (Francis et al., 2014) (Figure 2).
In the past couple of decades, studies have demonstrated the physiological role of kisspeptins and their receptor, KISS1R, in the neuroendocrine-reproductive axis (de Roux et al., 2003; Seminara et al., 2003), which thereafter revolutionized the field of reproductive physiology. In the meantime, plasma concentrations of kisspeptin-54 were also identified in men and women (pregnant and non-pregnant) (Horikoshi et al., 2003). Interestingly, the plasma concentration of kisspeptin increased dramatically throughout the gestational period (Horikoshi et al., 2003). The clinical data together with the evidence that the KISS1 gene is greatly expressed in the placenta (Lee et al., 1999; Kotani et al., 2001; Muir et al., 2001; Ohtaki et al., 2001) suggest that the plasma kisspeptin during pregnancy is mainly, if not completely, derived from the placental tissue. In the following sections, we gather the latest studies addressing the roles of kisspeptins in early pregnancy, including implantation and placentation, and discuss potential clinical applications.
Expression of kisspeptins and KISS1R in the embryo
Although previous studies have reported the expression of kisspeptins and KISS1R in the oocyte (Saadeldin et al., 2012; Dorfman et al., 2014; Cielesh et al., 2017), very few studies have documented the expression of this system in the embryo. Using single-cell RNA sequencing (RNA-Seq) analyses, a previous study showed that KISS1R is barely expressed in the human embryo (from the zygote stage to the late blastocyst stage) and in human embryonic stem cells (Yan et al., 2013). To the best of our knowledge, whether kisspeptin is expressed in the embryo has not been reported.
Kisspeptin/KISS1R system in the endometrium
Previous studies have indicated that the expression of the kisspeptin/KISS1R system in ovarian cells is affected by the estrous/menstrual cycle (Castellano et al., 2006; Gaytan et al., 2009; Cielesh et al., 2017; Tanyapanyachon et al., 2018) (for reviews, see Hu et al., 2017). It is most likely that the expression of the kisspeptin/Kiss1r system in the endometrium shares the cycle-dependent changes in different kinds of species. In the human endometrium, a previous study showed that immunostaining of kisspeptin peptides and KISS1R was strong and limited to epithelial cells, with no detection in stromal cells (Cejudo Roman et al., 2012). Additionally, another study demonstrated that the expression of kisspeptin and KISS1R was present in epithelial cells, though at weak expression levels and was nearly absent in endometrial stromal cells during the proliferative and early secretory phases (Baba et al., 2015) (Table I). However, the expression of kisspeptin was prominent in endometrial stromal cells in the late secretory phase (Baba et al., 2015), indicating that kisspeptin may function in the decidualized endometrium in preparation for adequate placentation. However, the endometrium showed no significant expression change in kisspeptin peptides and Kiss1r among different ovarian stages in domestic cats (Tanyapanyachon et al., 2018). Interestingly, it has been suggested that kisspeptin-10 expression is higher in the non-pregnant canine uterus compared to the gestational periods investigated (Schafer-Somi et al., 2017). The same study also suggested that the expression of Kiss1r is higher before embryo adhesion than during post-implantation and mid-gestation (Schafer-Somi et al., 2017), indicating a pregnancy-related down-regulation of the kisspeptin/Kiss1r system in the canine uterus. Additionally, no expression of kisspeptin or KISS1R was observed both in epithelial glands and stromal cells during the menopausal phase in humans (Baba et al., 2015), suggesting that the kisspeptin/KISS1R system may not have a fundamental role in the endometrium at this stage.
Expression of KISS1 and KISS1R at the utero-placental interface during mammalian pregnancy.
| Species | Cells or tissues reported | Trends across pregnancy | References |
|---|---|---|---|
| KISS1 | |||
| Human | vST, vCT, Decidua | Decreased in RSA compared with abortion | Wu et al. (2014) |
| Human | vST, vCT | Decreased in RSA compared with abortion | Park et al. (2012) |
| Human | vST; PB and DP (−) | Not investigated | Matjila et al. (2013) |
| Human | vCT | Not different between term pregnancy and early pregnancy | Janneau et al. (2002) |
| Human | Placenta (early and term pregnancy, PE) | Early pregnancy > Term pregnancy > PE | Cartwright and Williams (2012) |
| Human | Placenta (term pregnancy, PE); PB and DP (minimal) | Term pregnancy < PE | Matjila et al. (2016) |
| Human | Placenta (term pregnancy) | Circadian rhythm (two surges, 04:00 h and 12:00 h, respectively). | de Pedro et al. (2015) |
| Human | vST | Not investigated | Horikoshi et al. (2003) |
| Rat | Trophoblast giant cells | Gradually decreased | Terao et al. (2004) |
| Human | vST, vCT; EVTs (−) | First trimester > term pregnancy | Bilban et al. (2004) |
| Rat | Trophoblast giant cells (gestation Day 16 and Day 22) | Day 22 > Day 16 | Mark et al. (2013) |
| Human | Placenta | Preterm pregnancy > term pregnancy | Torricelli et al. (2008) |
| Cat | Luminal epithelial cells and uterine glands, stroma cells | Not investigated | Tanyapanyachon et al. (2018) |
| Dog | Superficial uterine glands, Syncytiotrophoblast | Not changed between pre-implantation and post-implantation | Schafer-Somi et al. (2017) |
| Mouse | Decidualizing stromal cells | Increased dynamically from D5 onward | Zhang et al. (2014) |
| Human | Endometrium | Glandular endometrium: ectopic > eutopic | Kolioulis et al. (2017) |
| Human | Epithelial cells (weak expression levels). ESCs (−) | Proliferative stage and early secretory phase | Baba et al. (2015) |
| ESCs (prominent expression levels) | Late secretory phase | ||
| Human | Endometrium | Glandular endometrium: Ectopic >eutopic, Stromal endometrium: no difference | Timologou et al. (2016) |
| Human | Luminal and glandular epithelial cells, stroma cells (−) | Not investigated | Cejudo Roman et al. (2012) |
| KISS1R | |||
| Human | vST, vCT | Decreased in RSA compared with abortion | Wu et al. (2014) |
| Deciduas | Not different between abortion and RSA | ||
| Human | vST, vCT | Not different between abortion and RSA | Park et al. (2012) |
| Human | vST, vCT, EVTs; PB and decidua basalis (−) | Not investigated | Matjila et al. (2013) |
| Human | vCT (early pregnancy) | First trimester > term pregnancy | Janneau et al. (2002) |
| Human | Placenta (early and term pregnancy, PE) | Early pregnancy > PE > term pregnancy | Cartwright and Williams (2012) |
| Human | Placenta, PB and DP (term pregnancy, PE) | Not different between term pregnancy and PE | Matjila et al. (2016) |
| Rat | Trophoblast giant cells | Low expression level | Terao et al. (2004) |
| Human | vST, vCT, EVTs | First trimester > term pregnancy | Bilban et al. (2004) |
| Rat | Placenta (gestation Day 16 and Day 22) | Day 22 > Day 16 | Mark et al. (2013) |
| Human | Umbilical vein endothelial cells | Not investigated | Ramaesh et al. (2010) |
| Mouse | Decidualizing stromal cells | Increased from D5 onward | Zhang et al. (2014) |
| Cat | Luminal epithelial cells and uterine glands. ESCs (−) | Not investigated | Tanyapanyachon et al. (2018) |
| Dog | Superficial and deep uterine glands | Higher in pre-implantation than post-implantation | Schafer-Somi et al. (2017) |
| Dog | Syncytiotrophoblast cells | Higher in mid-gestation than post-implantation | Schafer-Somi et al. (2017) |
| Human | Epithelial cells. ESCs (−) | Week expression levels | Baba et al. (2015) |
| Human | Luminal and glandular epithelial cells. ESCs (−) | Not investigated | Cejudo Roman et al. (2012) |
| Species | Cells or tissues reported | Trends across pregnancy | References |
|---|---|---|---|
| KISS1 | |||
| Human | vST, vCT, Decidua | Decreased in RSA compared with abortion | Wu et al. (2014) |
| Human | vST, vCT | Decreased in RSA compared with abortion | Park et al. (2012) |
| Human | vST; PB and DP (−) | Not investigated | Matjila et al. (2013) |
| Human | vCT | Not different between term pregnancy and early pregnancy | Janneau et al. (2002) |
| Human | Placenta (early and term pregnancy, PE) | Early pregnancy > Term pregnancy > PE | Cartwright and Williams (2012) |
| Human | Placenta (term pregnancy, PE); PB and DP (minimal) | Term pregnancy < PE | Matjila et al. (2016) |
| Human | Placenta (term pregnancy) | Circadian rhythm (two surges, 04:00 h and 12:00 h, respectively). | de Pedro et al. (2015) |
| Human | vST | Not investigated | Horikoshi et al. (2003) |
| Rat | Trophoblast giant cells | Gradually decreased | Terao et al. (2004) |
| Human | vST, vCT; EVTs (−) | First trimester > term pregnancy | Bilban et al. (2004) |
| Rat | Trophoblast giant cells (gestation Day 16 and Day 22) | Day 22 > Day 16 | Mark et al. (2013) |
| Human | Placenta | Preterm pregnancy > term pregnancy | Torricelli et al. (2008) |
| Cat | Luminal epithelial cells and uterine glands, stroma cells | Not investigated | Tanyapanyachon et al. (2018) |
| Dog | Superficial uterine glands, Syncytiotrophoblast | Not changed between pre-implantation and post-implantation | Schafer-Somi et al. (2017) |
| Mouse | Decidualizing stromal cells | Increased dynamically from D5 onward | Zhang et al. (2014) |
| Human | Endometrium | Glandular endometrium: ectopic > eutopic | Kolioulis et al. (2017) |
| Human | Epithelial cells (weak expression levels). ESCs (−) | Proliferative stage and early secretory phase | Baba et al. (2015) |
| ESCs (prominent expression levels) | Late secretory phase | ||
| Human | Endometrium | Glandular endometrium: Ectopic >eutopic, Stromal endometrium: no difference | Timologou et al. (2016) |
| Human | Luminal and glandular epithelial cells, stroma cells (−) | Not investigated | Cejudo Roman et al. (2012) |
| KISS1R | |||
| Human | vST, vCT | Decreased in RSA compared with abortion | Wu et al. (2014) |
| Deciduas | Not different between abortion and RSA | ||
| Human | vST, vCT | Not different between abortion and RSA | Park et al. (2012) |
| Human | vST, vCT, EVTs; PB and decidua basalis (−) | Not investigated | Matjila et al. (2013) |
| Human | vCT (early pregnancy) | First trimester > term pregnancy | Janneau et al. (2002) |
| Human | Placenta (early and term pregnancy, PE) | Early pregnancy > PE > term pregnancy | Cartwright and Williams (2012) |
| Human | Placenta, PB and DP (term pregnancy, PE) | Not different between term pregnancy and PE | Matjila et al. (2016) |
| Rat | Trophoblast giant cells | Low expression level | Terao et al. (2004) |
| Human | vST, vCT, EVTs | First trimester > term pregnancy | Bilban et al. (2004) |
| Rat | Placenta (gestation Day 16 and Day 22) | Day 22 > Day 16 | Mark et al. (2013) |
| Human | Umbilical vein endothelial cells | Not investigated | Ramaesh et al. (2010) |
| Mouse | Decidualizing stromal cells | Increased from D5 onward | Zhang et al. (2014) |
| Cat | Luminal epithelial cells and uterine glands. ESCs (−) | Not investigated | Tanyapanyachon et al. (2018) |
| Dog | Superficial and deep uterine glands | Higher in pre-implantation than post-implantation | Schafer-Somi et al. (2017) |
| Dog | Syncytiotrophoblast cells | Higher in mid-gestation than post-implantation | Schafer-Somi et al. (2017) |
| Human | Epithelial cells. ESCs (−) | Week expression levels | Baba et al. (2015) |
| Human | Luminal and glandular epithelial cells. ESCs (−) | Not investigated | Cejudo Roman et al. (2012) |
Abbreviations: RSA, recurrent spontaneous abortion; vST, villous syncytiotrophoblasts; vCT, villous cytotrophoblasts; EVTs, extravillous trophoblast cells; ESCs, endometrial stroma cells; DP, decidua parietalis; PB, placental bed.
Expression of KISS1 and KISS1R at the utero-placental interface during mammalian pregnancy.
| Species | Cells or tissues reported | Trends across pregnancy | References |
|---|---|---|---|
| KISS1 | |||
| Human | vST, vCT, Decidua | Decreased in RSA compared with abortion | Wu et al. (2014) |
| Human | vST, vCT | Decreased in RSA compared with abortion | Park et al. (2012) |
| Human | vST; PB and DP (−) | Not investigated | Matjila et al. (2013) |
| Human | vCT | Not different between term pregnancy and early pregnancy | Janneau et al. (2002) |
| Human | Placenta (early and term pregnancy, PE) | Early pregnancy > Term pregnancy > PE | Cartwright and Williams (2012) |
| Human | Placenta (term pregnancy, PE); PB and DP (minimal) | Term pregnancy < PE | Matjila et al. (2016) |
| Human | Placenta (term pregnancy) | Circadian rhythm (two surges, 04:00 h and 12:00 h, respectively). | de Pedro et al. (2015) |
| Human | vST | Not investigated | Horikoshi et al. (2003) |
| Rat | Trophoblast giant cells | Gradually decreased | Terao et al. (2004) |
| Human | vST, vCT; EVTs (−) | First trimester > term pregnancy | Bilban et al. (2004) |
| Rat | Trophoblast giant cells (gestation Day 16 and Day 22) | Day 22 > Day 16 | Mark et al. (2013) |
| Human | Placenta | Preterm pregnancy > term pregnancy | Torricelli et al. (2008) |
| Cat | Luminal epithelial cells and uterine glands, stroma cells | Not investigated | Tanyapanyachon et al. (2018) |
| Dog | Superficial uterine glands, Syncytiotrophoblast | Not changed between pre-implantation and post-implantation | Schafer-Somi et al. (2017) |
| Mouse | Decidualizing stromal cells | Increased dynamically from D5 onward | Zhang et al. (2014) |
| Human | Endometrium | Glandular endometrium: ectopic > eutopic | Kolioulis et al. (2017) |
| Human | Epithelial cells (weak expression levels). ESCs (−) | Proliferative stage and early secretory phase | Baba et al. (2015) |
| ESCs (prominent expression levels) | Late secretory phase | ||
| Human | Endometrium | Glandular endometrium: Ectopic >eutopic, Stromal endometrium: no difference | Timologou et al. (2016) |
| Human | Luminal and glandular epithelial cells, stroma cells (−) | Not investigated | Cejudo Roman et al. (2012) |
| KISS1R | |||
| Human | vST, vCT | Decreased in RSA compared with abortion | Wu et al. (2014) |
| Deciduas | Not different between abortion and RSA | ||
| Human | vST, vCT | Not different between abortion and RSA | Park et al. (2012) |
| Human | vST, vCT, EVTs; PB and decidua basalis (−) | Not investigated | Matjila et al. (2013) |
| Human | vCT (early pregnancy) | First trimester > term pregnancy | Janneau et al. (2002) |
| Human | Placenta (early and term pregnancy, PE) | Early pregnancy > PE > term pregnancy | Cartwright and Williams (2012) |
| Human | Placenta, PB and DP (term pregnancy, PE) | Not different between term pregnancy and PE | Matjila et al. (2016) |
| Rat | Trophoblast giant cells | Low expression level | Terao et al. (2004) |
| Human | vST, vCT, EVTs | First trimester > term pregnancy | Bilban et al. (2004) |
| Rat | Placenta (gestation Day 16 and Day 22) | Day 22 > Day 16 | Mark et al. (2013) |
| Human | Umbilical vein endothelial cells | Not investigated | Ramaesh et al. (2010) |
| Mouse | Decidualizing stromal cells | Increased from D5 onward | Zhang et al. (2014) |
| Cat | Luminal epithelial cells and uterine glands. ESCs (−) | Not investigated | Tanyapanyachon et al. (2018) |
| Dog | Superficial and deep uterine glands | Higher in pre-implantation than post-implantation | Schafer-Somi et al. (2017) |
| Dog | Syncytiotrophoblast cells | Higher in mid-gestation than post-implantation | Schafer-Somi et al. (2017) |
| Human | Epithelial cells. ESCs (−) | Week expression levels | Baba et al. (2015) |
| Human | Luminal and glandular epithelial cells. ESCs (−) | Not investigated | Cejudo Roman et al. (2012) |
| Species | Cells or tissues reported | Trends across pregnancy | References |
|---|---|---|---|
| KISS1 | |||
| Human | vST, vCT, Decidua | Decreased in RSA compared with abortion | Wu et al. (2014) |
| Human | vST, vCT | Decreased in RSA compared with abortion | Park et al. (2012) |
| Human | vST; PB and DP (−) | Not investigated | Matjila et al. (2013) |
| Human | vCT | Not different between term pregnancy and early pregnancy | Janneau et al. (2002) |
| Human | Placenta (early and term pregnancy, PE) | Early pregnancy > Term pregnancy > PE | Cartwright and Williams (2012) |
| Human | Placenta (term pregnancy, PE); PB and DP (minimal) | Term pregnancy < PE | Matjila et al. (2016) |
| Human | Placenta (term pregnancy) | Circadian rhythm (two surges, 04:00 h and 12:00 h, respectively). | de Pedro et al. (2015) |
| Human | vST | Not investigated | Horikoshi et al. (2003) |
| Rat | Trophoblast giant cells | Gradually decreased | Terao et al. (2004) |
| Human | vST, vCT; EVTs (−) | First trimester > term pregnancy | Bilban et al. (2004) |
| Rat | Trophoblast giant cells (gestation Day 16 and Day 22) | Day 22 > Day 16 | Mark et al. (2013) |
| Human | Placenta | Preterm pregnancy > term pregnancy | Torricelli et al. (2008) |
| Cat | Luminal epithelial cells and uterine glands, stroma cells | Not investigated | Tanyapanyachon et al. (2018) |
| Dog | Superficial uterine glands, Syncytiotrophoblast | Not changed between pre-implantation and post-implantation | Schafer-Somi et al. (2017) |
| Mouse | Decidualizing stromal cells | Increased dynamically from D5 onward | Zhang et al. (2014) |
| Human | Endometrium | Glandular endometrium: ectopic > eutopic | Kolioulis et al. (2017) |
| Human | Epithelial cells (weak expression levels). ESCs (−) | Proliferative stage and early secretory phase | Baba et al. (2015) |
| ESCs (prominent expression levels) | Late secretory phase | ||
| Human | Endometrium | Glandular endometrium: Ectopic >eutopic, Stromal endometrium: no difference | Timologou et al. (2016) |
| Human | Luminal and glandular epithelial cells, stroma cells (−) | Not investigated | Cejudo Roman et al. (2012) |
| KISS1R | |||
| Human | vST, vCT | Decreased in RSA compared with abortion | Wu et al. (2014) |
| Deciduas | Not different between abortion and RSA | ||
| Human | vST, vCT | Not different between abortion and RSA | Park et al. (2012) |
| Human | vST, vCT, EVTs; PB and decidua basalis (−) | Not investigated | Matjila et al. (2013) |
| Human | vCT (early pregnancy) | First trimester > term pregnancy | Janneau et al. (2002) |
| Human | Placenta (early and term pregnancy, PE) | Early pregnancy > PE > term pregnancy | Cartwright and Williams (2012) |
| Human | Placenta, PB and DP (term pregnancy, PE) | Not different between term pregnancy and PE | Matjila et al. (2016) |
| Rat | Trophoblast giant cells | Low expression level | Terao et al. (2004) |
| Human | vST, vCT, EVTs | First trimester > term pregnancy | Bilban et al. (2004) |
| Rat | Placenta (gestation Day 16 and Day 22) | Day 22 > Day 16 | Mark et al. (2013) |
| Human | Umbilical vein endothelial cells | Not investigated | Ramaesh et al. (2010) |
| Mouse | Decidualizing stromal cells | Increased from D5 onward | Zhang et al. (2014) |
| Cat | Luminal epithelial cells and uterine glands. ESCs (−) | Not investigated | Tanyapanyachon et al. (2018) |
| Dog | Superficial and deep uterine glands | Higher in pre-implantation than post-implantation | Schafer-Somi et al. (2017) |
| Dog | Syncytiotrophoblast cells | Higher in mid-gestation than post-implantation | Schafer-Somi et al. (2017) |
| Human | Epithelial cells. ESCs (−) | Week expression levels | Baba et al. (2015) |
| Human | Luminal and glandular epithelial cells. ESCs (−) | Not investigated | Cejudo Roman et al. (2012) |
Abbreviations: RSA, recurrent spontaneous abortion; vST, villous syncytiotrophoblasts; vCT, villous cytotrophoblasts; EVTs, extravillous trophoblast cells; ESCs, endometrial stroma cells; DP, decidua parietalis; PB, placental bed.
Endometriosis is considered a common benign gynecologic disease characterized by the ectopic presence of endometrial tissues (including glandular epithelium and stroma) outside the uterus. A previous study showed that the expression of KISS1 is significantly higher in the glandular endometrium of endometriosis lesions compared with the eutopic glandular endometrium in patients with or without endometriosis (Timologou et al., 2016). Interestingly, there was no significant difference in KISS1 expression in the stromal endometrium between patients with and patients without endometriosis (Timologou et al., 2016), indicating that kisspeptin, known as a suppressor of tumor metastasis, in the glandular endometrium, rather than the stromal endometrium, was potentially implicated in the pathogenesis or maintenance of endometriosis.
Expression of kisspeptins and KISS1R in the placenta
Human placenta is a highly specialized organ that forms the interface between the mother and fetus, supplying the fetus with nutrients and excreting waste products from the fetus. The placenta releases metabolic products into the maternal and fetal circulations. In addition, the placenta releases hormones into both the maternal and fetal circulations to modulate multiple gestational functions, including metabolism, fetal growth, parturition and other pregnancy-related functions (Gude et al., 2004). Immediately following implantation, the trophoblast stem cells proliferate and differentiate into two main cell types described as villous and extravillous (for reviews, see Gude et al., 2004; James et al., 2012). Non-migratory, villous cytotrophoblast cells (vCTs) fuse to form the multinucleated villous syncytiotrophoblast cells (vSTs), which reside the outer epithelial layer of chorionic villi. As the fundamental functional unit of the placenta, the chorionic villus is responsible for all nutritional-waste exchange between the maternal and the fetal circulation, acting to protect the fetus and exert various endocrine functions (for reviews, see Gude et al., 2004; James et al., 2012). Extravillous trophoblast cells (EVTs) eventually differentiate into the placental bed (PB) and endovascular trophoblasts. The PB is responsible for the migration of the embryo into the decidua, and the endovascular trophoblasts are responsible for the uterine spiral artery remodeling (Gude et al., 2004) (Figure 3). However, it is important to note that the differentiation of the placenta in rodents (particularly in the mouse placenta) is quite different from that in humans. Following implantation in mice, the non-polar trophectoderm proliferates and then differentiates into primary trophoblast giant cells, while the polar trophectoderm proliferates and then forms the extraembryonic ectoderm, which further differentiates into the labyrinth and secondary trophoblast giant cells. The labyrinth, named for the intricate network between the mother and fetus, shares similar functions with chorionic villi, such as the transportation of nutrients and endocrine regulation. Both primary and secondary trophoblast giant cells are invasive cells that lead the placenta to attach to the uterus and remodel the uterine vasculature. Therefore, the activities of these cells are similar to those of the EVTs in humans (see reviews Cross et al., 2002; James et al., 2012) (Figure 3).
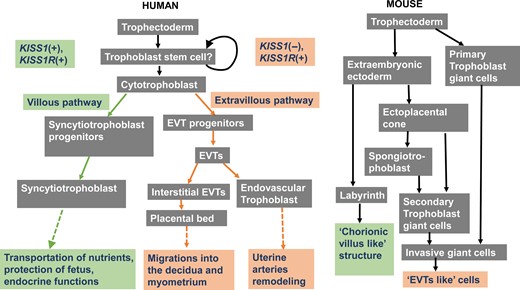
Flow diagram demonstrating the different cell lineage pathways involved in murine and human trophoblast differentiation and function.KISS1 is mainly expressed in cells of the villous pathway, while KISS1R is expressed in cells of both the villous and extravillous pathways. Modified from James et al. (2012).
Using Northern blot analysis, a study demonstrated that the expression of KISS1 mRNA was very weak in the kidney and pancreas but extremely high in the placenta (Lee et al., 1996). However, the expression of KISS1 mRNA was undetectable in the normal heart, brain, liver, lung and skeletal muscle (Lee et al., 1996). In humans, KISS1R was highly expressed in endocrine tissues, including the placenta, pituitary, pancreas and brain, suggesting a role for the kisspeptin/KISS1R system in the regulation of the endocrine function (Kotani et al., 2001; Muir et al., 2001). These early studies strongly suggested that the kisspeptin/KISS1R system may play a fundamental role in regulating the functions of the placenta. More recently, the expression of KISS1 and KISS1R in specific cells within the placenta has been identified, though some studies were inconsistent.
In early pregnancy in humans, the KISS1 gene is mainly expressed in vCTs (Janneau et al., 2002; Horikoshi et al., 2003; Park et al., 2012; Matjila et al., 2013; Wu et al., 2014) but not in EVTs (Bilban et al., 2004). As the placenta matures, the expression of KISS1 is detected in vSTs as well as in the PB (Horikoshi et al., 2003; Matjila et al., 2013, 2016) (Table I). Additionally, studies in rats suggested that Kiss1 is mainly expressed in trophoblast giant cells (similarly to vCTs in humans) and gradually decreases as the placenta matured (Terao et al., 2004; Mark et al., 2013). Nevertheless, many inconsistencies need to be addressed. In humans, it was first identified that there was no significant difference in KISS1 expression in the placenta between term pregnancy and early pregnancy (Janneau et al., 2002). However, later studies indicated that KISS1 expression is much higher in the placenta in early pregnancy than that in term pregnancy (Bilban et al., 2004; Cartwright and Williams, 2012). Another study also suggested that KISS1 expression in the placenta is higher in preterm pregnancy than in term pregnancy (Torricelli et al., 2008), indicating that KISS1 expression gradually decreases as the placenta matured. Similarly, Kiss1 expression gradually decreases during the maturation of the placenta in rats (Terao et al., 2004) (Table I). Interestingly, KISS1 expression in human term pregnancy shows a circadian rhythm, with two surges at 04:00 h and 12:00 h each day (de Pedro et al., 2015). Similar to the expression of the KISS1 gene in humans, KISS1R is also expressed in vCTs in early pregnancy (Janneau et al., 2002) and in vSTs during the maturation of the placenta (Bilban et al., 2004; Park et al., 2012; Matjila et al., 2013; Wu et al., 2014) (Table I). However, KISS1R is also located in EVTs (Bilban et al., 2004; Matjila et al., 2013), the sites where KISS1 expression was not detected (Bilban et al., 2004). The expression level of KISS1R in the placenta was higher in the first trimester than in the term pregnancy, indicating that KISS1R expression gradually decreases as the placenta matured (Janneau et al., 2002; Bilban et al., 2004; Cartwright and Williams, 2012). Additionally, KISS1R expression was not detected in PB (Matjila et al., 2013), and it showed a minimal expression level in human term pregnancy (Matjila et al., 2016). In combination with the expression pattern of KISS1 in the placenta, it may be speculated that the kisspeptin/KISS1R system in the placenta mainly functions in early pregnancy in humans in both autocrine and paracrine manners. However, the expression level of Kiss1r is very low in rats (Terao et al., 2004) and islower at Day 16 of pregnancy than at Day 22 of pregnancy Mark et al. (2013). Notably, Kiss1r expression levels in canine placenta is higher in mid-gestation than in post-implantation (Schafer-Somi et al., 2017) (Table I). These studies suggest that the role of the kisspeptin/KISS1R system in the placenta was not conserved in different species.
Circulating levels of kisspeptin during pregnancy
Because kisspeptin is produced by trophoblasts and because trophoblast invasion is underway 5 days after blastocyst transplantation (Norwitz et al., 2001), plasma kisspeptin concentrations during the peri-implantation period may reflect early developmental events associated with pregnancy outcome. In humans, the plasma concentration of kisspeptin increases dramatically over the course of pregnancy, with a 900-fold increase in the first trimester and further increases to over 7000-fold in the third trimester compared to the non-pregnant women (Horikoshi et al., 2003). Intriguingly, previous studies in humans have suggested that KISS1 expression levels in the early placenta and the term placenta do not differ significantly (Janneau et al., 2002) or that they are much higher in the early placenta (Bilban et al., 2004; Cartwright and Williams, 2012). When taking these published data together, it seems that the expression levels of KISS1 in the placenta of humans may not necessarily reflect the plasma levels of kisspeptin. It is most likely that the increase in the total number of trophoblast cells throughout the course of gestation is responsible for the dramatic increase in serum kisspeptin levels examined. Interestingly, peripheral kisspeptin levels were found to be very low and did not increase during pregnancy in sheep, cows, pigs, rabbits, horses, rhesus monkeys and marmosets, suggesting that the increase in plasma kisspeptin levels during pregnancy may be unique to humans (Babwah, 2015). However, by using the bovine-specific enzyme immunoassay, recent studies in cows suggested that plasma kisspeptin levels are consistently increased during pregnancy (Mondal et al., 2015, 2016), indicating that the increase of plasma kisspeptin levels during pregnancy is conserved in cows. Therefore, we may speculate that the detection methods currently used are simply failing to identify kisspeptin proteins in the circulation of certain animals. Future studies performed using species-specific enzyme immunoassay or radioimmunoassay to detect the circulation levels of kisspeptin of related animals will be of great interest. It needs to be noted that kisspeptins are a series of peptides, including kisspeptin-54, kisspeptin-14, kisspeptin-13 and kisspeptin-10 (Kotani et al., 2001) (for reviews, see Pinilla et al., 2012; Hu et al., 2018). These studies seemed to detect only one kisspeptin peptide in the plasma, and the possibility that the antibody used in these studies cross-reacted with other RF amide related-peptides (RFRP), including prolactin-releasing peptide, RFRP1, RFRP2, RFRP3, neuropeptide FF and neuropeptide AF, could not be excluded (Jayasena et al., 2014). Nevertheless, a previous study using a more specific and sensitive antibody to detect the plasma kisspeptins (including human kisspeptin-54, kisspeptin-14 and kisspeptin-10, rather than one kind of kisspeptin) suggested that plasma kisspeptin levels significantly correlate with gestational age (Jayasena et al., 2014) (Table II). Additionally, a very recent study that detected plasma kisspeptin-54 levels in pregnant women suggested the same results (Sullivan-Pyke et al., 2018) (Table II). Therefore, the evidence for the increase in plasma kisspeptin levels during gestation in women seems convincing, at least in humans (Table II). Future studies need to address the intriguing phenomenon of the increase in plasma kisspeptin levels and the decrease in placental KISS1 expression levels during gestation in humans.
Kisspeptin (KP) concentrations in human plasma or serum during early pregnancy.
| Pregnancy state | Serum/plasma | Kisspeptin (KP) | Gestation age | Concentration | Reference |
|---|---|---|---|---|---|
| UP | Serum | KP-54 | 6–10 Weeks (n = 20) | 1.50 (0.55–3.72) ng/ml | Sullivan-Pyke et al. (2018) |
| Plasma | KP-54 | 6–10 Weeks (n = 8) | 3.52 (1.09–21.7) ng/ml | ||
| SAB | Serum | KP-54 | 6–10 Weeks (n = 20) | 0.20 (0.07–0.37) ng/ml | |
| Plasma | KP-54 | 6–10 Weeks (n = 3) | 2.7 (0.46–2.7) ng/ml | ||
| UP | Plasma | KP-10 | 7–18 Weeks (n = 20) | 5783 (3168–9953) pg/ml | Kavvasoglu et al. (2012) |
| Miscarriage | Plasma | KP-10 | 7–18 Weeks (n = 20) | 391 (152–951) pg/ml | |
| UP | Plasma | KP-54, 14 and 10 | 11.3 ± 1.9 (n = 899) | 1.06 ± 0.42 pmol/ml | Jayasena et al. (2014) |
| Miscarriage | Plasma | KP-54, 14 and 10 | 9.8 ± 3.1 (n = 50) | 0.42 ± 0.39 pmol/ml | |
| UP (twin) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 20) | 1.64 ± 0.54 pmol/ml | |
| Miscarriage of one fetus (twin) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 10) | 1.15 ± 0.58 pmol/ml | |
| UP (triplet) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 2) | 2.09 ± 0.26 pmol/ml | |
| UP | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 25) | 4.51 ± 2.21 nmol/l | Cetkovic et al. (2012) |
| 21–25 Weeks (n = 25) | 10.33 ± 2.65 nmol/l | ||||
| 32–36 Weeks (n = 25) | 20.48 ± 7.60 nmol/l | ||||
| GD | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 20) | 4.51 ± 3.18 nmol/l | |
| 32–36 Weeks (n = 20) | 11.643 ± 7.65 nmol/l | ||||
| Type 1 | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 16) | 1.82 ± 1.47 nmol/l | |
| 21–25 Weeks (n = 16) | 7.12 ± 4.78 nmol/l | ||||
| 32–36 Weeks (n = 16) | 16.31 ± 12.10 nmol/l | ||||
| H | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 22) | 1.18 ± 0.36 nmol/l | |
| 21–25 Weeks (n = 22) | 3.42 ± 1.04 nmol/l | ||||
| 32–36 Weeks (n = 22) | 14.14 ± 10.44 nmol/l | ||||
| GH | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 18) | 8.46 ± 6.24 nmol/l | |
| 32–36 Weeks (n = 18) | 25.68 ± 9.23 nmol/l | ||||
| PE | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 28) | 4.46 ± 3.73 nmol/l | |
| 32–36 Weeks (n = 28) | 16.03 ± 10.09 nmol/l | ||||
| UP | Plasma | KP-54, 14 and 10 | FT (10.4 ± 0.5 Weeks) (n = 13) | 803 ± 125 pmol/l | Dhillo et al. (2006) |
| TT (38 ± 0.8 Weeks) (n = 7) | 2483 ± 302 pmol/l | ||||
| 15 d postpartum (n = 7) | <2 pmol/l | ||||
| Malignant GTN | Plasma | KP-54, 14 and 10 | Pre-therapy (n = 11) | 1363 ± 1076 pmol/l | |
| Post-therapy (n = 11) | <2 pmol/l | ||||
| UP | Plasma | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 13 783 ± 864 pmol/l | Jayasena et al. (2015) |
| Salivary | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 123 ± 34 pmol/l | ||
| Urine | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 301 ± 59 pmol/l | ||
| UP | Plasma | KP-54 | FT (n = 11) | 1230 ± 346 pmol/l | Horikoshi et al. (2003) |
| ST (n = 16) | 4590 ± 555 pmol/l | ||||
| TT (n = 12) | 9590 ± 1640 pmol/l | ||||
| 5 d postdelivery (n = 10) | 7.63 ± 1.33 pmol/l | ||||
| UP (singleton) | Serum | KP-54 | 11–14 Weeks (n = 31) | 1995 ± 375 pmol/l | Madazli et al. (2012) |
| PE (singleton) | Serum | KP-54 | 11–14 Weeks (n = 30) | 1554 ± 385 pmol/l | |
| UP (singleton) | Plasma | KP-54 | TT (n = 50) | 9.69 ± 1.35 ng/ml | Adali et al. (2012) |
| Mild PE | Plasma | KP-54 | TT (n = 15) | 2.61 ± 0.40 ng/ml | |
| Severe PE | Plasma | KP-54 | TT (n = 24) | 1.17 ± 0.24 ng/ml | |
| UP (singleton) | Plasma | KP-10 | 16 Weeks (n = 158) | 1044 ± 621 pmol/l | Logie et al. (2012) |
| 28 Weeks (n = 139) | 1714 ± 1306 pmol/l | ||||
| 36 Weeks (n = 99) | 2441 ± 2123 pmol/l | ||||
| UP | Serum | KP-10 | 16–20 Weeks (n = 317) | 1188 (494–2298) pg/ml | Armstrong et al. (2009) |
| UP | Serum | KP-10 | 16–20 Weeks (n = 118) | 1164 (442–3903) pg/ml | |
| PE | Serum | KP-10 | 16–20 Weeks (n = 57) | 1109 (605–1935) pg/ml | |
| UP | Serum | KP-10 | 38.03 ± 0.06 (n = 30) | 1.66 ± 0.59 ng/ml | Matjila et al. (2016) |
| Cord serum | 0.58 ± 0.39 ng/ml | ||||
| PE | Serum | KP-10 | 32.95 ± 0.53 (n = 19) | 2.03 ± 0.44 ng/ml | |
| Cord serum | 1.78 ± 0.38 ng/ml | ||||
| UP | Plasma | KP-10 | 8–14 Weeks (n = 31) | 2035 ± 1260 pmol/l | Smets et al. (2008) |
| SGA | Plasma | KP-10 | 8–14 Weeks (n = 31) | 1376 ± 1317 pmol/l |
| Pregnancy state | Serum/plasma | Kisspeptin (KP) | Gestation age | Concentration | Reference |
|---|---|---|---|---|---|
| UP | Serum | KP-54 | 6–10 Weeks (n = 20) | 1.50 (0.55–3.72) ng/ml | Sullivan-Pyke et al. (2018) |
| Plasma | KP-54 | 6–10 Weeks (n = 8) | 3.52 (1.09–21.7) ng/ml | ||
| SAB | Serum | KP-54 | 6–10 Weeks (n = 20) | 0.20 (0.07–0.37) ng/ml | |
| Plasma | KP-54 | 6–10 Weeks (n = 3) | 2.7 (0.46–2.7) ng/ml | ||
| UP | Plasma | KP-10 | 7–18 Weeks (n = 20) | 5783 (3168–9953) pg/ml | Kavvasoglu et al. (2012) |
| Miscarriage | Plasma | KP-10 | 7–18 Weeks (n = 20) | 391 (152–951) pg/ml | |
| UP | Plasma | KP-54, 14 and 10 | 11.3 ± 1.9 (n = 899) | 1.06 ± 0.42 pmol/ml | Jayasena et al. (2014) |
| Miscarriage | Plasma | KP-54, 14 and 10 | 9.8 ± 3.1 (n = 50) | 0.42 ± 0.39 pmol/ml | |
| UP (twin) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 20) | 1.64 ± 0.54 pmol/ml | |
| Miscarriage of one fetus (twin) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 10) | 1.15 ± 0.58 pmol/ml | |
| UP (triplet) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 2) | 2.09 ± 0.26 pmol/ml | |
| UP | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 25) | 4.51 ± 2.21 nmol/l | Cetkovic et al. (2012) |
| 21–25 Weeks (n = 25) | 10.33 ± 2.65 nmol/l | ||||
| 32–36 Weeks (n = 25) | 20.48 ± 7.60 nmol/l | ||||
| GD | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 20) | 4.51 ± 3.18 nmol/l | |
| 32–36 Weeks (n = 20) | 11.643 ± 7.65 nmol/l | ||||
| Type 1 | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 16) | 1.82 ± 1.47 nmol/l | |
| 21–25 Weeks (n = 16) | 7.12 ± 4.78 nmol/l | ||||
| 32–36 Weeks (n = 16) | 16.31 ± 12.10 nmol/l | ||||
| H | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 22) | 1.18 ± 0.36 nmol/l | |
| 21–25 Weeks (n = 22) | 3.42 ± 1.04 nmol/l | ||||
| 32–36 Weeks (n = 22) | 14.14 ± 10.44 nmol/l | ||||
| GH | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 18) | 8.46 ± 6.24 nmol/l | |
| 32–36 Weeks (n = 18) | 25.68 ± 9.23 nmol/l | ||||
| PE | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 28) | 4.46 ± 3.73 nmol/l | |
| 32–36 Weeks (n = 28) | 16.03 ± 10.09 nmol/l | ||||
| UP | Plasma | KP-54, 14 and 10 | FT (10.4 ± 0.5 Weeks) (n = 13) | 803 ± 125 pmol/l | Dhillo et al. (2006) |
| TT (38 ± 0.8 Weeks) (n = 7) | 2483 ± 302 pmol/l | ||||
| 15 d postpartum (n = 7) | <2 pmol/l | ||||
| Malignant GTN | Plasma | KP-54, 14 and 10 | Pre-therapy (n = 11) | 1363 ± 1076 pmol/l | |
| Post-therapy (n = 11) | <2 pmol/l | ||||
| UP | Plasma | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 13 783 ± 864 pmol/l | Jayasena et al. (2015) |
| Salivary | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 123 ± 34 pmol/l | ||
| Urine | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 301 ± 59 pmol/l | ||
| UP | Plasma | KP-54 | FT (n = 11) | 1230 ± 346 pmol/l | Horikoshi et al. (2003) |
| ST (n = 16) | 4590 ± 555 pmol/l | ||||
| TT (n = 12) | 9590 ± 1640 pmol/l | ||||
| 5 d postdelivery (n = 10) | 7.63 ± 1.33 pmol/l | ||||
| UP (singleton) | Serum | KP-54 | 11–14 Weeks (n = 31) | 1995 ± 375 pmol/l | Madazli et al. (2012) |
| PE (singleton) | Serum | KP-54 | 11–14 Weeks (n = 30) | 1554 ± 385 pmol/l | |
| UP (singleton) | Plasma | KP-54 | TT (n = 50) | 9.69 ± 1.35 ng/ml | Adali et al. (2012) |
| Mild PE | Plasma | KP-54 | TT (n = 15) | 2.61 ± 0.40 ng/ml | |
| Severe PE | Plasma | KP-54 | TT (n = 24) | 1.17 ± 0.24 ng/ml | |
| UP (singleton) | Plasma | KP-10 | 16 Weeks (n = 158) | 1044 ± 621 pmol/l | Logie et al. (2012) |
| 28 Weeks (n = 139) | 1714 ± 1306 pmol/l | ||||
| 36 Weeks (n = 99) | 2441 ± 2123 pmol/l | ||||
| UP | Serum | KP-10 | 16–20 Weeks (n = 317) | 1188 (494–2298) pg/ml | Armstrong et al. (2009) |
| UP | Serum | KP-10 | 16–20 Weeks (n = 118) | 1164 (442–3903) pg/ml | |
| PE | Serum | KP-10 | 16–20 Weeks (n = 57) | 1109 (605–1935) pg/ml | |
| UP | Serum | KP-10 | 38.03 ± 0.06 (n = 30) | 1.66 ± 0.59 ng/ml | Matjila et al. (2016) |
| Cord serum | 0.58 ± 0.39 ng/ml | ||||
| PE | Serum | KP-10 | 32.95 ± 0.53 (n = 19) | 2.03 ± 0.44 ng/ml | |
| Cord serum | 1.78 ± 0.38 ng/ml | ||||
| UP | Plasma | KP-10 | 8–14 Weeks (n = 31) | 2035 ± 1260 pmol/l | Smets et al. (2008) |
| SGA | Plasma | KP-10 | 8–14 Weeks (n = 31) | 1376 ± 1317 pmol/l |
Abbreviations: UP, uncomplicated pregnancy; GD, gestational diabetes mellitus; type 1, pre-gestational insulin-dependent diabetes mellitus; H, chronic hypertension; GH, gestational hypertension; PE, pre-eclampsia; SGA, restriction small for gestational age; FT, the first trimester; ST, the second trimester; TT, the third trimester.
Median ± SD or Median (quartile range) [range].
Kisspeptin (KP) concentrations in human plasma or serum during early pregnancy.
| Pregnancy state | Serum/plasma | Kisspeptin (KP) | Gestation age | Concentration | Reference |
|---|---|---|---|---|---|
| UP | Serum | KP-54 | 6–10 Weeks (n = 20) | 1.50 (0.55–3.72) ng/ml | Sullivan-Pyke et al. (2018) |
| Plasma | KP-54 | 6–10 Weeks (n = 8) | 3.52 (1.09–21.7) ng/ml | ||
| SAB | Serum | KP-54 | 6–10 Weeks (n = 20) | 0.20 (0.07–0.37) ng/ml | |
| Plasma | KP-54 | 6–10 Weeks (n = 3) | 2.7 (0.46–2.7) ng/ml | ||
| UP | Plasma | KP-10 | 7–18 Weeks (n = 20) | 5783 (3168–9953) pg/ml | Kavvasoglu et al. (2012) |
| Miscarriage | Plasma | KP-10 | 7–18 Weeks (n = 20) | 391 (152–951) pg/ml | |
| UP | Plasma | KP-54, 14 and 10 | 11.3 ± 1.9 (n = 899) | 1.06 ± 0.42 pmol/ml | Jayasena et al. (2014) |
| Miscarriage | Plasma | KP-54, 14 and 10 | 9.8 ± 3.1 (n = 50) | 0.42 ± 0.39 pmol/ml | |
| UP (twin) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 20) | 1.64 ± 0.54 pmol/ml | |
| Miscarriage of one fetus (twin) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 10) | 1.15 ± 0.58 pmol/ml | |
| UP (triplet) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 2) | 2.09 ± 0.26 pmol/ml | |
| UP | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 25) | 4.51 ± 2.21 nmol/l | Cetkovic et al. (2012) |
| 21–25 Weeks (n = 25) | 10.33 ± 2.65 nmol/l | ||||
| 32–36 Weeks (n = 25) | 20.48 ± 7.60 nmol/l | ||||
| GD | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 20) | 4.51 ± 3.18 nmol/l | |
| 32–36 Weeks (n = 20) | 11.643 ± 7.65 nmol/l | ||||
| Type 1 | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 16) | 1.82 ± 1.47 nmol/l | |
| 21–25 Weeks (n = 16) | 7.12 ± 4.78 nmol/l | ||||
| 32–36 Weeks (n = 16) | 16.31 ± 12.10 nmol/l | ||||
| H | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 22) | 1.18 ± 0.36 nmol/l | |
| 21–25 Weeks (n = 22) | 3.42 ± 1.04 nmol/l | ||||
| 32–36 Weeks (n = 22) | 14.14 ± 10.44 nmol/l | ||||
| GH | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 18) | 8.46 ± 6.24 nmol/l | |
| 32–36 Weeks (n = 18) | 25.68 ± 9.23 nmol/l | ||||
| PE | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 28) | 4.46 ± 3.73 nmol/l | |
| 32–36 Weeks (n = 28) | 16.03 ± 10.09 nmol/l | ||||
| UP | Plasma | KP-54, 14 and 10 | FT (10.4 ± 0.5 Weeks) (n = 13) | 803 ± 125 pmol/l | Dhillo et al. (2006) |
| TT (38 ± 0.8 Weeks) (n = 7) | 2483 ± 302 pmol/l | ||||
| 15 d postpartum (n = 7) | <2 pmol/l | ||||
| Malignant GTN | Plasma | KP-54, 14 and 10 | Pre-therapy (n = 11) | 1363 ± 1076 pmol/l | |
| Post-therapy (n = 11) | <2 pmol/l | ||||
| UP | Plasma | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 13 783 ± 864 pmol/l | Jayasena et al. (2015) |
| Salivary | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 123 ± 34 pmol/l | ||
| Urine | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 301 ± 59 pmol/l | ||
| UP | Plasma | KP-54 | FT (n = 11) | 1230 ± 346 pmol/l | Horikoshi et al. (2003) |
| ST (n = 16) | 4590 ± 555 pmol/l | ||||
| TT (n = 12) | 9590 ± 1640 pmol/l | ||||
| 5 d postdelivery (n = 10) | 7.63 ± 1.33 pmol/l | ||||
| UP (singleton) | Serum | KP-54 | 11–14 Weeks (n = 31) | 1995 ± 375 pmol/l | Madazli et al. (2012) |
| PE (singleton) | Serum | KP-54 | 11–14 Weeks (n = 30) | 1554 ± 385 pmol/l | |
| UP (singleton) | Plasma | KP-54 | TT (n = 50) | 9.69 ± 1.35 ng/ml | Adali et al. (2012) |
| Mild PE | Plasma | KP-54 | TT (n = 15) | 2.61 ± 0.40 ng/ml | |
| Severe PE | Plasma | KP-54 | TT (n = 24) | 1.17 ± 0.24 ng/ml | |
| UP (singleton) | Plasma | KP-10 | 16 Weeks (n = 158) | 1044 ± 621 pmol/l | Logie et al. (2012) |
| 28 Weeks (n = 139) | 1714 ± 1306 pmol/l | ||||
| 36 Weeks (n = 99) | 2441 ± 2123 pmol/l | ||||
| UP | Serum | KP-10 | 16–20 Weeks (n = 317) | 1188 (494–2298) pg/ml | Armstrong et al. (2009) |
| UP | Serum | KP-10 | 16–20 Weeks (n = 118) | 1164 (442–3903) pg/ml | |
| PE | Serum | KP-10 | 16–20 Weeks (n = 57) | 1109 (605–1935) pg/ml | |
| UP | Serum | KP-10 | 38.03 ± 0.06 (n = 30) | 1.66 ± 0.59 ng/ml | Matjila et al. (2016) |
| Cord serum | 0.58 ± 0.39 ng/ml | ||||
| PE | Serum | KP-10 | 32.95 ± 0.53 (n = 19) | 2.03 ± 0.44 ng/ml | |
| Cord serum | 1.78 ± 0.38 ng/ml | ||||
| UP | Plasma | KP-10 | 8–14 Weeks (n = 31) | 2035 ± 1260 pmol/l | Smets et al. (2008) |
| SGA | Plasma | KP-10 | 8–14 Weeks (n = 31) | 1376 ± 1317 pmol/l |
| Pregnancy state | Serum/plasma | Kisspeptin (KP) | Gestation age | Concentration | Reference |
|---|---|---|---|---|---|
| UP | Serum | KP-54 | 6–10 Weeks (n = 20) | 1.50 (0.55–3.72) ng/ml | Sullivan-Pyke et al. (2018) |
| Plasma | KP-54 | 6–10 Weeks (n = 8) | 3.52 (1.09–21.7) ng/ml | ||
| SAB | Serum | KP-54 | 6–10 Weeks (n = 20) | 0.20 (0.07–0.37) ng/ml | |
| Plasma | KP-54 | 6–10 Weeks (n = 3) | 2.7 (0.46–2.7) ng/ml | ||
| UP | Plasma | KP-10 | 7–18 Weeks (n = 20) | 5783 (3168–9953) pg/ml | Kavvasoglu et al. (2012) |
| Miscarriage | Plasma | KP-10 | 7–18 Weeks (n = 20) | 391 (152–951) pg/ml | |
| UP | Plasma | KP-54, 14 and 10 | 11.3 ± 1.9 (n = 899) | 1.06 ± 0.42 pmol/ml | Jayasena et al. (2014) |
| Miscarriage | Plasma | KP-54, 14 and 10 | 9.8 ± 3.1 (n = 50) | 0.42 ± 0.39 pmol/ml | |
| UP (twin) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 20) | 1.64 ± 0.54 pmol/ml | |
| Miscarriage of one fetus (twin) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 10) | 1.15 ± 0.58 pmol/ml | |
| UP (triplet) | Plasma | KP-54, 14 and 10 | 10.7 ± 1.5 (n = 2) | 2.09 ± 0.26 pmol/ml | |
| UP | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 25) | 4.51 ± 2.21 nmol/l | Cetkovic et al. (2012) |
| 21–25 Weeks (n = 25) | 10.33 ± 2.65 nmol/l | ||||
| 32–36 Weeks (n = 25) | 20.48 ± 7.60 nmol/l | ||||
| GD | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 20) | 4.51 ± 3.18 nmol/l | |
| 32–36 Weeks (n = 20) | 11.643 ± 7.65 nmol/l | ||||
| Type 1 | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 16) | 1.82 ± 1.47 nmol/l | |
| 21–25 Weeks (n = 16) | 7.12 ± 4.78 nmol/l | ||||
| 32–36 Weeks (n = 16) | 16.31 ± 12.10 nmol/l | ||||
| H | Plasma | KP-54, 14 and 10 | 9–12 Weeks (n = 22) | 1.18 ± 0.36 nmol/l | |
| 21–25 Weeks (n = 22) | 3.42 ± 1.04 nmol/l | ||||
| 32–36 Weeks (n = 22) | 14.14 ± 10.44 nmol/l | ||||
| GH | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 18) | 8.46 ± 6.24 nmol/l | |
| 32–36 Weeks (n = 18) | 25.68 ± 9.23 nmol/l | ||||
| PE | Plasma | KP-54, 14 and 10 | 21–25 Weeks (n = 28) | 4.46 ± 3.73 nmol/l | |
| 32–36 Weeks (n = 28) | 16.03 ± 10.09 nmol/l | ||||
| UP | Plasma | KP-54, 14 and 10 | FT (10.4 ± 0.5 Weeks) (n = 13) | 803 ± 125 pmol/l | Dhillo et al. (2006) |
| TT (38 ± 0.8 Weeks) (n = 7) | 2483 ± 302 pmol/l | ||||
| 15 d postpartum (n = 7) | <2 pmol/l | ||||
| Malignant GTN | Plasma | KP-54, 14 and 10 | Pre-therapy (n = 11) | 1363 ± 1076 pmol/l | |
| Post-therapy (n = 11) | <2 pmol/l | ||||
| UP | Plasma | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 13 783 ± 864 pmol/l | Jayasena et al. (2015) |
| Salivary | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 123 ± 34 pmol/l | ||
| Urine | KP-54, 14 and 10 | TT (34 ± 0.6) (n = 49) | 301 ± 59 pmol/l | ||
| UP | Plasma | KP-54 | FT (n = 11) | 1230 ± 346 pmol/l | Horikoshi et al. (2003) |
| ST (n = 16) | 4590 ± 555 pmol/l | ||||
| TT (n = 12) | 9590 ± 1640 pmol/l | ||||
| 5 d postdelivery (n = 10) | 7.63 ± 1.33 pmol/l | ||||
| UP (singleton) | Serum | KP-54 | 11–14 Weeks (n = 31) | 1995 ± 375 pmol/l | Madazli et al. (2012) |
| PE (singleton) | Serum | KP-54 | 11–14 Weeks (n = 30) | 1554 ± 385 pmol/l | |
| UP (singleton) | Plasma | KP-54 | TT (n = 50) | 9.69 ± 1.35 ng/ml | Adali et al. (2012) |
| Mild PE | Plasma | KP-54 | TT (n = 15) | 2.61 ± 0.40 ng/ml | |
| Severe PE | Plasma | KP-54 | TT (n = 24) | 1.17 ± 0.24 ng/ml | |
| UP (singleton) | Plasma | KP-10 | 16 Weeks (n = 158) | 1044 ± 621 pmol/l | Logie et al. (2012) |
| 28 Weeks (n = 139) | 1714 ± 1306 pmol/l | ||||
| 36 Weeks (n = 99) | 2441 ± 2123 pmol/l | ||||
| UP | Serum | KP-10 | 16–20 Weeks (n = 317) | 1188 (494–2298) pg/ml | Armstrong et al. (2009) |
| UP | Serum | KP-10 | 16–20 Weeks (n = 118) | 1164 (442–3903) pg/ml | |
| PE | Serum | KP-10 | 16–20 Weeks (n = 57) | 1109 (605–1935) pg/ml | |
| UP | Serum | KP-10 | 38.03 ± 0.06 (n = 30) | 1.66 ± 0.59 ng/ml | Matjila et al. (2016) |
| Cord serum | 0.58 ± 0.39 ng/ml | ||||
| PE | Serum | KP-10 | 32.95 ± 0.53 (n = 19) | 2.03 ± 0.44 ng/ml | |
| Cord serum | 1.78 ± 0.38 ng/ml | ||||
| UP | Plasma | KP-10 | 8–14 Weeks (n = 31) | 2035 ± 1260 pmol/l | Smets et al. (2008) |
| SGA | Plasma | KP-10 | 8–14 Weeks (n = 31) | 1376 ± 1317 pmol/l |
Abbreviations: UP, uncomplicated pregnancy; GD, gestational diabetes mellitus; type 1, pre-gestational insulin-dependent diabetes mellitus; H, chronic hypertension; GH, gestational hypertension; PE, pre-eclampsia; SGA, restriction small for gestational age; FT, the first trimester; ST, the second trimester; TT, the third trimester.
Median ± SD or Median (quartile range) [range].
Kisspeptins and embryo implantation
In endometrial tissues in mice, kisspeptin can trigger the phosphorylation of p38 and ERK1/2 in the uterus on D4 of pregnancy (Fayazi et al., 2015), suggesting that kisspeptin/KISS1R may affect the function of the endometrium in mice. Additionally, the expression of Kiss1 and Kiss1r mRNA gradually increases with the progression of stromal cell decidualization in the ovariectomized mouse endometrium (Zhang et al., 2014). Furthermore, the progression of stromal cell decidualization was significantly blocked when the expression of Kiss1 was attenuated by small interfering RNAs (Zhang et al., 2014). A previous study indicated that kisspeptin treatment increased mouse blastocyst adhesion to collagens (Taylor et al., 2014), possibly through the down-regulation of MMP (particularly MMP-2 and MMP-9) activity via the ERK1/2 and protein kinase C signaling pathways (Dey et al., 2004; McEwan et al., 2009; Taylor et al., 2014). These studies suggest that the kisspeptin/KISS1R system acts at both the blastocyst and endometrium to facilitate the process of embryo implantation (Figure 4). The ablation of kiss1 gene (Kiss1 -/-) led to hypogonadism and infertility in female mice (Funes et al., 2003; Lapatto et al., 2007), while acute replacement of gonadotropins and estradiol restored ovulation, mating and fertilization (Calder et al., 2014). However, these hormone-rescued mice were still unable to achieve pregnancy because the embryos (Kiss1 +/−) failed to implant (Calder et al., 2014). Furthermore, the same study demonstrated that embryos (Kiss1 +/−) successfully implanted in the uterus of a wild-type recipient female, suggesting that the inability to implant was due to a maternal factor but not an embryonic defect of the kisspeptin/KISS1R signaling (Calder et al., 2014). Leukemia inhibitory factor (LIF) is a cytokine that is required for implantation in mice (Bhatt et al., 1991; Stewart et al., 1992). The uterine gland-derived LIF initiates embryo-uterine communication, leading to embryo attachment and stromal cell decidualisation (Shuya et al., 2011; Kelleher et al., 2018). In a murine Kiss1 (−/−) model, LIF is weakly expressed in the uterine glands on the day of implantation, and exogenous LIF treatment in hormone-primed Kiss1 (−/−) female mice partially rescued implantation (Calder et al., 2014). These data suggest that kisspeptin is the upstream regulator of LIF; the mechanism by which kisspeptin affects the process of implantation is by up-regulating LIF levels. Furthermore, estrogen rapidly up-regulated uterine LIF expression, further demonstrating that supplementation with LIF restores decidualisation and implantation in uterine estrogen receptor-ablated mice (Curtis Hewitt et al., 2002; Pawar et al., 2015) and ovariectomized mice (Chen et al., 2000). However, in the Kiss1 knockout mouse, although LIF partially rescued implantation failure, E2 failed to do so (Calder et al., 2014), suggesting that Kiss1 may either lie downstream of E2 or cooperate with E2 in triggering Lif expression. The former is more likely, as the expression of Kiss1 mRNA was up-regulated after E2 treatment in the mouse endometrium (Zhang et al., 2014). Together, these studies indicated that the E2-kisspeptin-LIF pathway regulates the process of implantation. Interestingly, clinical studies indicated that deficiency of plasma kisspeptin on the hCG day in females with unexplained infertility was associated with a reduced likelihood of implantation after ICSI (Jamil et al., 2017; Mumtaz et al., 2017). However, it is obvious that plasma kisspeptin is not derived from placenta and that plasma kisspeptin levels on the hCG day barely reflect levels at the maternal-fetal interface. Nevertheless, it is most likely that the locally produced kisspeptin affects the level of LIF in the endometrium and the subsequent implantation rate (Babwah, 2015).
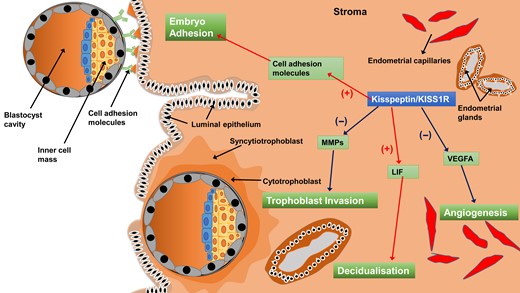
A schematic diagram showing how the kisspeptin/KISS1R system regulates the processes of embryo implantation and placentation. Kisspeptin promotes embryo adhesion to the endometrium and stroma decidualization by up-regulating cell adhesion molecules and LIF, respectively. Kisspeptin inhibits trophoblast invasion and angiogenesis via the down-regulation of MMPs and VEGFA, respectively. VEGFA, vascular endothelial growth factor A; MMPs, matrix metalloproteinases; LIF, leukemia inhibitory factor.
Kisspeptins and trophoblast invasion
The process of hemochorial placentation bears a striking resemblance to cancer invasion. However, placental development is well-organized and physiological, while cancer invasion is chaotic and pathological. There are several similarities in their respective capacities, including increased cellular proliferation, reduced cell death, increased blood supply and invasion of local tissues (summarized in Macklin et al., 2017). It was first reported in 2002 that the expression levels of KISS1 and KISS1R detected by quantitative polymerase chain reaction analysis of laser capture microdissection specimens are largely increased in human placenta (Janneau et al., 2002). Additionally, higher KISS1R expression levels were found in the early placenta, where the invasive capacity of trophoblasts needs to be tightly controlled compared to the term placenta (Janneau et al., 2002). This was the first evidence that suggested the potential role of the kisspeptin/KISS1R system in the invasive and migratory properties of trophoblast cells. One year later, a study demonstrated the localization of KISS1 mRNA and kisspeptin peptide immunoreactivity in the placental syncytiotrophoblasts of humans, suggesting that the dramatic increase in plasma kisspeptin levels during pregnancy was derived from placenta (Horikoshi et al., 2003). Intriguingly, among the four kisspeptins, only kisspeptin-10 resulted in an increase of intracellular Ca2+ in trophoblast cells of the early placenta and subsequently inhibited trophoblast migration but not proliferation, indicating that kisspeptin-10 is the physiological activator of KISS1R in human placenta (Bilban et al., 2004). The same study also demonstrated that kisspeptin-10 triggered a decrease in collagenase activity and reduced the activity of MMP-2 in a dose-dependent manner, which resulted in the attenuation of trophoblast migration (Bilban et al., 2004). In agreement, a recent study showed that kisspeptin-10 treatment reduced MMP-2 expression in primary trophoblast cells and subsequently reduced the migratory capability of trophoblast cells using a scratch-migration assay (Francis et al., 2014). The same study also demonstrated that kisspeptin-10 inhibited the expression of MMP-1, 3, 7, 9, 10 and 14 as well as VEGF and increased the expression of tissue inhibitors of metalloproteinases 1 and 3 in primary trophoblast cells (Francis et al., 2014). These effects were probably mediated via the ERK1/2 signaling pathway, as kisspeptin-10 treatment significantly induced ERK1/2 phosphorylation, while co-treating the cells with a kisspeptin antagonist almost completely blocked the activation of ERK1/2 (Francis et al., 2014). Interestingly, kisspeptin also exerted an anti-invasive effect on EVTs. A previous study indicated that kisspeptin induces the increased adhesion of human EVTs to type-I collagen in a dose- and time-dependent manner, which is a major component of the human placenta (Taylor et al., 2014). This effect was mediated via the PKC and ERK1/2 pathways, as the inhibition of both PKC and ERK1/2 attenuated the kisspeptin-dependent increase in EVT adhesion (Taylor et al., 2014). In the human placental cell line HTR8/SVneo derived from first trimester EVTs, the expression levels of KISS1 and KISS1R regarding both mRNA and protein levels are significantly lower than those in primary trophoblast cells (Martino et al., 2015). Nevertheless, treatment with kisspeptin-10 led to the phosphorylation of ERK1/2 in the HTR8/SVneo cells (Roseweir et al., 2012), suggesting that the kisspeptin-activated ERK1/2 signaling pathway is conserved in EVTs as well as in cell lines derived from EVTs. Additionally, kisspeptin-10 inhibited the migration of HTR8/SVneo cells by stimulating complex ERK1/2-p90rsk-GSK3 beta-FAK feedback interactions (Roseweir et al., 2012). Intriguingly, the kisspeptin/KISS1R system negatively regulated cell proliferation in bovine placental cotyledon cell lines isolated at the first trimester of pregnancy, which was not observed in human trophoblast cells (Martino et al., 2015). However, in human umbilical vein endothelial cells (HUVEC), kisspeptin-10 treatment induced the concentration-dependent inhibition of cell proliferation and migration but did not affect HUVEC cell viability or apoptosis (Ramaesh et al., 2010).
Taking these data together, we may draw a conclusion that kisspeptin/KISS1R signaling inhibits the migration of both trophoblast cells and EVTs via the activation of ERK1/2 and the suppression of MMP expression and activity. Therefore, kisspeptin/KISS1R signaling inhibits the proliferation of several cell types derived from placenta, but not that of human trophoblast cells.
Genetic mutations of KISS1 and KISS1R
In humans, kisspeptin was initially implicated in the onset of puberty as two independent research groups reported KISS1R gene mutations in patients with idiopathic hypogonadotropic hypogonadism (IHH) (de Roux et al., 2003; Seminara et al., 2003). Subsequently, heterozygous and homozygous KISS1 mutations have also been identified in patients with IHH (Chan et al., 2011; Topaloglu et al., 2012). Interestingly, women with heterozygous KISS1 or KISS1R mutations were able to deliver their babies with homozygous KISS1 or KISS1R mutations (Pallais et al., 2006; Topaloglu et al., 2012) (Table III). These results suggested that carrying heterozygous mutations in KISS1 or KISS1R does not appear to limit subsequent uterine and placental function. Additionally, a case report showed that a woman with homozygous KISS1R mutations had a normal vaginal delivery of a healthy child with a normal birthweight (Pallais et al., 2006). Similarly, Kiss1 (+/−) and Kiss1r (+/−) female mice were able to carry the normal weight of fetus with homozygous KISS1 mutations and there was no significant harmful effect on placental function (Herreboudt et al., 2015). These results indicate that the functional role of the kisspeptin/KISS1R system in the placenta could be compensated by other factors during early pregnancy. However, global ablation of Kiss1 or Kiss1r led to dramatically suppressed uterine growth and almost entirely prevented adenogenesis (Leon et al., 2016) and caused subsequent infertility (Funes et al., 2003; Lapatto et al., 2007), although Kiss1 (+/−) or Kiss1r (+/−) phenotypes were fertile (Seminara et al., 2003; Lapatto et al., 2007; Gaytan et al., 2014; Herreboudt et al., 2015). Even though E2 treatment in Kiss1 (−/−) mice and/or genetic restoration of kisspeptin signaling in GnRH neurons in Kiss1r (−/−) mice fully rescued the uterine growth, functional adenogenesis in the endometrium was only partially restored (Leon et al., 2016). Therefore, while uterine growth is mainly contingent on E2-output via kisspeptin signaling in GnRH neurons, local kisspeptin signaling in the endometrium is essential for normal endometrial adenogenesis and functions. Furthermore, embryos failed to implant in maternal mice with homozygous Kiss1 mutations (Calder et al., 2014). These results seem to support the fundamental role of maternal kisspeptin/Kiss1r system in preparation for embryo implantation, at least in mice.
Summary of the effect of Kiss1/Kiss1r in genetically modified or mutant models (placental specific knockout or overall knockout) on fertility or placental function.
| Knockout model | Species | Fertility or placental function | References |
|---|---|---|---|
| Kiss1+/− or Kiss1−/−; Kiss1r+/− or Kiss1r−/− (fetus) | Mouse | Normal weight of fetus and placenta, normal nutrient transfer function of placenta at E15.5 days | Herreboudt et al. (2015) |
| Kiss1+/− or Kiss1r+/− (maternal) | Mouse | Fertile | Herreboudt et al. (2015) |
| Kiss1−/− (maternal) | Mouse | Fail to implant and infertile | Calder et al. (2014) |
| Kiss1r−/− (maternal) | Mouse | Infertile | Funes et al. (2003) |
| Kiss1r+/− (maternal) | Mouse | Fertile | Seminara et al. (2003) |
| Kiss1r−/− (maternal) | Mouse | Infertile | Seminara et al. (2003) |
| Kiss1r+/− (maternal) | Mouse | Fertile before 48-week-old Infertile at 48-week-old | Gaytan et al. (2014) |
| Kiss1+/− (maternal) | Mouse | Fertile | Chan et al. (2011) |
| Kiss1+/− or Kiss1r+/− (maternal) | Mouse | Fertile | Lapatto et al. (2007) |
| Kiss1−/− or Kiss1r−/− (maternal) | Mouse | Infertile | Lapatto et al. (2007) |
| Kiss1−/− or Kiss1r−/− (maternal) | Mouse | Uterine growth suppressed and adenogenesis nearly stopped | Leon et al. (2016) |
| KISS1R+/− (maternal), KISS1R−/− (maternal) and KISS1R−/− (fetus) | Human | Fertile | Pallais et al. (2006) |
| KISS1+/− (maternal) and KISS1−/− (fetus) | Human | Fertile | Topaloglu et al. (2012) |
| Knockout model | Species | Fertility or placental function | References |
|---|---|---|---|
| Kiss1+/− or Kiss1−/−; Kiss1r+/− or Kiss1r−/− (fetus) | Mouse | Normal weight of fetus and placenta, normal nutrient transfer function of placenta at E15.5 days | Herreboudt et al. (2015) |
| Kiss1+/− or Kiss1r+/− (maternal) | Mouse | Fertile | Herreboudt et al. (2015) |
| Kiss1−/− (maternal) | Mouse | Fail to implant and infertile | Calder et al. (2014) |
| Kiss1r−/− (maternal) | Mouse | Infertile | Funes et al. (2003) |
| Kiss1r+/− (maternal) | Mouse | Fertile | Seminara et al. (2003) |
| Kiss1r−/− (maternal) | Mouse | Infertile | Seminara et al. (2003) |
| Kiss1r+/− (maternal) | Mouse | Fertile before 48-week-old Infertile at 48-week-old | Gaytan et al. (2014) |
| Kiss1+/− (maternal) | Mouse | Fertile | Chan et al. (2011) |
| Kiss1+/− or Kiss1r+/− (maternal) | Mouse | Fertile | Lapatto et al. (2007) |
| Kiss1−/− or Kiss1r−/− (maternal) | Mouse | Infertile | Lapatto et al. (2007) |
| Kiss1−/− or Kiss1r−/− (maternal) | Mouse | Uterine growth suppressed and adenogenesis nearly stopped | Leon et al. (2016) |
| KISS1R+/− (maternal), KISS1R−/− (maternal) and KISS1R−/− (fetus) | Human | Fertile | Pallais et al. (2006) |
| KISS1+/− (maternal) and KISS1−/− (fetus) | Human | Fertile | Topaloglu et al. (2012) |
Summary of the effect of Kiss1/Kiss1r in genetically modified or mutant models (placental specific knockout or overall knockout) on fertility or placental function.
| Knockout model | Species | Fertility or placental function | References |
|---|---|---|---|
| Kiss1+/− or Kiss1−/−; Kiss1r+/− or Kiss1r−/− (fetus) | Mouse | Normal weight of fetus and placenta, normal nutrient transfer function of placenta at E15.5 days | Herreboudt et al. (2015) |
| Kiss1+/− or Kiss1r+/− (maternal) | Mouse | Fertile | Herreboudt et al. (2015) |
| Kiss1−/− (maternal) | Mouse | Fail to implant and infertile | Calder et al. (2014) |
| Kiss1r−/− (maternal) | Mouse | Infertile | Funes et al. (2003) |
| Kiss1r+/− (maternal) | Mouse | Fertile | Seminara et al. (2003) |
| Kiss1r−/− (maternal) | Mouse | Infertile | Seminara et al. (2003) |
| Kiss1r+/− (maternal) | Mouse | Fertile before 48-week-old Infertile at 48-week-old | Gaytan et al. (2014) |
| Kiss1+/− (maternal) | Mouse | Fertile | Chan et al. (2011) |
| Kiss1+/− or Kiss1r+/− (maternal) | Mouse | Fertile | Lapatto et al. (2007) |
| Kiss1−/− or Kiss1r−/− (maternal) | Mouse | Infertile | Lapatto et al. (2007) |
| Kiss1−/− or Kiss1r−/− (maternal) | Mouse | Uterine growth suppressed and adenogenesis nearly stopped | Leon et al. (2016) |
| KISS1R+/− (maternal), KISS1R−/− (maternal) and KISS1R−/− (fetus) | Human | Fertile | Pallais et al. (2006) |
| KISS1+/− (maternal) and KISS1−/− (fetus) | Human | Fertile | Topaloglu et al. (2012) |
| Knockout model | Species | Fertility or placental function | References |
|---|---|---|---|
| Kiss1+/− or Kiss1−/−; Kiss1r+/− or Kiss1r−/− (fetus) | Mouse | Normal weight of fetus and placenta, normal nutrient transfer function of placenta at E15.5 days | Herreboudt et al. (2015) |
| Kiss1+/− or Kiss1r+/− (maternal) | Mouse | Fertile | Herreboudt et al. (2015) |
| Kiss1−/− (maternal) | Mouse | Fail to implant and infertile | Calder et al. (2014) |
| Kiss1r−/− (maternal) | Mouse | Infertile | Funes et al. (2003) |
| Kiss1r+/− (maternal) | Mouse | Fertile | Seminara et al. (2003) |
| Kiss1r−/− (maternal) | Mouse | Infertile | Seminara et al. (2003) |
| Kiss1r+/− (maternal) | Mouse | Fertile before 48-week-old Infertile at 48-week-old | Gaytan et al. (2014) |
| Kiss1+/− (maternal) | Mouse | Fertile | Chan et al. (2011) |
| Kiss1+/− or Kiss1r+/− (maternal) | Mouse | Fertile | Lapatto et al. (2007) |
| Kiss1−/− or Kiss1r−/− (maternal) | Mouse | Infertile | Lapatto et al. (2007) |
| Kiss1−/− or Kiss1r−/− (maternal) | Mouse | Uterine growth suppressed and adenogenesis nearly stopped | Leon et al. (2016) |
| KISS1R+/− (maternal), KISS1R−/− (maternal) and KISS1R−/− (fetus) | Human | Fertile | Pallais et al. (2006) |
| KISS1+/− (maternal) and KISS1−/− (fetus) | Human | Fertile | Topaloglu et al. (2012) |
Kisspeptins and early pregnancy viability
Since kisspeptins are mainly derived from placenta, the levels of plasma or serum kisspeptin can probably act as a marker for the function of the placenta and therefore pregnancy viability. Miscarriage is the most common early pregnancy complication as it affects 20% of recognized pregnancies, with a potentially devastating impact on pregnant women (Savitz et al., 2002; National Collaborating Centre for and Children’s, 2012) (for reviews, see Pillai et al., 2016). As miscarriages (except threatened miscarriage) are sometimes irreversible, prevention is probably the only way to intervene in this problem. One possible approach is to develop biochemical markers that have high accuracy to predict or diagnose the occurrence of abortion. Accumulating evidence has suggested that plasma or serum kisspeptin (kisspeptin-10 or kisspeptin-54) levels show excellent accuracy to discriminate confirmed miscarriage from intrauterine pregnancy at the time of diagnosis (Kavvasoglu et al., 2012; Jayasena et al., 2014; Savaris, 2018; Sullivan-Pyke et al., 2018). To discriminate miscarriage from viable intrauterine pregnancy, one study detected kisspeptin-54 in both serum and plasma samples (Sullivan-Pyke et al., 2018), while another study detected kisspeptin-54, kisspeptin-14 and kisspeptin-10 in plasma samples (Jayasena et al., 2014). Nevertheless, both studies performed adequate statistical analysis to control for possible confounders, including gestational and maternal age. Both studies suggested that kisspeptin levels were significantly lower in the miscarriage group than in the intrauterine pregnancy group. Compared with the serum β-hCG levels, the plasma kisspeptin levels had a higher diagnostic performance for miscarriage. Furthermore, combined kisspeptin and β-hCG measurements had similar diagnostic accuracy compared with kisspeptin measurement alone (Jayasena et al., 2014). It is interesting to determine whether there is a relationship between serum or plasma kisspeptin and β-hCG levels depending on the pregnancy state. Although plasma kisspeptin levels were positively associated with several placenta-derived hormones (including progesterone, estradiol and human prolactin), plasma kisspeptin levels and β-hCG levels were not significantly associated (Jayasena et al., 2014; Horikoshi et al., 2003). Consonantly, a most recent linear regression analysis performed using clinical samples showed that serum levels of kisspeptin and β-hCG were positively associated in women with miscarriages (at estimated gestational age 6–10 weeks), whereas there was no significant association between serum levels of kisspeptin and β-hCG in women with viable pregnancies (Sullivan-Pyke et al., 2018). These data strongly suggest that kisspeptin levels can be considered as a new potential marker for early pregnancy viability and may have clinical utility in developing an accurate test of early pregnancy outcome in the future. A prospective cohort study is necessary to confirm the applicability of the serum or plasma kisspeptin assay for predicting or diagnosing miscarriage. Furthermore, it is also necessary to compare the kisspeptin assay with other potential biomarkers, including β-hCG (Jayasena et al., 2014), or their combination in predicting early pregnancy outcome. It should also be noted that the expression of KISS1 in the placenta or plasma kisspeptin levels may be affected by the adverse uterine environment that could lead to abortion (Xu et al., 2015; Sullivan-Pyke et al., 2018). Therefore, it needs to be further determined whether plasma kisspeptin levels could be considered as a marker to evaluate early pregnancy outcome in abnormal conditions.
Kisspeptins and gestational trophoblastic neoplasia
The highest KISS1 expression levels in the placenta were found during the first trimester in humans (Bilban et al., 2004; Cartwright and Williams, 2012) and during embryo Day 12.5 in rodents (Terao et al., 2004; Babwah, 2015). The expression pattern of KISS1 coincides with the time of peak trophoblast invasion when the regulation of this process is of critical importance (Babwah, 2015). In humans, KISS1 is mainly expressed in the villous trophoblast and has an inhibitory role in trophoblast migration through a paracrine/autocrine mechanism within the placental tissue (Bilban et al., 2004; Roseweir et al., 2012; Taylor et al., 2014). Therefore, the change in the local expression of KISS1 in the placenta may be involved in some invasion-dysregulated diseases.
Gestational trophoblastic neoplasia (GTN) is a spectrum of both benign and malignant gestational tumors, including complete mole, partial hydatidiform mole, invasive mole, choriocarcinoma, placental site trophoblastic tumor and epithelioid trophoblastic tumor (Lurain, 2011; Biscaro et al., 2015). Plasma β-hCG levels have been widely used to predict the prognosis of malignant GTN before, during or after chemotherapy (Yang et al., 2006, 2008). Previous studies showed that the expression levels of KISS1 and KISS1R were significantly increased in molar pregnancies (a benign GTN), but the expression levels of KISS1 and KISS1R were decreased in choriocarcinoma cells (malignant GTN cells) (Janneau et al., 2002). These results strongly indicate that down-regulation of the placental expression of KISS1 is involved in the malignant invasion. Intriguingly, plasma kisspeptin levels (including kisspeptin-54, kisspeptin-14 and kisspeptin-10) in patients with malignant GTN were elevated (Dhillo et al., 2006). This study supported the notion that the expression levels of KISS1 in the placenta may not necessarily reflect the plasma levels of kisspeptin, and the increase in serum kisspeptin levels could be explained by the dramatic increase of malignant trophoblast cells. Additionally, plasma kisspeptin levels in GTN patients fell during and after treatment with chemotherapy (Dhillo et al., 2006), indicating that plasma kisspeptin levels could be used to predict the prognosis of malignant GTN during or after chemotherapy. Interestingly, the plasma kisspeptin levels strongly and positively correlated with plasma β-hCG levels (r2 = 0.99, P < 0.0001) in patients with malignant GTN before, during or after chemotherapy (Dhillo et al., 2006), suggesting that kisspeptin and β-hCG in the blood were derived from the same tissue and showed similar secretion pulses in cases of malignant GTN. It would be of great interest for future studies to compare plasma or serum kisspeptin levels with serum β-hCG levels regarding their sensitivity and specificity of predicting the prognosis of malignant GTN (before, during or after chemotherapy).
Unanswered questions and future perspectives
Although the anti-invasive effect of kisspeptin and kisspeptin levels during early pregnancy have been thoroughly discussed in this review, many formidable questions remain to be addressed. The expression of KISS1 or KISS1R was increased in some tumors of advanced stage (Ikeguchi et al. 2003; Schmid et al., 2007; Kostakis et al., 2015, 2018; Savvidis et al., 2015). It is likely that the kisspeptin/KISS1R system is increased to prevent further tumor invasiveness and the formation of local or distant metastasis. In support of this hypothesis, the expression of KISS1 or KISS1R in placental tissues was higher in early pregnancy when the invasion was higher than that in term pregnancy (Janneau et al., 2002; Bilban et al., 2004; Torricelli et al., 2008; Cartwright and Williams, 2012) (Table I). Nevertheless, until now, no direct evidence has confirmed this hypothesis. Future studies are required to investigate whether overexpression of the kisspeptin/ KISS1R system can attenuate the metastasis and/or invasiveness of tumor cells as well as trophoblast cells.
The concentrations of kisspeptin detected in serum samples are significantly lower than those detected in plasma (Ramachandran et al., 2008). Additionally, the collection tube type, processing time and storage condition of samples can also affect the kisspeptin concentrations examined. Therefore, a standardized sample collection method for the measurement of serum or plasma kisspeptin levels should be established if kisspeptin levels are to be used as markers for pregnancy or for other use. It remains possible that the plasma kisspeptin (kisspeptin-10 or kisspeptin-54) measured in pregnant patients is not, in fact, kisspeptin but several cross-reacting peptides, such as RFRP1, RFRP2, RFRP3, neuropeptide FF and neuropeptide AF. Furthermore, since all kisspeptins, including kisspeptin-54, kisspeptin-14, kisspeptin-13 and kisspeptin-10, have high-affinity binding and activation of KISS1R (Kotani et al., 2001; Ohtaki et al., 2001), it is better to detect all these peptides rather than one single peptide. A measurement method of great sensitivity and specificity has been shown to detect kisspeptin levels, including kisspeptin-54, kisspeptin-14 and kisspeptin-10 (Dhillo et al., 2005; Jayasena et al., 2014). In fact, kisspeptin-13 shares the C-terminal decapeptide motif with other isoforms of kisspeptin, which is most likely to have cross-reaction with kisspeptin-10. Future studies performed using mass spectrometry analyses or other proteomics to differentiate different kisspeptin isoforms will be of great interest. At present, the ratio of these kisspeptins in the blood is still unknown.
Although plasma or serum kisspeptin levels are significantly increased in early pregnancy compared to non-pregnant controls (Horikoshi et al., 2003; Jayasena et al., 2014; Sullivan-Pyke et al., 2018), the time at which plasma or serum kisspeptin levels start to increase has not been investigated. It would be of great interest to compare the sensitivity and specificity of the diagnosis of early pregnancy between plasma or serum kisspeptin levels and serum β-hCG levels. It has been suggested that the urine kisspeptin:creatinine ratios are higher in pregnant women compared to non-pregnant women in the third trimester (Jayasena et al., 2015), indicating that kisspeptin could be released in the urine and be measured as a potential marker. Whether the urine kisspeptin:creatinine ratio is up-regulated in the early pregnancy has not been investigated. If it is true, could this ratio or kisspeptin levels be more specific and sensitive than the urine β-hCG levels to detect early pregnancy? Most likely, urine kisspeptin levels or urine kisspeptin:creatinine ratio may be used as a new screening method to detect early pregnancy.
The incompatible change in plasma or serum kisspeptin levels and the expression of KISS1 in placental tissue during pregnancy in humans are still unexplained based on previous studies. It seems that the expression of KISS1 in the placenta may not necessarily reflect the plasma levels of kisspeptin, at least in humans. Most likely, the placenta-derived kisspeptins are not immediately secreted in the bloodstream after they are produced and modified within the cells in the placenta. It is also likely that the kisspeptins in the blood are derived from other organs or tissues during late pregnancy. More studies are needed to elucidate this interesting phenomenon in humans or other animals, if this phenomenon does exist.
With regard to miscarriage and GTN, a prospective cohort study is necessary to explore the sensitivity and specificity of plasma kisspeptin levels in predicting these diseases or newborn health. A large-scale study to define the normal range of plasma and serum kisspeptin levels during early pregnancy will be greatly helpful. Furthermore, it is also necessary to compare the kisspeptin assay with other potential biomarkers, such as β-hCG, or their combination in predicting early pregnancy outcome. Additional studies should investigate the kisspeptin levels in other placenta-related diseases, such as placenta previa and placenta accreta/increta/percreta. Future studies focused on the kisspeptin levels in women with assisted reproduction will also be of great importance. To the best of our knowledge, the applicability of the serum or plasma kisspeptin assay in other pregnancy outcomes, such as multiple pregnancy and ectopic pregnancy, has not been investigated.
Pre-eclampsia, a pregnancy-induced complication characterized by the de-novo development of concurrent hypertension and proteinuria, remains the second leading cause of maternal death (Steegers et al., 2010). Recent studies implicated a possible role of kisspeptin in predicting and diagnosing pre-eclampsia (Adali et al., 2012; Cetkovic et al., 2012; Matjila et al., 2016; Ziyaraa et al., 2016). Plasma kisspeptin-10 levels were negatively correlated with the severity of disease (Adali et al., 2012), but the potential mechanism for kisspeptin signaling in this disease has not been fully investigated. The defect of placental angiogenesis has been thought to be involved in pre-eclampsia (Liu et al., 2015; Stevens et al., 2015) and related to the severity of the disease as well as perinatal outcome (Stevens et al., 2013, 2012). It is likely that the inhibition of new vessel sprouting from placental vessels by kisspeptin is involved in the pathogenesis of pre-eclampsia (Ramaesh et al., 2010; Francis et al., 2014). Indeed, a clinical study showed that plasma kisspeptin-10 levels were lower in women with pre-eclampsia and inversely correlated with the severity of pre-eclampsia (Ziyaraa et al., 2016). The same study also showed that the maternal plasma kisspeptin-10 levels were correlated with the estimated fetal weight in the uterus during the second and the third trimesters (Ziyaraa et al., 2016). Furthermore, kisspeptin levels (16 weeks) in maternal plasma were positively correlated with the birthweight of fetuses in uncomplicated pregnancies (Smets et al., 2008; Logie et al., 2012). These studies strongly suggested that plasma kisspeptin levels were an emerging biomarker for impaired utero-placental perfusion and intrauterine growth restriction.
Conclusion
We conducted literature reviews regarding the coordinated spatial and temporal expression patterns of kisspeptins and their functional receptors during embryo implantation and early human gestation. We also provided an overview of the available evidence, indicating a potential role of kisspeptin as an emerging marker in early pregnancy and a diagnostic or therapeutic target in pregnancy-related diseases. Admittedly, the conclusive demonstration of plasma, serum or urine kisspeptin levels as potential markers for early pregnancy-related diseases is still pending. In addition, the diagnostic utility and therapeutic target of kisspeptin in early pregnancy or placenta-related diseases have not been fully investigated. Thus, there are many unsolved problems that exist and are far from elucidated.
Authors’ roles
K.L.H. reviewed the literature, wrote the manuscript and designed the figures and tables. H.M.C. wrote the manuscript and revised the draft. H.C.Z. reviewed the literature. Y.Y. and R.L. conceived the idea and provided input into manuscript content and composition. J.Q. provided the key idea for this manuscript.
Funding
This work was supported, in part, by the National Key Research & Development Program of China (2016YFC1000201 to R.L, 2016YFC1000601 to Y.Y) and the National Natural Science Funds for general program (31501201 to H.C.Z, 81471427 to R.L, 81771650 to R.L, 81571400 to Y.Y, 81771580 to Y.Y).
Conflict of interest
All authors declare no conflict of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}