





Abstract
Uterine leiomyomas (fibroids) are highly prevalent benign smooth muscle tumors of the uterus. In the USA, the lifetime risk for women developing uterine leiomyomas is estimated as up to 75%. Except for hysterectomy, most therapies or treatments often provide only partial or temporary relief and are not successful in every patient. There is a clear racial disparity in the disease; African-American women are estimated to be three times more likely to develop uterine leiomyomas and generally develop more severe symptoms. There is also familial clustering between first-degree relatives and twins, and multiple inherited syndromes in which fibroid development occurs. Leiomyomas have been described as clonal and hormonally regulated, but despite the healthcare burden imposed by the disease, the etiology of uterine leiomyomas remains largely unknown. The mechanisms involved in their growth are also essentially unknown, which has contributed to the slow progress in development of effective treatment options.
A comprehensive PubMed search for and critical assessment of articles related to the epidemiological, biological and genetic clues for uterine leiomyoma development was performed. The individual functions of some of the best candidate genes are explained to provide more insight into their biological function and to interconnect and organize genes and pathways in one overarching figure that represents the current state of knowledge about uterine leiomyoma development and growth.
In this review, the widely recognized roles of estrogen and progesterone in uterine leiomyoma pathobiology on the basis of clinical and experimental data are presented. This is followed by fundamental aspects and concepts including the possible cellular origin of uterine fibroids. The central themes in the subsequent parts are cytogenetic aberrations in leiomyomas and the racial/ethnic disparities in uterine fibroid biology. Then, the attributes of various in vitro and in vivo, human syndrome, rodent xenograft, naturally mutant, and genetically modified models used to study possible molecular mechanisms of leiomyoma development and growth are described. Particular emphasis is placed on known links to fibrosis, hypertrophy, and hyperplasia and genes that are potentially important in these processes.
Menstrual cycle-related injury and repair and coinciding hormonal cycling appears to affect myometrial stem cells that, at a certain stage of fibroid development, often obtain cytogenetic aberrations and mutations of Mediator complex subunit 12 (MED12). Mammalian target of rapamycin (mTOR), a master regulator of proliferation, is activated in many of these tumors, possibly by mechanisms that are similar to some human fibrosis syndromes and/or by mutation of upstream tumor suppressor genes. Animal models of the disease support some of these dysregulated pathways in fibroid etiology or pathogenesis, but none are definitive. All of this suggests that there are likely several key mechanisms involved in the disease that, in addition to increasing the complexity of uterine fibroid pathobiology, offer possible approaches for patient-specific therapies. A final model that incorporates many of these reported mechanisms is presented with a discussion of their implications for leiomyoma clinical practice.
Introduction
In eutherian mammals, the uterus supports the development of the embryo and fetus to term and is essential for propagation of the species. The uterus is derived from the embryonic Müllerian ducts and is classically divided into the inner endometrium and the outer myometrium (Teixeira et al., 2008; Spencer et al., 2005). The inner endometrium is composed of luminal epithelium, glandular epithelium and endometrial stroma. The myometrium consists mostly of smooth muscle cells, which are organized for maximum efficiency of contractions during labor and supported by an extensive network of large arteries, veins and lymphatic vessels. Uterine leiomyomas (or uterine fibroids) are benign smooth muscle tumors that develop in the myometrium. These tumors have an estimated incidence of up to 75% in reproductive age women and are usually composed of areas of disordered fascicles of smooth muscle characterized by an excess of acellular extracellular matrix (ECM; Cramer and Patel, 1990; Borgfeldt and Andolf, 2000; Baird et al., 2003).
The most common symptoms for these sex steroid hormone-responsive tumors include: heavy and prolonged menstrual bleeding, anemia (secondary to excessive menstrual blood loss), pelvic pain and pressure, dysmenorrhea (painful menses), urinary incontinence, constipation, lower back pain, sexual dysfunction, infertility, recurrent pregnancy loss and compression of adjacent pelvic organs by large uterine fibroids. Contemporary medical treatment options are gonadotrophin-releasing hormone (GnRH) agonists, progestins, oral contraceptives, levonorgestrel intrauterine devices, selective progesterone receptor modulators and procedural interventions such as myomectomy (uterine preservation with removal of uterine fibroids), uterine artery embolization and MR-guided focused ultrasound treatments. All of these have variable side effect profiles and may not consistently relieve symptoms or prevent disease recurrence (Gorny et al., 2014). Owing to the significant degree of fibroid-associated morbidities and to the often-ineffective, above-mentioned therapies, hysterectomy may be the only option for many women. Fibroids are the most common indication for gynecologic surgery in the USA and accounts for ∼600 000 hysterectomies and 60 000 myomectomies annually (Farquhar and Steiner, 2002; Wu et al., 2007). The total costs (including lost productivity and associated morbidities) related to surgical and non-surgical leiomyoma treatment are estimated to be between 6 and 35 billion US dollars annually (Cardozo et al., 2012).
These benign tumors occur throughout the uterus but are classified by their location in the uterus. Intramural fibroids are located in the myometrial wall; submucosal fibroids are located at the endometrium–myometrium interface; and subserosal fibroids are located under the serosa of the uterus. Both submucosal and subserosal fibroids can be pedunculated and extend into the uterine or abdominal cavity, respectively. Intramural, submucosal and subserosal fibroids can and often exist within the same uterus. The growth rate of multiple tumors in one uterus is not associated with their location or size and concurrent progression and regression of different fibroids may occur under the same hormonal conditions (Peddada et al., 2008). With magnetic resonance imaging or high-resolution ultrasound, it is possible to identify two different myometrial layers: an inner junctional zone adjacent to the endometrium and an outer myometrial layer (Brosens et al., 1998b; Tetlow et al., 1999). On a histological level there is a gradual decrease in both cellular density and the cytoplasmic-nuclear ratio starting from the junctional zone to the outermost layers of the outer myometrium. The distinction between both myometrial layers can only be made in reproductive age women and thickness differs during the menstrual cycle, indicating a different response to female sex hormones. The intrinsic differences between the junctional zone and outer myometrium, including cellular density, estrogen and progesterone receptor expression and vascularization could coincide with a partially different pathogenesis (Marugo et al., 1989; Tetlow et al., 1999). In clinical practice, submucosal and intramural fibroids could also be considered as two different manifestations of the same disease because they partially differ in symptoms and surgical treatment: submucosal fibroids generally show a stronger response to GnRHa treatment and can often be surgically removed in contrast to intramural fibroids (Brosens et al., 2003). While this interesting concept of different etiologies of uterine fibroids based on their location is still under debate because of the lack of highly distinctive and unique features of different tumors, submucosal uterine fibroids do typically contain fewer chromosomal abnormalities compared with intramural or subserosal located tumors (Brosens et al., 1998a).
The heterogeneity of fibroid location and growth characteristics in the same patient illustrates the complex biological mechanisms involved in their development and growth. Several predisposing factors, such as hormonal status and exposure, age, parity, ethnicity and family history, are among the most widely recognized (Flake et al., 2003; Walker and Stewart, 2005). There are also many minor histological subtypes of the disease (Wilkinson and Rollason, 2001) that could provide important clues for their specific etiology, but we will not be addressing these specifically here, primarily because these are not normally identified in uterine fibroid studies, making their ability to inform the discussion difficult.
Despite the large-scale medical and financial burden posed by fibroids, the functional roles of the various factors and genes implicated in their etiology and growth remain unclear. To this end, an expanded understanding of the molecular mediators implicated in the pathobiology of uterine fibroids may facilitate the future development of targeted therapeutic options. Several studies have compared the gene expression profile of normal myometrium with leiomyoma tissue from the same patient to assess altered pathways that could be driving leiomyoma development or progression. Multiple groups have published microarray, and more recently, exome sequencing and microRNA studies. We further address some of the exome sequencing results in this review and refer to other reviews for information about microarray results and microRNA in uterine fibroid biology (Arslan et al., 2005; Karmon et al., 2014). Herein, we focus on several genes that have been proposed to play a role in the disease because of their association with fibrosis or hyperplasia and hypertrophy. Some of these genes are responsible for human syndromes associated with fibroid development; others are differentially expressed in uterine leiomyomas, or associated with in vivo or in vitro models that are phenotypically similar to human leiomyomas. Still others are associated with cytogenetic aberrations or are differentially expressed among different ethnic groups. This review describes the known functions of some of these genes, with an emphasis on those with known roles in fibrosis and hyperplasia, and discusses their theoretical and experimental relevance to leiomyoma biology. We briefly describe what is currently known about hormonal regulation and the possible cellular source of fibroids. We end by introducing a molecular model that encompasses the genes and factors implicated in the development and progression of leiomyomas.
Methods
An outline for this review was sketched by making a list of known clinical and epidemiological observations about uterine leiomyoma development and growth. A comprehensive PubMed search of English literature was performed to gather more detailed information about derived individual genes and molecular processes. Every gene was used as a search term in combination with the search term ‘uterine fibroid’. Additionally, genes and molecular processes were identified by the opposite approach, in this case not symptoms, but genes that were part of cytogenetic aberrations and animal models were the starting point for further assessment and discussion in relation to leiomyoma biology and the interaction with other genes. Again, a PubMed search of the English literature with the search term ‘uterine fibroid’ in combination with each gene was performed. We have assessed the relevance of each article in relation to our selection of main subjects based on the title and abstract. Our goal was to be as comprehensive as possible and to discuss the pros and cons of each article in our review.
Clinical observations relevant to fibroid biology
There are numerous clinical observations indicating that development of leiomyomas is related to hormonal status (Ross et al., 1986). For example, leiomyomas do not normally occur in prepubertal girls and are rarely observed in adolescent women (Fields and Neinstein, 1996). The cumulative incidence increases until menopause, after which the fibroid burden of disease decreases in almost all women. On a histological level, fibroids may persist after menopause, although usually in smaller numbers and size (Cramer and Patel, 1990). Complicating interpretations of these observations is the fact that, although the levels of sex steroid hormones are very high during pregnancy, paradoxically fibroids may decrease in size during pregnancy and can become undetectable by post-partum ultrasound examination (Laughlin et al., 2010). It has been surmised that this may be attributed to a combined effect of hormonal status during pregnancy and the reorganization of the uterus after parturition (Cesen-Cummings et al., 2003). Epidemiologic studies suggest that there is a reduced prevalence of leiomyomas in multiparous women and women with a later age of menarche (Marshall et al., 1998; Terry et al., 2010). Potential concerns associated with epidemiologic studies linking leiomyoma development with gravidity are selection bias and other possible confounding factors, but some epidemiologic studies that control for several confounders still demonstrate a significant association between hormonal status and the presence of this disease (Marshall et al., 1998; Cesen-Cummings et al., 2003; Baird, 2004; Terry et al., 2010).
Clinical effects of estrogen and progesterone receptor targeted therapies
Despite ample clinical evidence that hormonal status is associated with leiomyoma development (see the recent comprehensive review articles: Kim et al., 2013; Moravek et al., 2015), the precise functional roles of estrogens and progestins in fibroid biology are still incompletely understood. For example, the proliferation of human uterine smooth muscle-derived cells and uterine leiomyoma-derived cells is stimulated dose dependently by 17β-estradiol, which can be inhibited by either LY294002, a phosphatidylinositol-3 kinase (PI3K) inhibitor or rapamycin, a mammalian target of rapamycin (mTOR, aka FRAP1) inhibitor. These findings suggest that estrogens exert their pro-mitogenic effect by transcriptional mechanisms (Andersen et al., 1995; Pedeutour et al., 1998; Yin et al., 2007). There seems to be no difference in estrogen receptor-α (ERα) expression in GnRH agonist-sensitive and -resistant uterine fibroids (Kim et al., 2014). Proliferation of Eker rat-derived leiomyoma cell lines, which carry a germline mutation in the Tsc2 gene, is also stimulated by 17β-estradiol and inhibited by the selective estrogen receptor modulator (SERM) tamoxifen in vitro (Howe et al., 1995). Additionally, supraphysiologic estrogen levels are also required for proliferation and development of fibroids in guinea pigs and also for multiple mouse xenograft models, further implicating estrogens in fibroid development and growth (Porter et al., 1995; Hassan et al., 2008; Ishikawa et al., 2010; Tsuiji et al., 2010). However, a recent meta-analysis concluded that there is currently no consistent evidence that SERMs either reduce the size of fibroids or have beneficial effects on clinical outcomes (Deng et al., 2012).
There are conflicting experimental and clinical results about a possible stimulatory, inhibitory or facilitating role for progesterone in uterine fibroid development (Kim and Sefton, 2012). Progesterone appears to be essential for the growth of human leiomyoma xenografts, which is both counterintuitive, given the propensity of pregnancy to mitigate leiomyoma development and/or growth, and consistent with improvement in tumor burden observed with antiprogestin therapy (Ishikawa et al., 2010). A number of antiprogestin drugs have been developed and tested in clinical trails for the treatment of leiomyoma, including Mifepristone, Asoprisnil and Ulipristal acetate. Ulipristal acetate can be effective in reducing fibroid size, as well as clinical symptoms, and is United States Food and Drug Administration (FDA) approved for short-term use to reduce fibroid size before surgery (Donnez et al., 2012a, b). One small clinical trial indicated that Asoprisnil could be effective in reducing uterine blood loss and fibroid size, but it is not FDA approved for this use (Chwalisz et al., 2007). There are several published studies suggesting that Mifepristone may act similarly (Murphy et al., 1993; Eisinger et al., 2005, 2009; Fiscella et al., 2006; Carbonell Esteve et al., 2008; Bagaria et al., 2009; Engman et al., 2009; Esteve et al., 2012; Kulshrestha et al., 2013; Yerushalmi et al., 2014). However, a recent meta-analysis of three studies concluded that, although Mifepristone therapy significantly reduces uterine bleeding, it does not impact fibroid size (Tristan et al., 2012).
GnRH agonists are used for preoperative reduction of fibroid size, the temporary induction of amenorrhea and for treatment of endometriosis. However, because of their side effects, mainly osteoporosis, it is only recommended for short-term use (Lethaby et al., 2001). GnRH agonists impact sex hormone production via pituitary down-regulation and desensitization, which reduces the levels of circulating LH and FSH and leads to a hypogonadal state, anovulation and hypoestrogenism, the last of which could contribute to a reduction of tumor size (Maruo et al., 2004). These contradictory results, coupled with the fact that multiple fibroids are clonal and can have different growth characteristics in the same uterus, suggests that local and cell autonomous mechanisms, perhaps with input from their microenvironment, play key roles in leiomyoma development and growth (Peddada et al., 2008; Hanahan and Coussens, 2012).
Clues for a role of genetics in leiomyoma development
There are several clinical and epidemiological observations that suggest that genetic or chromosomal alterations play a significant role in the development of uterine leiomyomas. For example, women with first-degree relatives diagnosed with leiomyomas have a greater risk of developing leiomyomas compared with women without affected relatives (Vikhlyaeva et al., 1995; Sato et al., 2002). Additionally, it has been reported that monozygotic twins have a higher concordance of developing leiomyomas compared with dizygotic twins (Treloar et al., 1992; Luoto et al., 2000).
Furthermore, ethnicity has a major influence on the development and clinical severity of uterine leiomyomas. African-American women are two to three times more likely to develop symptomatic leiomyomas compared with Caucasian women. These studies also demonstrate that African-American women have more severe clinical symptoms at an earlier age, which may be related to the significantly larger tumor burden observed in these patients (Velebil et al., 1995; Kjerulff et al., 1996; Marshall et al., 1997; Baird et al., 2003). Since these ethnic disparities cannot be fully explained by socio-economic or other environmental factors, the search for genetic contributors led to a recent genome wide association study comparing large cohorts of African women with leiomyomas to women from other ethnic backgrounds and controls. However, that study failed to identify any specific loci correlated to a higher incidence of leiomyoma (Wise et al., 2012). Thus, it appears that the increased incidence and severity of the disease in African-American women might be attributable to a combination of specific genetic and environmental factors that are not independent risk factors for disease.
There are several genetic syndromes in which smooth muscle tumor development in the uterus and other organ systems is characteristic of the phenotype (Table I). It is generally accepted that leiomyomas develop in the Alport and Reed's syndromes (Launonen et al., 2001; Hudson et al., 2003). However, it is not so clear whether leiomyomas occur as a symptom of the Proteus or Cowden syndromes (Pilarski and Eng, 2004; Cohen, 2005). The scarcity of patients with these syndromes makes it difficult to perform appropriately powered studies to further investigate this proposed relationship. In either case, the genetics of these leiomyoma-generating syndromes do inform our discussion.
List of human syndromes associated with leiomyoma development.
| Syndrome | OMIM entry number | Gene mutation | Gene ontology |
|---|---|---|---|
| Alport syndrome | 308940 | COL4A5, COL4A6 | Structure of basement membranes |
| Proteus syndrome | 176920 | AKT1 | Metabolism, proliferation, cell survival, growth and angiogenesis (up-regulation of mTOR) |
| Reed's syndrome | 150800 | FH | Tricarboxylic acid cycle (aerobic cellular respiration) |
| Cowden syndrome | 158350 | PTEN | Tumor suppression (up-regulation of AKT1/ PI3K and mTOR) |
| Syndrome | OMIM entry number | Gene mutation | Gene ontology |
|---|---|---|---|
| Alport syndrome | 308940 | COL4A5, COL4A6 | Structure of basement membranes |
| Proteus syndrome | 176920 | AKT1 | Metabolism, proliferation, cell survival, growth and angiogenesis (up-regulation of mTOR) |
| Reed's syndrome | 150800 | FH | Tricarboxylic acid cycle (aerobic cellular respiration) |
| Cowden syndrome | 158350 | PTEN | Tumor suppression (up-regulation of AKT1/ PI3K and mTOR) |
Alport syndrome, Proteus syndrome, Reed′s syndrome and Cowden syndrome listed with the Online Mendelian Inheritance in Man id (OMIM), the corresponding gene mutation and main function.
AKT1, v-akt murine thymoma viral oncogene homolog 1; COL4A5/6, collagen type IV α5 and α6 chain; FH, fumarate hydratase; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol-3 kinase; PTEN, phosphatase and tensin homolog.
List of human syndromes associated with leiomyoma development.
| Syndrome | OMIM entry number | Gene mutation | Gene ontology |
|---|---|---|---|
| Alport syndrome | 308940 | COL4A5, COL4A6 | Structure of basement membranes |
| Proteus syndrome | 176920 | AKT1 | Metabolism, proliferation, cell survival, growth and angiogenesis (up-regulation of mTOR) |
| Reed's syndrome | 150800 | FH | Tricarboxylic acid cycle (aerobic cellular respiration) |
| Cowden syndrome | 158350 | PTEN | Tumor suppression (up-regulation of AKT1/ PI3K and mTOR) |
| Syndrome | OMIM entry number | Gene mutation | Gene ontology |
|---|---|---|---|
| Alport syndrome | 308940 | COL4A5, COL4A6 | Structure of basement membranes |
| Proteus syndrome | 176920 | AKT1 | Metabolism, proliferation, cell survival, growth and angiogenesis (up-regulation of mTOR) |
| Reed's syndrome | 150800 | FH | Tricarboxylic acid cycle (aerobic cellular respiration) |
| Cowden syndrome | 158350 | PTEN | Tumor suppression (up-regulation of AKT1/ PI3K and mTOR) |
Alport syndrome, Proteus syndrome, Reed′s syndrome and Cowden syndrome listed with the Online Mendelian Inheritance in Man id (OMIM), the corresponding gene mutation and main function.
AKT1, v-akt murine thymoma viral oncogene homolog 1; COL4A5/6, collagen type IV α5 and α6 chain; FH, fumarate hydratase; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol-3 kinase; PTEN, phosphatase and tensin homolog.
The characteristic symptoms of the Alport syndrome are progressive renal disease, hearing loss and visual impairment (Hudson et al., 2003). All of these symptoms are a consequence of structural deficiency of the basal membranes due to a lack of one or more specific collagen chains. Most cases of Alport syndrome are caused by mutations of the collagen type IV α5 chain gene (COL4A5) (Cochat et al., 1988; Antignac et al., 1992; Anker et al., 2003; Hudson et al., 2003). A very small minority of the women with Alport syndrome also develop diffuse leiomyomatosis or countless growing fibroids outside of the uterus (Kashtan, 1999). Diffuse leiomyomatosis as part of the Alport syndrome is virtually always associated with a variable deletion of the COL4A5 gene and a breakpoint in the second exon of the COL4A6 gene (Kashtan, 1999). It is unclear what the specific effect is of this breakpoint for diffuse leiomyomatosis or for the development of uterine fibroids.
Biomechanical stress on the structurally impaired glomerular filtration membranes in the kidneys of patients with Alport syndrome leads to accumulating local damage, the onset of local inflammation and, eventually, fibrosis (Noone and Licht, 2013). Multiple factors that are assumed to be important in this process have also been associated with uterine fibroid formation, including transforming growth factor β (TGFβ1) and Peroxisome proliferator-activated receptor gamma (PPARγ) (Tsibris et al., 1999; Catherino et al., 2004). In addition, hypertension is an important risk factor for the progression of Alport syndrome, and there are indications that there is also a positive association between the presence of fibroids and hypertension (Faerstein et al., 2001a; Noone and Licht, 2013).
Proteus syndrome is extremely rare and is characterized by excessive and asymmetrical growth of numerous tissues and organs, including skin, adipose tissue, vascular structures and parts of limbs (Cohen and Hayden, 1979; Cohen, 2014). The underlying cause is a mosaic activating mutation of the v-akt murine thymoma viral oncogene homolog 1 (AKT1) (Lindhurst et al., 2011). Leiomyoma development can also be seen in these patients (Cohen, 2005). Interestingly, AKT1 is an important upstream stimulator of the mTOR pathway, which is proposed to play a central role in the pathogenesis of uterine fibroids (Kovacs et al., 2003; Crabtree et al., 2009).
Reed's syndrome, also named hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome, is an autosomal dominant disease characterized by smooth muscle tumors of the skin and uterus, as well as renal cancer (Reed et al., 1973; Launonen et al., 2001; Tomlinson et al., 2002). Both uterus and kidneys are derived from the embryonic urogenital ridge, suggesting similar genetic mediators of development in this disease. The underlying cause is a germline mutation of the fumarate hydratase (FH) gene, which is involved in the tricarboxylic acid cycle (Tomlinson et al., 2002). The accumulation of fumarate has been linked to the induction of hypoxia inducible factor 1, subunit alpha (HIF-1α) (Pollard et al., 2005; O'Flaherty et al., 2010; Ooi and Furge, 2012; Yang et al., 2013), leading to a pseudohypoxic state, which is another characteristic of uterine leiomyomas (Casey et al., 2000; Mayer et al., 2008). Activation of AMP-activated protein kinase (AMPK; Bardella et al., 2012; Fleming et al., 2014) has been found in kidney tumors of patients with Reed's syndrome (Bardella et al., 2012), which in turn inhibits the mTOR pathway (Shaw, 2009), the activity of which has been associated with ∼50% of leiomyomas (Crabtree et al., 2009). The possible mechanisms by which either accumulation of fumarate or loss of fumarate hydratase activity induces these smooth muscle tumors are unclear (Ricketts et al., 2012).
Cowden syndrome is an autosomal dominant disease characterized by typical mucocutaneous lesions, the growth of multiple hamartomas, and an increased risk for breast, thyroid, endometrial and renal cancers (Pilarski and Eng, 2004). A mutation of the tumor suppressor gene, phosphatase and tensin homolog (PTEN), is the underlying cause of this syndrome (Lloyd and Dennis, 1963; Liaw et al., 1997). The development of leiomyoma is a minor criterion for the diagnosis of this syndrome (Pilarski and Eng, 2004), but this is not definitive since the incidence of uterine fibroids was only 26% in a cohort of 69 women older than 18 years with a confirmed PTEN mutation (Pilarski et al., 2011) and is within the range of what is observed in the general population. Mutations of PIK3CA or AKT1 can be detected in ∼10% of the patients with Cowden syndrome who are not carriers of a germline PTEN mutation (Orloff et al., 2013). The common denominator in these mutations is a possible downstream up-regulation of the mTOR pathway.
Tuberous sclerosis complex (TSC) is caused by mutation of the tumor suppressor genes, TSC1 or TSC2, whose protein products, hamartin and tuberin, respectively, heterodimerize and negatively regulate the activation of mTOR (Curatolo et al., 2008). It shares a number of major diagnostic features with Reed's syndrome, Proteus syndrome and Cowden syndrome including hamartomas and the development of multiple types of benign and malignant tumors (Eng, 1993; Curatolo et al., 2008; Orloff et al., 2013). Also on a molecular level, TSC is very closely related to Proteus and Cowden syndrome. The mutated AKT1 and PTEN proteins in these two syndromes are both upstream of TSC1 and TSC2 (Shaw, 2009; Laplante and Sabatini, 2013). Despite the similarities on the clinical and molecular level, TSC does not coincide with fibroid formation. The growth of hamartomas in the skin, brain and kidneys are the main symptoms of this disease, as is renal cell cancer in some patients (Curatolo et al., 2008). Of note concerning leiomyomas in this disease is that the most widely used animal model for human leiomyoma studies is the Eker rat, which has a germline mutation in the Tsc2 gene (Eker et al., 1981). These rats develop both renal cell cancer and leiomyoma-like lesions; this will be discussed in more detail in the animal models section.
The cellular origin of leiomyomas
Human uterine leiomyomas are clonal in origin, which has been confirmed by analysis of the inactivation status of heterozygous alleles on the X chromosome (Linder and Gartler, 1965; Townsend et al., 1970; Nilbert and Strömbeck, 1992; Mashal et al., 1994; Hashimoto et al., 1995; Baschinsky et al., 2000; Wang et al., 2002; Canevari et al., 2005; Zhang et al., 2006; Cai et al., 2007). This technique exploits the fact that in each cell one of the X chromosomes is randomly inactivated during gastrulation. Thus, the alleles of the androgen receptor or phosphoglycerokinase genes can differ in methylation status or the presence of a single nucleotide polymorphism in a specific enzyme restriction site. Another method that is used to determine clonal origin is the separation of two functional isoenzymes of the X-linked glucose-6-phosphate dehydrogenase gene by gel electrophoresis. Only one of the alleles has been found in almost all of the human leiomyoma samples, suggesting that the whole tumor originates from one cell. Additionally, while the average length of telomere repeats differs in multiple fibroids in hysterectomy samples, within a fibroid they are the same, supporting a monoclonal origin for tumor formation (Rogalla et al., 1995; Bonatz et al., 1998). Another recent study has suggested that the different cell types in fibroid tumors, mainly smooth muscle cells and fibroblasts, are all clonally derived from one parental cell (Holdsworth-Carson et al., 2014). The observation that multiple cell types isolated by flow cytometry from the same fibroids are derived from one parental cell implies that the parental cell has multipotent stem cell properties.
The high plasticity and regenerative capacity of the uterus during menstrual cycles and pregnancy suggests that there are stem cells present in the uterus (Morrione and Seifter, 1962; Teixeira et al., 2008). Multiple groups have focused on this subject and have shown the presence of myometrial stem cells first in a rodent model and later also in human. The first in vivo evidence to support a myometrial stem cell population was observed when conditional deletion of β-catenin in the embryonic Müllerian duct mesenchyme of mice resulted in the progressive replacement of smooth muscle cells of the uterus with adipose tissue (Arango et al., 2005). This strongly suggested the presence of multipotent stem cells, which normally differentiate into smooth muscle cells to maintain smooth muscle cell homeostasis but in the absence of β-catenin lead to adipocyte differentiation.
Specific cell populations with multipotent stem cell properties have been identified in mouse and human myometrium using two distinct techniques to identify possible somatic stem cells. The first utilizes pulse-chase experiments with the nucleotide analog 5-bromo-2′-deoxyuridine (BrdU), which exploits the immortal strand hypothesis and the ability of a slowly dividing stem cell population to preferentially retain label (Cairns, 1975). A detectable level of BrdU is present in 10% of the initial stained nuclei of mouse uterine sections after a chase period of 8–12 weeks (Szotek et al., 2007). These label-retaining cells are usually positive for α-smooth muscle actin (αSMA), ERα and β-catenin by immunohistochemistry and are in close proximity to cells positive for the stem cell markers, c-Kit and Abcg2, but are negative for Sca-1. The cells are also negative for the hematopoietic markers (CD45.1, CD31, pan-NK, Gr-1, Ter-119, Mac-1, CD34, Thy-1/CD90.1, CD44 and Tie-2) and are thus unlikely to be of bone marrow origin suggesting an origin in the uterine mesenchyme. The second method is done by flow cytometry and relies on the ‘side population’ (SP) technique, the propensity of stem cells to be multidrug resistant and efflux Hoechst 33342 dye through the ABC transporter Abcg2/Bcrp1 (Goodell et al., 1996). Approximately 0.2% of the cells isolated from mouse uteri were found in the SP by using this technique, and ∼30% of the SP cells had previously expressed, or still express, the MIS type 2 receptor, which is another way to prove that they are not of hematopoietic origin, but truly derived from the Müllerian duct mesenchyme (Szotek et al., 2007).
In humans, ∼3% of myometrial cells were shown to have stem cell properties as determined by flow cytometry based on the SP criteria (Ono et al., 2007). Virtually all cells in this SP were positive for the hematopoietic and endothelial stem cell marker CD34 and negative for the hematopoietic lineage marker CD45 and endothelial cell markers CD106, VEGF receptor 1 and factor VIII-related antigen. In accordance with their more undifferentiated state, the expression levels of ERα, PR, and the smooth muscle cell markers calponin and smoothelin were relatively low compared with the main population of myometrial cells (Ono et al., 2007). Additionally, 98% of human myometrial SP cells were in G0 as would be expected from a quiescent stem cell population. The SP cells spontaneously expressed smooth muscle cell markers that were absent at the start of culture, and under appropriate conditions were able to differentiate and express osteogenic and adipose-specific markers. In vitro experiments also showed that xenografted SP cells were able to differentiate into αSMA-expressing cells and that they contribute to the growth of the uterus in pregnant mice (Ono et al., 2007). An interesting observation from that study was the ability of the side population of human myometrium to grow only under hypoxic conditions in vitro. The authors of that study speculated that hypoxia during normal menstruation could be a trigger for myometrial stem cell proliferation ultimately leading to uterine fibroid formation (Ono et al., 2007; Ono et al., 2013). Another group has more recently used the SP technique to isolate putative leiomyoma stem cells for a comprehensive analysis of their stem cell properties (Mas et al., 2012). They showed that the leiomyoma SP cells expressing pluripotency markers were able to differentiate in vitro along mesenchymal lineages, and grow into leiomyomas in xenotransplants. Leiomyoma SP cells co-cultured with either myometrial or leiomyoma primary cells appear to respond to mitogenic signaling by estradiol and progesterone via a paracrine mechanism that involves WNT/β-catenin signaling (Ono et al., 2013). A possible role for dysregulated WNT/β-catenin signaling is discussed further in the Animal Models section description of the gain-of-function β-catenin mice. A study from our group has shown that while CD90+ cells sorted from human myometrium can differentiate into CD90+ and CD90− cells, those from leiomyoma only produce CD90+ cells, suggesting that pluripotency in leiomyoma cells is more limited compared with normal myometrium (Chang et al., 2010). Thus, several laboratories have now shown that cells with classical stem or progenitor cell characteristics can be isolated from myometrial and leiomyoma tissues. A model for how these stem/progenitor cells can go awry and lead to fibroid development has been developed by Dr Bulun's laboratory that illustrates the important steps likely to be involved (Fig. 1). Further characterization of these cells and their roles in fibroid biology needs to be done. However, the prospect of targeting these stem/progenitor cells for fibroid therapy or even preventative strategies for recurrence provides a new and compelling approach to be explored.

The stem/progenitor cell dysregulation theory for the etiology and growth of uterine leiomyomas. (A) Normal myometrial stem/progenitor cells respond to ovarian steroid hormones by self-renewal and differentiation of new myometrial smooth muscle cells during normal cycling, pregnancy and post-partum repair. (B) The myometrial stem/progenitor cells become dysregulated, perhaps after a genetic event, and become a fibroid smooth muscle cell and ECM-producing stem/progenitor cell. (C) The fibroid smooth muscle stem/progenitor cell no longer responds to local factors correctly to maintain tissue homeostasis and begins to form tumors (Used with permission (Bulun, 2013)).
MED12 and interacting proteins
A genome wide DNA exome sequencing study demonstrated that 70% of the 225 investigated fibroids from 80 individual patients contained a mutation of the mediator complex subunit 12 (MED12) gene (Makinen et al., 2011). In women, one allele of MED12 is randomly inactivated in each cell because the gene is located on the X chromosome. MED12 is part of the ∼30 subunit mediator complex, which functions as a central scaffold within the preinitiation complex. It relays signals from regulatory transcription factors directly to the RNA polymerase II enzyme and is necessary for expression of both coding and non-coding RNAs (Malik and Roeder, 2010). Approximately 50% of the identified MED12 mutations in the samples were missense mutations of the 44th codon of the second exon, indicating the specific importance of this amino acid for MED12 function in relation to leiomyoma and suggesting these mutations could represent gain-of-function alleles. Analysis of tumor derived MED12 cDNA confirmed the predominant expression of the mutated allele. Despite the preponderance of MED12 mutations in leiomyomas, the determination of whether mutated MED12 is a driver of leiomyoma development or growth awaits definitive proof.
The presence of MED12 mutation is significantly associated with smaller tumor size, suggesting an inhibitory effect on global gene expression due to MED12 dysfunction (Makinen et al., 2011; de Graaff et al., 2013). These results could also suggest different etiologies for tumors of different sizes, with some larger tumors developing by a mechanism that is not associated with MED12 mutation. Alternatively, fibroids might be MED12-deficient when small and growth could involve contribution of outside fibroblast cells, in a decidedly non-dogmatic mechanism of tumor progression. Indeed, leiomyoma cells in culture that originally carried a MED12 mutation tend to not proliferate after more than a few passages (Markowski et al., 2014), suggesting that in primary cell cultures, the MED12 mutant cells become overwhelmed by wild-type fibroblasts.
MED12 mutations are mainly found in fibroids with a normal karyotype (Markowski et al., 2012) and are not associated with patient age or ethnicity (Mäkinen et al., 2011; McGuire et al., 2012). Sequencing of MED12 in a large variety and number of tumors, including ovarian cancer, breast cancer, colon cancer, leiomyosarcoma and smooth muscle tumors of uncertain malignant potential, indicates that MED12 mutations are highly specific for leiomyomas, and only occur in a small proportion of leiomyosarcoma and smooth muscle tumors of uncertain malignant potential (Huang et al., 2012; Kämpjärvi et al., 2012; Pérot et al., 2012; de Graaff et al., 2013; Markowski et al., 2013; Matsubara et al., 2013; Ravegnini et al., 2013; Schwetye et al., 2014). Leiomyosarcomas are malignant, highly aggressive uterine tumors causing a poor survival rate and having a much lower incidence compared with benign leiomyoma of the uterus (Chiang and Oliva, 2013). Historically, leiomyosarcomas are not thought to develop from fibroids; hence, it is an interesting observation that 10–20% of them contain MED12 mutations (Ravegnini et al., 2013; Schwetye et al., 2014).
MED12 has been shown to physically and functionally interact with β-catenin, the key effector of canonical WNT signaling, to activate transcription of target genes (Kim et al., 2006). MED12 is also essential for embryonic development and its deletion disrupts both canonical WNT signaling and WNT/planar cell polarity (Rocha et al., 2010). Mice that express a constitutively active form of β-catenin in the mesenchymal cells of the uterus develop myometrial hyperplasia and leiomyoma-like tumors (Tanwar et al., 2009). These studies suggest that dysfunction of MED12 and dysregulated β-catenin expression could play a central role in some human leiomyoma formation. This hypothesis is supported by recent in vitro studies that demonstrate that normal myometrial cells are able to stimulate nuclear translocation of β-catenin and proliferation of myometrial smooth muscle stem cells upon addition of estrogen and progesterone (Ono et al., 2013).
RE1-silencing transcription factor and prolactin-releasing hormone receptor
MED12 also interacts directly with repressor element 1 (RE1)-silencing transcription factor (REST, also known as neuron restrictive silencer factor, NRSF) (Ding et al., 2008). REST is able to bind to ∼1900 short RE1 sites in the human genome via its eight zinc finger domains (Bruce et al., 2004). In addition to its role in relation to the mediator complex and transcriptional machinery, it also serves as a hub for the recruitment of multiple chromatin-modifying enzymes, including histone H3 lysine 9 methylase and histone deacetylases (Ooi and Wood, 2007). REST has been shown to play critical roles during embryonic development and neurogenesis by inhibition of differentiation. It may also function as a tumor suppressor gene or proto-oncogene, depending on the tissue type (Westbrook et al., 2005, 2008; Weissman, 2008). A study performed in chick embryos showed that REST is a direct target of canonical Wnt signaling during the process of neural stem cell differentiation (Nishihara et al., 2003). Thus, it might be possible that REST is also a direct downstream target of β-catenin during fibroid formation. Another interesting observation derived from neurodevelopment research is that REST is able to modulate the degradation of retinoic acid, which has been proposed to be involved in fibroid formation (Singh et al., 2011).
Global transient ischemia of rat brains by occlusion of the vertebral and common carotid arteries leads to up-regulation of REST at both the mRNA and protein levels in hippocampal neurons (Calderone et al., 2003; Noh et al., 2012). The preferential target region of REST is the RE1 element of glutamate receptor, ionotropic, AMPA 2 (GRIA2), where it recruits corepressors that lead to epigenetic remodeling and gene silencing. In vitro inhibition of REST by antisense RNA significantly inhibits ischemia-induced neuronal cell death, which shows that in neurons epigenetic remodeling by REST is an essential factor in ischemia-induced cell death (Noh et al., 2012). Multiple microarray studies show that GRIA2 is among the most consistently up-regulated genes in human leiomyoma (Arslan et al., 2005), which supports the hypothesis that ischemia is a relevant factor in the formation of fibroids. Varghese et al. demonstrated that a reduction in REST protein could play a significant role in human fibroid formation by transcriptional release of inhibition of the prolactin-releasing hormone receptor (PRLHR, aka GPR10), resulting in up-regulation of the PI3K/AKT/mTOR pathway and its downstream activities (Fig. 2; Varghese et al., 2013). GPR10 is highly expressed in more than 90% of human leiomyomas at both the mRNA and protein level, while it is present at a very low level or completely absent in normal myometrium (Varghese et al., 2013). The expression level of GPR10 has been positively correlated with the amount of phosphorylated AKT in leiomyoma cell culture. Activation of GPR10 by its ligand, prolactin-releasing peptide, in cultures of human leiomyoma cells leads to an increase in p-AKT, p-mTOR, p-p70S6 kinase and p-4EBP1, which are all indicative of up-regulation of the PI3K/AKT/mTOR pathway and subsequent activation of the cell cycle, cell growth and survival pathways (Figs 2 and 3). Prolactin-releasing peptide also has a dose-dependent stimulatory effect on the cell cycle, which is reflected by an increase of BrdU incorporation. Normal myometrial cells in culture do not up-regulate the mTOR pathway constituents in response to prolactin-releasing peptide as expected from their negligible expression of the receptor. The preferential effect of GPR10 stimulation on leiomyoma cell makes it a compelling potential pharmacological target for future treatment of human leiomyomas (Varghese et al., 2013). Mice that overexpress GPR10 in the myometrium show a phenotype comparable to human leiomyoma. They show a significant increase in the transverse cross-section area of the myometrium, with a clear but modestly increased mitotic index and an overexpression of a number of ECM components (Varghese et al., 2013).
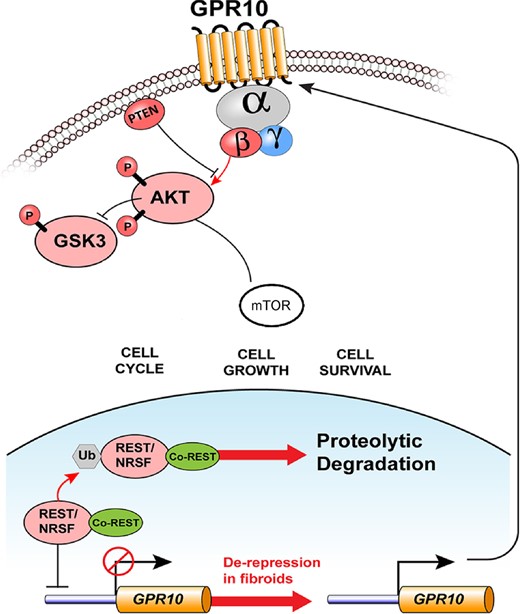
Loss of REST in leiomyomas leads to overexpression of GPR10. In the myometrium, expression of the transcriptional repressor and tumor suppressor, REST, normally represses expression of GPR10. In uterine leiomyomas, REST protein is down-regulated, which leads to GPR10 expression becoming de-repressed and overexpression at the cell surface. Increased GPR10 activity leads to downstream signaling via PI3K/AKT/mTOR and other mechanisms of cellular proliferation, growth and survival (Used with permission (Varghese et al., 2013)).
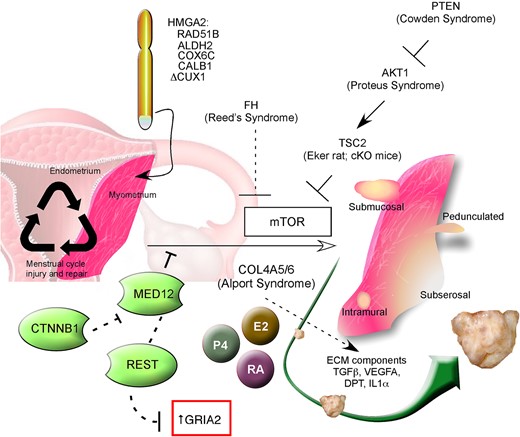
Schematic composite representation of factors thought to be involved in leiomyoma development and growth. Menstrual cycle-related injury and repair, and coinciding hormonal cycling, affects myometrial stem cells that at a certain stage of fibroid development can obtain cytogenetic aberrations mostly involving HMGA2 and RAD51B. MED12 mutations are highly prevalent and specific for uterine fibroids; this transcription factor interacts with mTOR, which itself appears to be a central component of the pathogenesis of fibroids. Possible control of MED12 activity by REST and CTNNB1 are indicated. In fibroids, local dysregulation of extra cellular matrix composition and interaction occurs in which dermatopontin and TGF-β are essential actors. Estrogens, progestins and retinoic acid appear to be involved in both the etiology of the disease as well as progression. The fibrosis syndromes (Reed's, Cowden, Proteus) which affect mTOR activity are shown, along with Alport Syndrome and the TSC2 rodent models. CUX1, cut-like homeobox 1; AKT1, v-akt murine thymoma viral oncogene homolog 1; ALDH2, aldehyde dehydrogenase 2; CALB1, calbindin 1; COL4A5/6, collagen type IV α5 and α6 chain; COX6C, cytochrome C oxidase subunit VIc; CTNNB1, catenin (cadherin-associated protein), beta 1, 88 kDa; DPT, dermatopontin; E2, estradiol, ECM, extracellular matrix; FH, fumarate hydratase; GRIA2, glutamate receptor, ionotropic; AMPA 2HMGA2, high-mobility group AT-hook 2; IL-1α, interleukin-1 alpha; MED12, mediator complex subunit 12; mTOR, mammalian target of rapamycin; P4, progesterone; PTEN, phosphatase and tensin homolog; RA, retinoic acid; RAD51B, RAD51 recombinase; REST, RE1-silencing transcription factor; TGFβ, transforming growth factor beta; TSC2, tuberous sclerosis 2; VEGFA, vascular endothelial growth factor A.
In women, the mRNA levels of REST are similar in normal myometrium and fibroid tissue, but there is significantly less protein present in fibroids (Varghese et al., 2013). During neuronal differentiation, REST is degraded by ubiquitin-mediated proteosomal degradation, but the β-transducin repeat-containing E3 ubiquitin protein ligase (β-TRCP) that is responsible for ubiquitination is not differentially expressed between fibroid and normal myometrium (Westbrook et al., 2008; Varghese et al., 2013). This indicates that there might be a unique mechanism in fibroids responsible for the degradation of REST, possibly due to a change of MED12 function. REST is also important for maintenance of chromosomal integrity by regulation of the expression of MAD2 mitotic arrest deficient-like 1 (MAD2L1), which is an essential component of the mitotic spindle assembly checkpoint (Dobles et al., 2000; Guardavaccaro et al., 2008). In light of these novel recent findings implicating REST in the genesis of fibroids, additional investigation will be required to assess a causal relation between loss of REST and the occurrence of cytogenetic aberrations in fibroids.
Interleukins
Interleukin-1 alpha (IL-1α), is a cytokine that is stored within various types of immune and non-immune cells in precursor form and immediately released upon tissue damage, rapidly initiating a cascade of inflammatory cytokines (Sims and Smith, 2010; Garlanda et al., 2013). Human bone marrow derived mesenchymal stem cells (MSCs) are able to transdifferentiate in vitro into functional neurons after treatment with retinoic acid (Cho et al., 2005). This transdifferentiation of MSCs into neuronal cells is enhanced by addition of IL-1 by inhibiting REST expression. In the context of neuronal transdifferentiation of MSCs, it has been shown that REST is affected by and also plays a central role during tissue damage and inflammation (Greco and Rameshwar, 2007; Heinrich et al., 2009). A number of studies suggest that the same mechanism might also be relevant in fibroid formation (Faerstein et al., 2001b; Wegienka, 2012; Plewka et al., 2013; Wegienka et al., 2013). In vitro wound healing assays of cultured primary human myometrial cells showed that cells migrating to the scratched area express significantly lower amounts of REST compared with those in the intact area (Varghese et al., 2013). Moreover, knockdown of REST with siRNA accelerated the migration of cells to the scratched area. There is some limited evidence in several human studies that support the hypothesis that polymorphisms or different expression levels of interleukin genes are significantly associated with human leiomyoma. There is a significant difference in IL-1β polymorphisms between women with a histological diagnosis of leiomyoma and a control group (Pietrowski et al., 2009). Distributions of the polymorphisms in the IL-12 receptor-β subunit are significantly different between women with and without leiomyoma, while several other polymorphisms do not significantly differ in distribution (Hsieh et al., 2007). These observations lend further credence for leiomyoma development sharing similarities with dysregulated wound healing (Leppert et al., 2006), which is discussed in more detail below.
Cytogenetic aberrations in leiomyomas
A recent comprehensive study reported the results of whole genome sequencing and gene expression profiling of 38 human uterine leiomyomas and the corresponding normal myometrium from 30 women (Mehine et al., 2013). The selection of the fibroids was based on the presence of a MED12 mutation in 16 samples or presence of a FH mutation in 4 samples, while 18 samples contained neither MED12 nor FH mutations. The average age of the included patients was 49.5 years, while the average age of menopause was 51.3 years. Several potential confounders may have been present within the population and samples analyzed. Given the perimenopausal mean of the patient ages and homogenous population (samples were obtained from women undergoing hysterectomy at the Helsinki University Central Hospital of Finland), the results of the study may not be generalizable. Multiple uteri contained fibroids that were clonally related to each other; in one patient, five separate fibroids were clonally related to each other, suggesting local seeding from one primary tumor. This is an intriguing observation because it is in direct contradiction to the central hallmark of uterine leiomyomas, which only show local expansion. It may explain, in part, the frequent occurrence of multiple tumors of the same size in one uterus (Cramer and Patel, 1990). Cytogenetic analysis of fibroids had earlier indicated that individual leiomyoma might be clonally derived from one original tumor (Nibert and Heim, 1990; Nilbert et al., 1990). There are also three extremely rare diseases in which smooth muscle tumors grow outside the uterus and are histologically similar to uterine fibroids namely, disseminated peritoneal leiomyomatosis, benign metastasizing leiomyoma and intravenous leiomyomatosis. In particular, a specific number of genes were affected by rearrangements, including HMGA2, RAD51B, COL4A5, COL4A6 and CUX1 (Fig. 3; Mehine et al., 2013). These genes had been identified earlier by cytogenetic research and are discussed in the section below. A large number (∼42%) of the fibroids contained multiple chromosomal rearrangements that are likely the result of a single event of multiple double-strand breaks and subsequent random repair. Such an event, on a larger scale, is considered as a causal factor in tumorigenesis (Forment et al., 2012), but further investigation is needed to determine a possible causal relation in fibroid formation.
Despite their benign nature, ∼40–50% of the fibroids contain chromosomal abnormalities (Nibert and Heim, 1990; Mark et al., 1991; Rein et al., 1991; Vanni et al., 1991; Ligon and Morton, 2001; Sandberg, 2005). Whether this is a consequence of deregulation of DNA repair mechanisms in vivo or a causal factor itself is unknown. The latter automatically implies that there are at least two or more pathogenic mechanisms responsible for fibroid formation since 50% of leiomyomas have a normal karyotype.
In most cases, fresh human fibroid tissue is first cultured for a number of days in vitro to obtain cells in metaphase suitable for karyotype analysis (Medikare et al., 2011). Microarray and xenograft studies have shown that the growth properties of leiomyoma tissue significantly change during in vitro culture (Zaitseva et al., 2006) as does MED12 mutational status (Markowski et al., 2014). With comparative genomic hybridization (CGH), it is possible to investigate chromosomal aberrations in fresh, uncultured cells. One CGH study identified chromosome aberrations in 8 out of 12 (67%) of the cases while a second study only found chromosomal aberrations in 2 out of 14 cases (14%) (Packenham et al., 1997; Levy et al., 2000). When combined, these two studies show approximately the same incidence as found in karyotype analysis of cultured fibroid cells, suggesting that chromosomal aberrations are not an artifact of in vitro culture (Sandberg, 2005). Mosaicism in leiomyomas that are also of clonal origin, but contain a mixture of karyotypically normal and abnormal cells, supports the hypothesis that chromosomal instability is a secondary event occurring during leiomyoma growth (Mashal et al., 1994). Fibroids with a normal karyotype are typically smaller than fibroids with chromosomal abnormalities (Kataoka et al., 2003), and non-mosaic fibroids are usually larger than mosaic fibroids (Rein et al., 1991; Hennig et al., 1999). However, there are multiple clear exceptions to this general rule, for instance fibroids with deletions of chromosome 7 are as large as karyotypically normal fibroids (Hennig et al., 1999).
Translocation t(12;14)(q14–15;q23–24), HMGA2 and RAD51B
Approximately 200 different chromosomal abnormalities have been described in leiomyomas, but there are a limited number that have been reported by several investigators as prevalent (Sandberg, 2005). About 20% of the karyotypically abnormal leiomyoma contain a translocation between chromosome 12 and 14 (t(12;14)(q15;q24)). A wide variety of benign and malignant tumors also contain a breakpoint on chromosome 12q15 (Schoenmakers et al., 1995). For instance, the 12q14–15 translocation has been observed in ∼70% of pulmonary chondroid hamartomas, and ∼70% of all hamartomas contain chromosomal aberrations. Hamartomas have multiple similarities with leiomyoma (Kazmierczak et al., 1999). They are benign tumors thought to be derived from undifferentiated mesenchymal cells in the submucosa of bronchi (Bateson, 1973). Also several human hamartomous syndromes are associated with leiomyoma development (Table I).
The high-mobility group AT-hook 2 (HMGA2) on chromosome 12 has been identified as the target gene and RAD51B is the preferential translocation partner on chromosome 14 (Ingraham et al., 1999; Schoenmakers et al., 1999; Quade et al., 2003). HMG proteins are architectural transcription factors that regulate transcription by changing DNA conformation through binding to AT-rich regions on DNA via their so-called AT-hooks (Pfannkuche et al., 2009). Normally, HMGA2 is highly expressed during embryonic development, however, expression is also observed under pathological conditions in a wide variety of malignant and benign tumors (Cleynen and Van de Ven, 2008). It is also overexpressed in fibroids containing the t(12;14)(q14–15;q23–24) translocation (Gattas et al., 1999; Gross et al., 2003; Hodge et al., 2012), which has also been associated with the largest leiomyoma size when compared with leiomyomas containing other translocations or none (Rein et al., 1991). In vitro, HMGA2 expression, which can be regulated by Let-7 microRNAs that are deleted in many cancers (Peng et al., 2008), is also associated with inhibiting fibroid cell senescence (Markowski et al., 2011). Because of their ability to bind to AT-rich DNA sequences, it is not surprising that HMG proteins are involved in several important cellular processes, including cell growth, proliferation, differentiation and cell death. Accumulating evidence indicates that HMGA2 plays an essential role in the renewal of stem cells and functions as a proto-oncogene (Ligon et al., 2005; Nishino et al., 2008; Pfannkuche et al., 2009; Copley et al., 2013). Mice that overexpress a truncated form of the HMGA2 gene, but still containing the AT hooks exhibit a giant and obese phenotype with predominantly abdominal and pelvic lipomatosis (Battista et al., 1999; Arlotta et al., 2000). Several female mice develop numerous genitourinary symptoms, such as clitoral gland hyperplasia, dermoid cysts and/or hydrosalpinx, but not leiomyomas (Battista et al., 1999). These latter results suggest that HMGA2 truncation does not play a central role in the development of uterine fibroids, at least in mice.
Although HMGA2 is not expressed in normal myometria of Eker rats, expression has been observed in ∼85% of leiomyomas (Hunter et al., 2002). One in vitro culture study of human leiomyoma tissue showed that senescence of cultured cells is accompanied by a decrease in HMGA2 expression (Markowski et al., 2011). It is unclear why the t(12;14)(q14–15;q23–24) translocation is the most common cytogenetic aberration in leiomyoma and is associated with larger fibroids. From this perspective, it is interesting to hypothesize that the t(12;14)(q14–15;q23–24) translocation is actually not a secondary event, but an important causal factor in the development of fibroids. In support of that theory, overexpression of HMGA2 induces the expression of bFGF (aka FGF2) mRNA (Helmke et al., 2011), a factor with widespread mitogenic and angiogenic effects that is a principal matrix component in a xenograft model discussed below. A more recent report also suggests that overexpression of a truncated form of HMGA2 in putative myometrial stem cells induces a tumor-initiating phenotype in those cells (Mas et al., 2015).
RAD51 recombinase (RAD51) is essential for double-strand DNA break repair and homologous recombination during meiosis (Krejci et al., 2012). Thus, a disturbance of RAD51 function is more likely to be a cause instead of an effect of chromosomal aberrations elsewhere. PTEN, which is mutated in Cowden syndrome, directly controls the expression of RAD51 (Liu et al., 2008). There were no RAD51L1-HMGA2 transcripts and four HMGA2-RAD51L1 transcripts detected in 10 uterine leiomyoma with a translocation between chromosomes 12 and 14 (Quade et al., 2003). Another study identified in 2 out of 81 human fibroid samples a fusion protein containing RAD51L1 nucleotide-binding domains and the HMGA2 protein lacking the N-terminal AT hook motifs (Takahashi et al., 2001). This strongly suggests that the expression of a RAD51L1-HMGA2 fusion protein is not the principle pathophysiological mechanism in leiomyoma development. It is very likely that the interruption of a complex regulation network of HMGA2 and other factors that associate with this complex are actually involved in leiomyoma biology (Ingraham et al., 2006). In rare cases, HMGA2 can also be fused to several other genes, including, ALDH2, COX6C and CALB1. Below are brief discussions of each and their possible relationships to fibroids.
Aldehyde dehydrogenase 2
The main function of aldehyde dehydrogenase 2 (ALDH2) is the oxidation of aldehydes into carboxylic acids. It has been shown that the human ALDH2 promoter contains a retinoid response element (Pinaire et al., 2003). Moreover, a fusion transcript between HMGA2 and ALDH2 has been detected in human fibroid tissue (Kazmierczak et al., 1995). Thus, it could function in conjunction with ADH1 and CRABP2 to deregulate the retinoic acid pathway and subsequent myometrial stem cell differentiation. In several types of cancers, ALDHs have been shown to be up-regulated and are also associated with differentiation and/or expansion of the cancer stem cell population. For example, ALDH1 expression or activity is often used to identify cancer stem cells (Douville et al., 2009) and a majority of fibroid cells are ALHDH1+ by flow cytometry (Holdsworth-Carson et al., 2014).
Cytochrome C oxidase subunit VIc
Cytochrome C oxidase subunit VIc (COX6C) is the terminal enzyme of the mitochondrial respiratory chain and catalyzes the electron transfer from reduced cytochrome C to oxygen (Hofmann et al., 1998). A possible dysfunction in this activity in leiomyoma might lead to metabolic stress. Results from one study in an Eker rat-derived cell line suggest a role for cytochrome C in induction of apoptosis (Raymond et al., 2006).
Calbindin 1
Calbindin 1 (CALB1) (aka RTLV-H) functions as a calcium sensor and buffer (Kojetin et al., 2006) and its expression is strongly related to the quantity of vitamin D in the duodenum (Wasserman and Fullmer, 1989). In light of the associations of African-American ethnicity with both increased incidence of uterine fibroids and low vitamin D status, a number of studies have investigated a possible role for vitamin D in fibroid biology. For example, fibroid tumor size in xenotransplants of rat leiomyoma cells and ECM protein of human leiomyoma cells in vitro are both significantly diminished by vitamin D treatment (Halder et al., 2013, 2014). While it is not clear if CALB1-HMGA2 fusion transcripts are affected by vitamin D status, it would appear that both low vitamin D and disrupted CALB1 expression could play a role in leiomyoma development and warrant further investigation. A more complete discussion of vitamin D deficiency and leiomyoma development is provided below in the ethnic disparities section.
Del (7) (q22q32), Cut-like homeobox 1
Translocations of the long arm of chromosome 7 occur in ∼17% of the karyotypically abnormal leiomyomas (Sandberg, 2005), and loss of heterozygosity of 7q22, quite possibly involving Cut-like homeobox 1 (CUX1), occurs in ∼10–35% of unselected cases of human fibroids (Zeng et al., 1997; van der Heijden et al., 1998; Patrikis et al., 2003). It is likely that this gene is also involved in leiomyoma cases with chromosome 7 deletions (Schoenmakers et al., 2013). Normally, CUX1 plays an important role during development, cell cycle progression, cell proliferation, cell migration and invasion (Sansregret and Nepveu, 2008; Hulea and Nepveu, 2012). Recently, CUX1 was identified as the target gene in two individual fibroids containing a closely related pericentric and paracentric chromosomal inversion of band 7q22 (Schoenmakers et al., 2013), which effectively leads to a monoallelic knockout of this gene. One study did not find any somatic mutations in all coding exons of CUX1 in 42 fibroids (Patrikis et al., 2003). A possible explanation is that CUX1 is a tumor suppressor gene in which loss of one allele is sufficient to facilitate tumor growth (Zeng et al., 1999; Cook and McCaw, 2000). For example, CUX1 haploinsufficiency has been correlated with increased tumorigenesis in human hematopoietic stem cells (McNerney et al., 2013). A tumor suppressor role of CUX1 in fibroids is supported by two studies, which indicate that sequestration of both CUX1 and retinoblastoma through interaction with the large T antigen of the polyoma virus, frequently leads to fibroid formation in transgenic mice (Romagnolo et al., 1996; Webster et al., 1998).
CUX1 is a ubiquitously expressed transcription factor that contains a Cut homeobox domain and two or three Cut repeats. It can be spliced into five different isoforms with distinct DNA binding and transcriptional activating or repressing properties (Sansregret and Nepveu, 2008). In addition, CUX1 also undergoes extensive post-translational modification by means of phosphorylation, dephosphorylation, acetylation and proteolytic processing (Sansregret and Nepveu, 2008). These issues make it rather complicated to investigate the precise role of each isoform of the CUX1 gene. Additionally, studies of different aspects of CUX1 functions have been reportedly linked to other pathways or genes that are, to a varying degree, associated with human fibroid development. For instance PI3K/AKT, Wnt/β-catenin, TGF-β and vitamin D receptor have all been linked to CUX1 (Ripka et al., 2010; Hulea and Nepveu, 2012; Liu et al., 2013).
Racial/ethnic disparities in uterine fibroid biology
As mentioned above, there are several significant differences in leiomyoma disease among different ethnic groups. African-American women develop the disease at a higher frequency and with more severe uterine fibroid-related symptoms. Hispanic women have an intermediate disease profile and Caucasian women are the least severely affected ethnic group (Velebil et al., 1995; Baird et al., 2003; Wise et al., 2012). Several studies have tried to assess an association between specific gene polymorphisms and the severity of uterine fibroids. There are conflicting results of case–control studies regarding the possible association between gene polymorphisms related to estrogen and progesterone metabolism and uterine fibroids. This can be attributed to several complicating factors including multiple etiologies among different ethnic groups or inconsistencies in the studied ethnic groups. For instance, the definition of an ethnic group is not straightforward. In the context of recent or past admixture of different ancestries in families, approximately one-third of people consider themselves to belong to more than one ethnic group (Parra et al., 2004; Owen et al., 2013). This raises concern for the validity of the often used, self-reported ethnicity to make a distinction between different ethnic groups. The use of specific single nucleotide polymorphism seems to provide a more precise classification of ethnicity, but introduces significant cost and infrastructure needs to large epidemiological cohort studies.
Nonetheless, there are several studies that have identified candidate factors and genetic differences that are believed to play a role in fibroid development in different ethnic groups, including vitamin D status, polymorphisms of genes involved in estrogen synthesis, retinoic acid pathway-related genes and miRNA expression levels. Below we discuss the relevance of each factor related to uterine fibroid biology.
Vitamin D status
The prevalence of vitamin D deficiency is 10-fold greater in African-American women compared with Caucasian women, making it an obvious potential candidate for further investigation (Nesby-O'Dell et al., 2002). Vitamin D is important for protection from several aspects of malignant tumor formation, including regulation of cell cycle proteins, angiogenesis and inflammation (Mocellin, 2011). An increasing number of recent studies suggest that vitamin D could also play comparable roles in uterine fibroid formation. Firstly, there are several reports that show an inverse association between serum vitamin D levels and the incidence of uterine fibroids (Baird et al., 2013; Paffoni et al., 2013; Sabry et al., 2013). Additionally, in vitro studies demonstrate a growth inhibitory effect of vitamin D in several different models of human uterine fibroids (Blauer et al., 2009; Sharan et al., 2011; Halder et al., 2012, 2014). The proposed downstream effectors of vitamin D-mediated inhibition are a reduction of ECM-associated proteins, matrix metalloproteinases, TGFβ3 and catechol-O-methyltransferase (COMT) (Kojetin et al., 2006; Raymond et al., 2006; Baird et al., 2013; Paffoni et al., 2013). As a result, vitamin D therapy or downstream activation has interesting potential for future leiomyoma treatment.
COMT expression
Several studies have investigated the genotype distribution of a number of genes related to estrogen metabolism among different ethnic groups to see if this may explain the observed differences in disease phenotype. For example, the activity of the COMT enzyme is significantly higher in human leiomyoma tissue compared with surrounding normal myometrium (Reddy et al., 1981). This enzyme is essential for estrogen metabolism and this observed association imposes a possible causal role for COMT in fibroid formation (Axelrod and Tomchick, 1958). An allele of the COMT gene (Val158) that produces a highly active protein is found significantly more often in African-American women compared with Caucasian women, strengthening the proposed causal association (Al-Hendy and Salama, 2006). Additionally, in vitro cell culture experiments show that there is a relation between a highly active COMT genotype and cellular proliferation and that this effect is independent of ethnicity (Al-Hendy and Salama, 2006). In contrast to the latter culture experiment results, several case-controlled studies have found no differences in genotype distribution of the COMT gene in African-American, Caucasian, Japanese, Brazilian and Turkish women. In the Brazilian and Turkish populations, there was a significant association between the presence of large fibroids and a Val158 polymorphism of COMT (de Oliveira et al., 2008; Ates et al., 2013). Notably, a recent meta-analysis including the above-mentioned case-controlled studies concluded that there is a significant association between specific COMT gene polymorphisms and uterine fibroids (Feng et al., 2013).
CYP17A1 polymorphisms
A T/C SNP polymorphism found 27 base pairs downstream of the transcription start site in the 5′ untranslated region of the CYP17A1 gene, which codes for a cytochrome P450 enzyme required for the synthesis of several steroid hormones, might also be associated with leiomyoma development (Catherino et al., 2013). The C (or A2) CYP17A1 polymorphism has been associated with increased estrogen and progesterone levels, providing a possible link with uterine fibroid development (Feigelson et al., 1998). One study found that the frequency of homozygous A2 CYP17A1 polymorphism is significantly higher in black African women compared with white African women who underwent hysterectomy for uterine fibroid disease (Amant et al., 2004). Another case–control study of Caucasian women who underwent hysterectomy for uterine fibroid disease could not identify an association between the A2 CYP17A1 polymorphism and the occurrence of fibroids (Denschlag et al., 2006). Two additional case–control studies in Japanese and Brazilian women also failed to find an association between either A1 or A2 CYP17A1 polymorphisms and the risk of having uterine fibroids (Tsujino et al., 2006; Vieira et al., 2008). However, a fourth case–control study of the CYP17A1 polymorphisms in Slovenian women found the A1 CYP17A1 polymorphism to have a protective effect against multiple uterine fibroid development (Pakiz et al., 2010). Possible comorbidities and confounding factors among the women used for these studies (age, smoking, family history, etc.) could account for the discrepancies in the study results.
CYP19 (aromatase) expression
Aromatase, encoded by the CYP19A1 gene, is a key enzyme necessary for the synthesis of estrogens from androgens. The local aromatase activity, mRNA and protein expression of CYP19 in uterine fibroids are significantly higher compared with adjacent normal myometrium in women of different ethnic groups (Folkerd et al., 1984; Bulun et al., 1994). However, the mRNA expression level is ∼2-fold higher in African-American women compared with Caucasian and Japanese women (Ishikawa et al., 2009). High local aromatase levels are associated with a local increase in estrogen production and proposed to be part of a positive autocrine feedback loop in fibroid tumors (Sumitani et al., 2000). This is supported by the observation that women who are treated with the GnRH analog leuprolide acetate for 12–24 weeks show a significant decrease in aromatase mRNA, protein and function in fibroid tissue (Shozu et al., 2001). GnRH analogs can also inhibit the production of local growth factors such as epidermal growth factor (EGF), insulin-like growth factor and TGFβ (Maruo et al., 2004).
Predisposing, facilitating and initiating factors
There are epidemiological, histological and experimental results that support the hypothesis that repeated injury and repair of the myometrium are factors driving leiomyoma formation. The number of normal menstruations and uterine infection are both associated with the occurrence of fibroids (Faerstein et al., 2001a, b). There are conflicting epidemiological, experimental and clinical results about the role of estrogen and progesterone (Howe et al., 1995; Rivera-Gonzalez et al., 1998; Lethaby et al., 2001; Deng et al., 2012; Tristan et al., 2012; Sangkomkamhang et al., 2013). Possibly, non-hormonal factors are responsible for the initiation of fibroid development and timely hormonal stimulation could very well become necessary for further growth. Interestingly, the local effect of estrogen and progesterone can be stimulatory or inhibitory (Kim et al., 2013), stressing the importance of the local extra cellular matrix composition.
Stress from chronic inflammation can set preconditions facilitating the development of several types of cancer (Coussens and Werb, 2002). Clinical observations and experimental evidence support the plausibility that the classical signs of inflammation such as increased vascularization and vascular permeability and fibroblast proliferation are also important for leiomyoma formation (Faerstein et al., 2001b). Abnormal bleeding is the most common symptom of fibroids, and can be present in the setting of a large solitary fibroid as well as in multiple small fibroids. Often the symptom of abnormal bleeding can be resolved by myomectomy, suggesting the local secretion of proangiogenic factors. Moreover, multiple animal model studies stress the utmost importance of sufficient neovascularization for xenograft survival. Xenotransplantation under the highly vascularized renal capsule or overexpression of angiogenic factors such as VEGF are essential for leiomyoma survival (described in more detail in the animal model section). One mouse xenograft model shows the importance of inflammation status and a proangiogenic environment by COX-2 and VEGF-A overexpression (Hassan et al., 2008).
A unique underlying facet of the pathobiology of disease includes similarities between fibroid development and keloid formation. Keloids, or hypertrophic scarring, are characterized by disordered wound healing with abnormal remodeling and the aberrant persistence of differentiated myofibroblasts. Similar to the disproportionately higher prevalence of uterine fibroids in African-American women, the formation of keloids is most common in people of African descent. Notably, Leppert et al. reported a markedly similar morphologic ultrastructure and aberrant ECM formation for fibroids and keloids (Leppert et al., 2006). Dysregulated ECM-related gene expression profiles in fibroids and keloids have also been described (Catherino et al., 2004). Dermatopontin, a collagen-binding protein was found to be expressed at reduced levels in both fibroids and keloids, and was associated with increased TGFB3 expression in fibroid/myometrium matched tissue cohort. These findings suggest a possible common underlying molecular mechanism for the fibroid and keloid phenotype via dysregulation of cell/tissue turnover and remodeling.
Animal models for the study of leiomyomas
There are several animal models utilized for in vivo investigation of the pathobiology of leiomyoma development and progression. The main methods used to establish these models are genetic engineering, hormonal modulation and xenotransplantation of human fibroid tissue. Herein, we will describe the advantages and limitations of some of the models, as well as fibroid relevant factors and genes involved (Table II).
Overview of animal models used in leiomyoma research.
| Animal model | Techniques | Model requirements/properties | Reference |
|---|---|---|---|
| Guinea pig | Surgery, drugs | Ovariectomy, estrogen supplementation, 9-cis or all-trans retinoic acid administration | Porter et al. (1995), Tsibris et al. (1999) |
| Mouse xenograft | Surgery, drugs | Hormone supplementation, transfected cells | Hassan et al. (2008), Suo et al. (2009), Ishikawa et al. (2010), Tsuiji et al. (2010), Drosch et al. (2013) |
| Rat | Selective breeding | Heterozygous germline TSC2 mutation | Eker et al. (1981) |
| Mouse | Genetically modified | Conditional Tsc2 knockout | Prizant et al. (2013), Kaneko-Tarui et al. (2014) |
| Mouse | Genetically modified | GPR10 overexpression | Varghese et al. (2013) |
| Mouse | Genetically modified | Conditional, constitutive β-catenin expression | Tanwar et al. (2009) |
| Animal model | Techniques | Model requirements/properties | Reference |
|---|---|---|---|
| Guinea pig | Surgery, drugs | Ovariectomy, estrogen supplementation, 9-cis or all-trans retinoic acid administration | Porter et al. (1995), Tsibris et al. (1999) |
| Mouse xenograft | Surgery, drugs | Hormone supplementation, transfected cells | Hassan et al. (2008), Suo et al. (2009), Ishikawa et al. (2010), Tsuiji et al. (2010), Drosch et al. (2013) |
| Rat | Selective breeding | Heterozygous germline TSC2 mutation | Eker et al. (1981) |
| Mouse | Genetically modified | Conditional Tsc2 knockout | Prizant et al. (2013), Kaneko-Tarui et al. (2014) |
| Mouse | Genetically modified | GPR10 overexpression | Varghese et al. (2013) |
| Mouse | Genetically modified | Conditional, constitutive β-catenin expression | Tanwar et al. (2009) |
For each model, the animal, used techniques, requirements/properties and reference are indicated. The oldest and most widely reported animal model in leiomyoma research is the Eker rat model containing a heterozygous germline Tsc2 mutation.
GPR10, official name PRLHR: prolactin-releasing hormone receptor; TSC2, tuberous sclerosis 2.
Overview of animal models used in leiomyoma research.
| Animal model | Techniques | Model requirements/properties | Reference |
|---|---|---|---|
| Guinea pig | Surgery, drugs | Ovariectomy, estrogen supplementation, 9-cis or all-trans retinoic acid administration | Porter et al. (1995), Tsibris et al. (1999) |
| Mouse xenograft | Surgery, drugs | Hormone supplementation, transfected cells | Hassan et al. (2008), Suo et al. (2009), Ishikawa et al. (2010), Tsuiji et al. (2010), Drosch et al. (2013) |
| Rat | Selective breeding | Heterozygous germline TSC2 mutation | Eker et al. (1981) |
| Mouse | Genetically modified | Conditional Tsc2 knockout | Prizant et al. (2013), Kaneko-Tarui et al. (2014) |
| Mouse | Genetically modified | GPR10 overexpression | Varghese et al. (2013) |
| Mouse | Genetically modified | Conditional, constitutive β-catenin expression | Tanwar et al. (2009) |
| Animal model | Techniques | Model requirements/properties | Reference |
|---|---|---|---|
| Guinea pig | Surgery, drugs | Ovariectomy, estrogen supplementation, 9-cis or all-trans retinoic acid administration | Porter et al. (1995), Tsibris et al. (1999) |
| Mouse xenograft | Surgery, drugs | Hormone supplementation, transfected cells | Hassan et al. (2008), Suo et al. (2009), Ishikawa et al. (2010), Tsuiji et al. (2010), Drosch et al. (2013) |
| Rat | Selective breeding | Heterozygous germline TSC2 mutation | Eker et al. (1981) |
| Mouse | Genetically modified | Conditional Tsc2 knockout | Prizant et al. (2013), Kaneko-Tarui et al. (2014) |
| Mouse | Genetically modified | GPR10 overexpression | Varghese et al. (2013) |
| Mouse | Genetically modified | Conditional, constitutive β-catenin expression | Tanwar et al. (2009) |
For each model, the animal, used techniques, requirements/properties and reference are indicated. The oldest and most widely reported animal model in leiomyoma research is the Eker rat model containing a heterozygous germline Tsc2 mutation.
GPR10, official name PRLHR: prolactin-releasing hormone receptor; TSC2, tuberous sclerosis 2.
Guinea pig
Approximately 8% of guinea pigs spontaneously develop leiomyomas by the age of 4 years (Field et al., 1989). This innate tendency to develop uterine tumors has led to the search for a feasible guinea pig model. All guinea pigs that are ovariectomized and exposed to chronic estradiol alone develop tumors predominantly on the inside of the abdominal wall and smaller uterine leiomyomas within 3 months (Porter et al., 1995). In contrast, only 1 out of 12 non-ovariectomized animals supplemented with estradiol in that study developed fibroid-like tumors, suggesting a protective effect of an ovarian factor on leiomyoma development. In a follow-up study, ovariectomized guinea pigs treated with both estradiol and retinoic acid developed uterine tumors (Tsibris et al., 1999). Desmin immunostaining and electron microscopy was used to confirm the leiomyoma-like identity of the tumors. Interestingly, the uterine leiomyomas were even larger when the PPARγ agonist, Troglitazone, was administered in combination with estrogen and all-trans retinoic acid. No tumor formation occurred when Troglitazone was administered alone or combined with only estradiol or estradiol plus 9-cis retinoic acid (Tsibris et al., 1999), implying that the retinoic acid pathway is an important part of the guinea pig model, but only effective under the appropriate steroid hormone conditions and in combination with other factors.
Mouse xenograft
The first reported xenograft model was based on subcutaneous xenotransplantation of human fibroid tissue in severe combined immunodeficient (SCID) mice (Hassan et al., 2008). The mice were supplemented with subcutaneous 17β-estradiol pellets to obtain chronic high levels of serum estrogen. Before transplantation, the fibroid tissue was transduced with two adenoviruses containing either the cyclooxygenase-2 (COX2) or vascular endothelial growth factor A (VEGFA) genes, with expression driven by the cytomegalovirus (CMV) promoter. COX-2 is the rate-limiting enzyme that converts arachidonic acid into prostaglandin H2, the precursor of all prostaglandins (Smith et al., 1996). Prostaglandins are most well known for their pro-inflammatory effect and role in blood coagulation. COX-2 also plays an essential role during ovulation, fertilization, implantation and decidualization (Lim et al., 1997). Knockout or selective inhibition of COX-2 results in a significant decrease of tumor formation in an APCΔ716 knockout mouse model for human familial adenomatous polyposis (Oshima et al., 1996). The xenotransplanted tissue survived for 30 days (8/8 mice) and maintained key leiomyoma features, i.e. tissue morphology, apoptotic and proliferation indices, ER, PR, αSMA, desmin and vimentin expression. The major highlight of this model is that it is possible to engraft multiple xenografts in one mouse, which makes it possible to sequentially harvest grafts for analysis at multiple time points during an experiment. Additional advantages are the relative low costs and a preserved benign leiomyoma phenotype (Hassan et al., 2008).
The need for adenoviral transduction with proangiogenic factors can be circumvented by implanting fibroid tissue beneath the renal capsule of adult female non-obese diabetic, severe combined immunodeficient (NOD/SCID) IL2Rγnull mice (Ishikawa et al., 2010). The neovascularization potential of the kidney is far greater than in subcutaneous locations in this model. Mice were ovariectomized and supplemented with subcutaneous slow release estrogen, progesterone and/or Mifepristone capsules. In this model, whole fibroid tissue or primary cells from 2 to 3 days old human fibroid cultures embedded in collagen matrix were xenotransplanted. The xenotransplanted tissue (14/29 mice) and single cell collagen matrix (13/17 mice) only increased significantly in size if both estrogen and progesterone were administered. Administration of Mifepristone blocked the effects of progesterone, and the resulting tumors had a higher proliferation index and a lower cell density compared with tissue from mice treated with only estrogen or progesterone. The expression of estrogen and progesterone receptor in tumors was comparable to the original tissue. In contrast to subcutaneous models, a possible disadvantage of xenotransplantation under the renal capsule is that the growth of the tumors cannot be readily measured over time.
One group has transduced fresh fibroid cells with fluorescent and bioluminescent proteins to better track growth of xenotransplanted tumors in vivo (Suo et al., 2009) and injected the cells in the tail vein, intraperitoneally or subcutaneously into immunocompromized mice. The Rag2−/−γ−/− knockout female mice were supplemented with a subcutaneous estradiol pellet (Shinkai et al., 1992). Only subcutaneously injected uncultured fibroid cells in Matrigel maintained their tumor size for 20 days.
A fourth xenotransplant model was established in NOD/SCID/γcnull mice (Tsuiji et al., 2010). These mice were implanted with only 17β-estradiol pellets and fresh human fibroid tissue was implanted subcutaneously in the flanks of mice. The xenotransplants maintained a stable size up to 8 weeks after transplantation. There was a small amount of growth when both estrogen and progesterone were administered. In accordance with the previously described model (Ishikawa et al., 2010), the proliferation index was increased. The apoptotic index, ER, PR and αSMA expression remained the same at 4 and 8 weeks after transplantation. The growth rate in this subcutaneous NOD/SCID/γc-null model is considerably slower compared with the renal capsule xenotransplant NOD/SCID IL2Rγnull model.
Recently, a different approach was used to establish a human xenograft mouse model. Collagenized cell suspensions obtained from human fibroid tissue were transfected with lentivirus containing the Simian Virus 40 early region of large and small T antigen (SV40ER) (Drosch et al., 2013). The large T protein forms a complex with the tumor suppression genes p53 and Retinoblastoma protein resulting in cell immortalization (Ozer et al., 1996). SV40ER-transfected cells were able to form anchorage-independent colonies in a soft agar petri dish, whereas normal, untransfected myometrial cells did not grow in this condition. However, cultured SV40ER-transfected myometrial cells underwent apoptosis when transplanted into SCID/beige mice. Fresh fibroid cells in Matrigel supplemented with EGF, basic fibroblast growth factor (bFGF) and insulin formed tumors when they were transplanted into the dorsal flanks of SCID/beige mice. As in the previous models, the mice were first ovariectomized and implanted with subcutaneous slow release estrogen and progesterone pellets. The tumors were stable in size up to 60 days after transplantation. The histology was comparable to the original human fibroid tissue; the proliferation rate was low, and the cells were positive for αSMA. Fluorescence in situ hybridization analyses for mouse and human centromeric sequences showed that the majority of the tumor cells were of human origin and that a limited number of peripheral tumor cells were mouse cells. Fresh fibroid cells in Matrigel supplemented with EGF, bFGF and insulin were also able to survive when they were directly injected into the uterine horn of SCID/beige mice. The disadvantage of this location is that there was not a clear growth of tumors that can be easily visualized and compared in an experimental setting.
What can be concluded from the xenograft models is that supraphysiologic estrogen and progesterone serum concentrations as well as vascularization and angiogenesis are crucial for a successful fibroid xenotransplantation.
Rat Tsc2 mutation
The most widely reported in vivo animal model to study leiomyoma biology is the Eker rat model, which has a germ line mutation in Tsc2 (Eker et al., 1981; Yeung et al., 1994), resulting in a loss of function allele of the Tsc2 lacking its catalytic domain. The most profound effect in rats with this heterozygous mutation is that virtually all develop chronic and progressive nephropathy and renal cell carcinomas by the age of 1 year (Everitt et al., 1992). In addition, the heterozygous carriers also frequently develop uterine tumors, pituitary adenomas and hemangiosarcomas of the spleen (Hino et al., 1994; Everitt et al., 1995). The adverse effects of renal failure on the immune system and possible dysfunction of the endocrine system may be contributory to the emergence of leiomyomas in the uterus (Manucha and Valles, 2012). At the age of 14 months, 43% of the Eker rats developed gross tumors in the reproductive tract, and in 72% of the cases microscopic lesions are detectable. Most of the cases were diagnosed as benign leiomyomas and sometimes as malignant leiomyosarcomas (Everitt et al., 1995). These lesions were typically located at the dorsal side of the cervical–uterine junction. It proved to be difficult to determine if the tumors originate from the uterine myometrium or cervical stroma (Everitt et al., 1995). To a lesser degree, the lesions could form in the region of the cervix and fused uterine body, and even less frequently in the uterine horns. Because all the rats die due to renal failure, it is not possible to observe the long-term behavior of these tumors. Of five cell lines derived from Eker rat tumors, one was capable of inducing tumors in nude mice and four of the five cell lines expressed a functional estrogen receptor (Howe et al., 1995). Nonetheless, tumor growth could not be stimulated by estradiol supplementation. Analysis of progesterone receptor expression showed inconclusive results because the expression levels were highly variable among different cell lines.
Mouse conditional Tsc2 knockout
Recently, two closely related, but distinct conditional Tsc2 knockout mouse models have been developed by mating mice with a floxed allele of Tsc2 (Hernandez et al., 2007) with mice that have Cre recombinase knocked into either the progesterone receptor (Pgr) (Soyal et al., 2005) locus or the Müllerian-inhibiting substance/Anti-Müllerian hormone type 2 receptor promoter (Amhr2) locus (Jamin et al., 2002). Uterine Pgr-Cre activity starts in mice as early as 1–2 weeks post-natal (Soyal et al., 2005) and Amhr2-Cre activity is detected in the Müllerian duct mesenchyme from embryonic Day 12.5 onwards (Arango et al., 2008). Perhaps more importantly, whereas Pgr-Cre is ubiquitously expressed in the endometrial epithelium and stroma, as well as the myometrium, Amhr2-Cre is expressed in the Mullerian duct mesenchyme-derived endometrial stroma and myometrium but not in the epithelium. The Pgr-Cre;Tsc2 mice develop significant myometrial expansion at the age of 12 weeks, which can be inhibited by Sirolimus (Prizant et al., 2013). The growth inhibitive effect of this mTORC1 inhibitor and the observed up-regulation of phosphorylated S6 protein in Pgr-Cre;Tsc2 mice indicates that the myometrial expansion could be the result of up-regulation of mTOR activity. The authors of that study observed that at the age of 24 weeks, the Pgr-Cre;Tsc2 conditional knockout mice developed uterine tumors that were similar to human fibroids. Myometrial expansion also reached a significant level after 12 weeks in Amhr2-Cre;Tsc2 conditional knockout mice with a concomitant increase in phosphorylated S6 protein, indicating activated mTOR activity (Kaneko-Tarui et al., 2014). Similarly, administration of Sirolimus significantly counteracted the up-regulation of the mTOR pathway and the myometrial hyperplasia. Evidence of myometrial fibrosis was observed, but in contrast to the Pgr-Cre;Tsc2 knockout mice, uterine fibroids were not observed in Amhr2-Cre;Tsc2 knockout mice. The difference in Cre promotor or another factor might be responsible for this observed difference. The central assumption of the widely used Eker rat model is that loss of TSC2 plays a central role in the pathobiology of uterine fibroids. This is supported by the Pgr-Cre;Tsc2 model, but somehow contradicted by the results of our own Amhr2-Cre;Tsc2 model, which suggests that additional yet unidentified factors might be necessary for fibroid formation. Additionally, a recent report investigating human leiomyoma tissue found increased instead of diminished levels of TSC2 (Cui et al., 2011). It should also be noted that when the gene for TSC1, which partners with TSC2 in a heterodimer to control mTOR activity, is deleted by either Pgr-Cre (Daikoku et al., 2013) or Amhr2-Cre (Tanaka et al., 2012), neither mice develop detectable uterine leiomyomas.
GPR10 overexpression
Above we described how a reduction in REST protein could play a significant role in human fibroid formation by possibly activating expression of GPR10, which is highly expressed in human leiomyomas but present at a very low level or completely absent in normal myometrium (Varghese et al., 2013). This group produced a mouse that was genetically engineered to overexpress the GPR10 transgene specifically in the myometrium using the proximal promoter sequence from space the rat calbindin-D9K (CaBP9K) to test whether unregulated expression of GPR10 could induce uterine leiomyoma development. This promoter has been used to drive expression of SV40 Large T in mouse myometria that also develop smooth muscle tumors (Romagnolo et al., 1996). Histological analyses of uterine cross-sections from the GPR10-overexpressing mice showed an increase in the myometrial area and with significant, widespread ECM deposition but not focal tumorigenesis, similar to that which we observed in the conditional Tsc2 knockout mice (Kaneko-Tarui et al., 2014). A trivial explanation for the lack of tumors in these mice could be because both models use a promoter-driven expression system that is active in virtually all the myometrial cells, leading us to speculate that if overexpression of GPR10 or Tsc2 deletion could be targeted to a few cell, tumors more akin to those observed in humans could form.
Mouse conditional β-catenin overexpression
Another genetically engineered mouse model is based on expression of a constitutively active (or gain-of-function allele) of β-catenin in uterine mesenchyme driven by Amhr2 promoter (Tanwar et al., 2009). β-Catenin is the canonical signaling component of the WNT signaling pathway (Clevers and Nusse, 2012). This signal transduction cascade controls a wide variety of processes during embryonic development and tissue homeostasis in adult animals. In the context of fibroid development, its involvement in uterine development, stem cell renewal and carcinogenesis are essential. A floxed β-catenin allele has been developed that contains loxP sites surrounding the third exon of this gene, which codes for the N-terminal phosphorylation sites (Harada et al., 1999). Following Cre recombinase activity, this in frame exon 3 β-catenin protein is unable to be phosphorylated by GSK3β in the adenomatosis polyposis coli (APC) complex and ubiquitinated for subsequent degradation. As a consequence, β-catenin accumulates, translocates to the nucleus, and regulates expression of its target genes, essentially mimicking chronic WNT signaling activity. All the Amhr2-Cre constitutive β-catenin-expressing mice develop myometrial hypertrophy and can develop uterine tumors that share multiple characteristics with human leiomyomas and endometrial stromal sarcomas (Tanwar et al., 2009). The whole myometrium is hyperplastic and additionally polyps develop at the antimesometrial side of the uterus. These polyps are observed in prepubertal anestrous 4-week-old mice and are more pronounced in older as well as multiparous mice. The fact that the polyps develop in prepubertal mice suggests a hormone-independent start to their development. The myometria of these mutants consists of hypertrophic cells with a low mitotic index within fascicles. TGFB3 expression and mTOR activity are elevated in mutant mice compared with wild type, which is also the case in human leiomyoma (Arici and Sozen, 2000; Crabtree et al., 2009). The major advantage of the constitutive active β-catenin mouse model is that all mice develop the antimesometrial hyperplastic myometrial polyp and most mice develop significant leiomyoma-like lesions within several months.
Conclusion
Despite the rapid expansion of the field of fibroid biology, the fundamental mechanisms of leiomyoma development and recurrence are incompletely understood. As reported by several recent comprehensive reviews in this journal (Ciarmela et al., 2011; Karmon et al., 2014; Tal and Segars, 2014), the inherent limitations of contemporary medical therapy have prompted a prolific rate of investigation for novel therapeutic targets for uterine leiomyoma. Although the mainstream FDA approved medications, Lupron™ and Ulipristal acetate have focused on the primary symptoms of fibroids, pelvic pressure and heavy menses, they are only effective as short-term agents, and were not originally targeted toward the specific molecular mediators of disease. To this end, this review of molecular clues for the genesis of fibroids is timely, and underscores the necessary focus on the various possible fibroid-specific mechanisms (Fig. 3) for the development and growth of disease with the promise of developing patient-specific, precision medicine for their treatment.
When devising an approach to clinically applicable therapeutic targets, one must first consider the feasibility of primary versus secondary prevention. In light of the extended growth period and predominance of fibroids prior to menopause, primary prevention is challenging since the majority of fibroids are subclinical, and are not detected until large in size (pain symptoms) or located in the endometrium (abnormal uterine bleeding). Since uterine fibroids do not pose significant mortality, are frequently treated via routine procedural and/or surgical means, and can have a significant recurrence rates following removal, secondary prevention or reduction in the risk of recurrence is a prime area of focus for translational investigation and clinical application.
In light of the signature dysregulated ECM phenotype of fibroids and the overlap of several fibroid relevant molecular mediators related to ECM production, signaling pathways regulated by TGFβ, GRP10/REST, PI3K-AKT-mTOR, and β-catenin represent the most promising targets. A clinically relevant approach would be specific targeted approaches to concurrently inhibit essential pathways for proliferation and ECM formation. As seen in both Proteus and Cowden's syndrome, alterations of PIK3CA or AKT, may represent significant upstream stimulators of the mTOR pathways (Kovacs et al., 2003). As a therapeutic target, alteration of these regulators to serve as upstream inhibitors of mTOR, may induce leiomyoma senescence and/or regression, decrease progression of subclinical fibroids, and reduce the risk of fibroid recurrence following surgery. Within the murine fibroid model, the REST/GPR10 paradigm also highlights the probable significance of PI3K/AKT/mTOR in the genesis of fibroids. Interestingly, GPR10 up-regulation is associated with increased expression of a variety of ECM proteins and proliferation (Varghese et al., 2013). Since GPR10 is up-regulated in fibroids, minimally expressed in the myometrium, and has been correlated to phosphorylated AKT in leiomyoma cells in vitro, the GPR10 mouse is a promising in vivo model to investigate the pathobiology of fibroids.
Although a major element of tumorigenesis is proliferation, the signature characteristic of fibroids, ECM dysregulation, is noteworthy. The TGFβ/SMAD model has been the most widely described signaling pathway to account for abnormal collagen formation within the ECM (Ciarmela et al., 2011). However, common mediators of the TGFβ and ECM dysregulation have been difficult to delineate. Findings of increased expression of TGFβ3 and mTOR signaling in the setting of hypertrophic cells in the mouse models described above require further investigation of the synergistic mechanisms of cell proliferation and aberrant ECM formation, and may also represent clinically relevant targets to reduce the risk of fibroid recurrence.
Authors' roles
A.E.C., A.K.S. and J.M.T. drafted the original manuscript, revised the manuscript and approved the final version.
Funding
This worked was funded by: National Institutes of Health Grants R01OD012206 and R01HD072489 (J.M.T.); and Office of Diversity Inclusion and Community Partnership/Harvard Catalyst Faculty Fellowship, Harvard Medical School (A.K.S.). A.E.C. is the recipient of an Academic Medical Center (AMC) PhD Scholarship.
Conflicts of interest
A.E.C., A.K.S. and J.M.T. declare that they have no conflict of interest.
Acknowledgements
We thank Dr Jan Kooter for his input in the early phase of manuscript drafting. We thank Drs Amanda Patterson, John Risinger and Vargheese Chennathukuzhi for reviewing the manuscript.

{kind=link}
{kind=link}
{kind=link}