





Abstract
Can whole-exome sequencing (WES) reveal new genetic factors responsible for male infertility characterized by oligozoospermia?
We identified biallelic missense variants in the Potassium Channel Tetramerization Domain Containing 19 gene (KCTD19) and confirmed it to be a novel pathogenic gene for male infertility.
KCTD19 is a key transcriptional regulator that plays an indispensable role in male fertility by regulating meiotic progression. Kctd19 gene-disrupted male mice exhibit infertility due to meiotic arrest.
We recruited a cohort of 536 individuals with idiopathic oligozoospermia from 2014 to 2022 and focused on five infertile males from three unrelated families. Semen analysis data and ICSI outcomes were collected. WES and homozygosity mapping were performed to identify potential pathogenic variants. The pathogenicity of the identified variants was investigated in silico and in vitro.
Male patients diagnosed with primary infertility were recruited from the Reproductive and Genetic Hospital of CITIC-Xiangya. Genomic DNA extracted from affected individuals was used for WES and Sanger sequencing. Sperm phenotype, sperm nuclear maturity, chromosome aneuploidy, and sperm ultrastructure were assessed using hematoxylin and eosin staining and toluidine blue staining, FISH and transmission electron microscopy. The functional effects of the identified variants in HEK293T cells were investigated via western blotting and immunofluorescence.
We identified three homozygous missense variants (NM_001100915, c.G628A:p.E210K, c.C893T:p.P298L, and c.G2309A:p.G770D) in KCTD19 in five infertile males from three unrelated families. Abnormal morphology of the sperm heads with immature nuclei and/or nuclear aneuploidy were frequently observed in individuals with biallelic KCTD19 variants, and ICSI was unable to rescue these deficiencies. These variants reduced the abundance of KCTD19 due to increased ubiquitination and impaired its nuclear colocalization with its functional partner, zinc finger protein 541 (ZFP541), in HEK293T cells.
The exact pathogenic mechanism remains unclear, and warrants further studies using knock-in mice that mimic the missense mutations found in individuals with biallelic KCTD19 variants.
Our study is the first to report a likely causal relationship between KCTD19 deficiency and male infertility, confirming the critical role of KCTD19 in human reproduction. Additionally, this study provided evidence for the poor ICSI clinical outcomes in individuals with biallelic KCTD19 variants, which may guide clinical treatment strategies.
This work was supported by the National Key Research and Developmental Program of China (2022YFC2702604 to Y.-Q.T.), the National Natural Science Foundation of China (81971447 and 82171608 to Y.-Q.T., 82101961 to C.T.), a key grant from the Prevention and Treatment of Birth Defects from Hunan Province (2019SK1012 to Y.-Q.T.), a Hunan Provincial Grant for Innovative Province Construction (2019SK4012), and the China Postdoctoral Science Foundation (2022M721124 to W.W.). The authors declare no conflicts of interest.
N/A.
Introduction
Infertility is a worldwide reproductive health problem that affects ∼15% of reproductive-aged couples (Agarwal et al., 2021), with male factors contributing to half of the infertility cases (Agarwal et al., 2021). Oligozoospermia and azoospermia, characterized by quantitative spermatogenic defects, are two common infertility phenotypes (Agarwal et al., 2015; Barratt et al., 2017). Oligozoospermia and azoospermia exhibit high genetic heterogeneity, including karyotype abnormalities, Y chromosome microdeletions, and monogenic mutations (Asero et al., 2014; Houston et al., 2021; Xie et al., 2022). Therefore, it is important to elucidate the underlying genetic defects, which would help to provide precise genetic counseling for infertile couples and to decide the optimal therapeutic approach to use.
In the past decade, whole-exome sequencing (WES) has proved to be a powerful research and diagnostic tool for male infertility and has permitted a major step forward in identifying genetic causes of male infertility (Pena et al., 2020; Houston et al., 2021). Using WES, dozens of genes causing male infertility associated with oligozoospermia and azoospermia, such as TEX11 (Yatsenko et al., 2015), RPL10L (Tu et al., 2020), SHOC1 (Yao et al., 2021; Wang et al., 2022), and M1AP (Tu et al., 2020; Wyrwoll et al., 2020), have been identified. However, despite these plethoric findings, the etiology of oligozoospermia and azoospermia remains unknown in more than 70% of cases, and the discovery of the remaining genes is now a major challenge (Oud et al., 2017; Cannarella et al., 2019). Although ARTs, especially ICSI, have revolutionized the treatment of infertile men with oligozoospermia and azoospermia, the outcomes lack efficient evaluation of patients harboring different gene mutations, leading to limited support for precise clinical guidance.
The POZ/BTB domain-containing KCTD19 is a member of the KCTD family, which consists of 26 members (Oura et al., 2021). The KCTD family is an emerging class of proteins that is involved in important biological processes (Angrisani et al., 2021), including ion channel regulation, transcriptional regulation, interaction with ubiquitin ligase complexes such as cullin 3 (CUL3), and degradation of various proteins such as histone deacetylases (HDACs) (Tong et al., 2014; Smaldone et al., 2015). To date, KCTD genes have been associated with several diseases (Teng et al., 2019), including neurocognitive disorders (KCTD3), neurodevelopmental disease (KCTD7), bipolar disorder (KCTD12), autism and schizophrenia (KCTD13), movement disorders (KCTD17), cancer (KCTD11), and obesity (KCTD15). Unlike the widely expressed pattern of these proteins, KCTD19 is specifically expressed in the testis. Recent studies have reported that Kctd19 knockout male mice have reduced testicular size and are infertile due to meiotic metaphase I arrest. KCTD19 forms a complex with zinc finger protein 541 (ZFP541) and HDAC1 (Horisawa-Takada et al., 2021; Li et al., 2022), which is essential for chromatin remodeling and transcriptional regulation of meiotic genes during meiotic I prophase in mice (Oura et al., 2021). These findings indicate that KCTD19 is essential for meiosis and male fertility. However, the role of KCTD19 in human reproduction is poorly understood, and whether KCTD19 deficiency causes oligozoospermia and azoospermia remains to be established.
Here, we performed WES to investigate the genetic causes of oligozoospermia in patients. We identified three homozygous missense variants of KCTD19 in five men with oligoasthenoteratozoospermia from three unrelated Han Chinese families. We also investigated the effects of these three missense variants in HEK293T cells. These findings establish a causal relationship between KCTD19 mutations and male infertility, confirming the critical role of KCTD19 in human reproduction.
Materials and methods
Human subjects
A total of 536 infertile males with oligozoospermia were recruited to participate in this study from 2014 to 2022. All subjects (Han Chinese origin) visited the Reproductive and Genetic Hospital of CITIC-Xiangya for ART. All the individuals had normal somatic karyotypes. Azoospermia factor microdeletions were not detected on the Y chromosome. Other causes of infertility, such as drug use and exposure to gonadotoxic factors, were also excluded. Three independent semen evaluations were performed, and semen parameters were evaluated according to the World Health Organization (WHO, 2010) guidelines (Sanchez-Alvarez et al., 2012).
This study was approved by the institutional ethics committees of the Central South University and Reproductive Genetic Hospital of CITIC-Xiangya, China (LLSC-2017). All methods were performed in accordance with approved guidelines. Written informed consent was obtained from all the participants at the beginning of the study.
WES, bioinformatics analysis, and Sanger sequencing
Genomic DNA was extracted from peripheral blood samples of infertile individuals using a QIAamp® DNA Blood Midi kit (QIAGEN, Hilden, Germany) and sequenced on the HiSeq2000 sequencing platform (Illumina, San Diego, CA, USA). WES data were analyzed as described previously (Tan et al., 2019). Candidate pathogenic variants were screened using the following criteria: (i) minor allele frequency below 1% in public databases: the 1000 Genomes Project and Genome AD (https://gnomad.broadinstitute.org); (ii) predicted to be deleterious using the following software: Polyphen-2 (genetics.bwh.harvard.edu/pph2), MutationTaster (www.mutationtaster.org/), SIFT (sift.jcvi.org/), or combined annotation-dependent depletion (cadd.gs.washington.edu/); (iii) homozygous or compound heterozygous variants, and (iv) phenotype relevance using comprehensive expression data (high expression in testis) and model organism data (Eppig et al., 2015). Homozygosity analysis was performed using Homozygosity Mapper (Seelow et al., 2009), and variants located in homozygous regions >5.0 Mb were preferentially considered. Additionally, to predict the functional consequences of the variants, 3D structures of both the wild type (WT) and candidate variant polypeptides were built using Alphafold (https://alphafold.com/), and the PyMol software (V.1.5) was used to visualize the protein structures.
PCR primers were designed to specifically amplify variants in KCTD19. The primer sequences were as follows:
KCTD19-F1 (M1): 5ʹ-CATGGACACACCCCTGTTAGAC-3ʹ;
KCTD19-R1 (M1): 5ʹ-GTCATGCTTCCGCTACCTC-3′;
KCTD19-F2 (M2): 5ʹ-AAGCCTCCAATCCAGCTATGACC-3′;
KCTD19-R2 (M2): 5ʹ-GCTGAAAGAGGACGCCATTCC-3′;
KCTD19-F3 (M3): 5′-AGCCCCCTCCCTATGTTGT-3′;
KCTD19-R3 (M3): 5′-CTGGCTGTGGGTGTTGTGTG-3′.
PCR products were sequenced using a 3730XL sequencer (Applied Biosystems, Foster City, USA) according to the manufacturer’s instructions.
Semen parameter and sperm morphology analyses
Semen samples were collected by masturbation after 3–5 days of sexual abstinence, and the samples were analyzed according to the World Health Organization Standards, 5th Edition (Cooper et al., 2010). Hematoxylin and eosin (H&E) staining was used to assess sperm morphology. Multiple head and flagellar malformations, including round cells, isolated tails, abnormal tails, large-headed spermatozoa, and multi-tails, were assessed (Ben Khelifa et al., 2014). The maturity of sperm nuclei was assessed via acidic aniline staining. At least 100 spermatozoa per subject were examined to determine the percentage of abnormal spermatozoa.
Transmission electron microscopy
Semen samples from the proband (F3: II-1) and the fertile control were washed and fixed overnight in 2.5% glutaraldehyde (Sigma-Aldrich, Saint Louis, MO, USA) in 0.1 mol l:1 phosphate buffer (pH 7.4), and then in 1.0% osmium tetroxide (OsO4, HalingBio, Shanghai, China) for 2 h. Post-fixation processing was done as previously described (Tu et al., 2021). Finally, ultrathin 80-nm-thick sections were contrasted with uranyl acetate (Chemos, Altdorf, Germany) and lead citrate (Chemos) and were examined using a H7700 Hitachi electron microscope (Hitachi, Tokyo, Japan). Digital images were captured using a MegaView III digital camera (MegaView, Munster, Germany).
Fluorescence in situ hybridization
Two fluorescence probe sets for chromosomes 18 (white)/X(green)/Y(red) and chromosomes 3(red)/8(white)/12(green) were purchased from Abbott-Vysis, Inc. (Downers Grove, IL, USA). FISH analysis for sperm sample was performed as previously described (Cheng et al., 2012). Briefly, the semen from patients and normal control were washed and dropped onto clean microscopic slides, and then fixed by fresh fixative (methanol:glacial acetic acid, 3:1). The sperm chromatin was decondensed by 0.25 mol/l dithiothreitol solution for 8 min. After being dried, the slides were denatured in denaturing solution (70% formamide, 10% 20× sodium citrate buffer, and 20% distilled water) for 5 min. Meanwhile, the probe was incubated at 75°C for 5 min and immediately added to each denatured slide for hybridization overnight. The slides were then washed and counterstained with 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI). The FISH signals were observed using an Olympus BX-51 fluorescence microscope. Images were captured using the VideoTesT-FISH 2.0 software (version 5.0.74.4803, VideoTesT Ltd, Russia).
Cell culture
HEK293T cells were grown in high-glucose DMEM containing 10% FBS (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) at 37°C with 5% CO2. Cells were transiently transfected with plasmids encoding FLAG- or MYC-tagged cDNA constructs using the Lipofectamine 3000 reagent (Thermo Fisher Scientific) based on the manufacturer’s instructions. The cells were harvested after 48 h of incubation and homogenized using the NP40 lysis buffer (P0013F, Beyotime Biotechnology, China) supplemented with a protease inhibitor cocktail (Thermo Fisher Scientific). The lysates were centrifuged (12 000g, 10 min at 4°C) and subsequently used for immunoblotting analysis.
Immunofluorescence
The HEK293T cells were fixed onto slides with 4% paraformaldehyde for 20 min, permeabilized using 0.5% Triton X-100 (Sigma-Aldrich, T8787) for 10 min, and blocked with 5% bovine serum albumin (Sigma-Aldrich, V900933) for 1 h at 25°C. The slides were sequentially incubated overnight at 4°C with the following primary antibodies: KCTD19 antibody (1:100 dilution, ABclonal, A17274), FLAG antibody (1:100 dilution, Abways, AB0008), MYC antibody (1:100 dilution, Abways, AB0001), and SPACA1 antibody (1:100 dilution, Abcam, ab191843). Then, the slides were incubated with Alexa Fluor 488- or 555-conjugated secondary antibodies (1:400; Thermo Fisher, A21121 and A31572) for 1 h at 37°C. Finally, the slides were stained with DAPI to label the nuclei for 5 min at 25°C. Images of stained cells were obtained using a Leica SP5 II scanning confocal microscope (Leica, Bannockburn, USA).
Co-immunoprecipitation assay
For the detection of KCTD19 and ZFP541 interactions, HEK293T cells co-transfected with FLAG-tagged KCTD19 and MYC-tagged ZFP541 constructs were prepared and lysed with NP-40 Lysis Buffer (#P0013F, Beyotime Biotechnology, China) containing a protease inhibitor cocktail (#P1005, Beyotime Biotechnology, China). The cell lysates were incubated with antibodies overnight at 4°C and cross-linked to Protein G beads (#20399, Thermo Fisher Scientific) for 4 h at 4°C on an orbital shaker using standard methods. After elution and neutralization, the eluents were subjected to western blotting analysis.
Western blotting
Proteins were denatured and separated using the sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were blotted onto polyvinylidene difluoride membranes and incubated overnight at 4°C with anti-KCTD19 antibody (1:1000 dilution, ABclonal, A17274), FLAG antibody (1:2000 dilution, Abways, AB0008), MYC antibody (1:2000 dilution, Abways, AB0001), ubiquitin antibody (1:2000 dilution, Abcam, CY5520), and GAPDH antibody (1:2000; Affinity, AB_2833041). The sections were then incubated with goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibodies for 1.5 h at 25°C and visualized using enhanced chemiluminescence (Pierce). The relative levels of the test proteins based on GAPDH (control) were analyzed using the ImageJ2 software (Madison). The assays were performed in triplicate.
ICSI procedures
Ovarian stimulation was performed on the partner of the affected individuals. Follicular maturation was triggered by HCG (Livzon), and oocyte retrieval was performed 36 h after HCG injection. Oocytes were then subjected to ICSI, and cultured in 6% CO2, 5% O2, and 89% N2 at 37°C in a humidified incubator.
Statistical analysis
Statistical analysis was performed using Student’s t-test or one-way analysis of variance using GraphPad PRISM version 5.01 (GraphPad Software, Inc., La Jolla, CA, USA; RRID: SCR_002798). All experiments were repeated at least three times, and the results are presented as the mean ± SD. Differences were considered significant when the P-value was <0.05 (*), 0.01 (**), or 0.001(***).
Results
Clinical characteristics of the probands
Three affected probands from three unrelated Han Chinese families were diagnosed with primary infertility of unknown cause for more than 2 years. They did not suffer from any diseases related to the reproductive system, and female factors were excluded. No abnormalities were observed in their somatic karyotypes, nor were there Y chromosome deletions, and hormone levels and testicular volume were normal (Table 1). In Family 1, the proband (F1: II-5, 29 years old) was born to first-degree consanguineous parents. Routine semen analyses of the proband revealed normal semen volume, but a highly reduced number of spermatozoa, and no progressive motile sperm were observed (Table 1). In addition, the wives of his eldest brother (F1: II-1, 38 years old) and his second brother (F1: II-4, 33 years old), who had normal fertility, failed to conceive during >8 years of marriage. However, routine semen analyses of his brothers were not available. In Family 2, the proband (F2: II-1) was 35 years old during his first visit to our hospital. Spermatozoa were seldom observed in the semen analyses of F2: II-1 (Table 1), indicating a severe oligospermia phenotype. In Family 3, the proband (F3: II-1, 40 years old) in another consanguineous family also suffered from severe oligoasthenospermia. His sperm concentration and progressive motile sperm were both highly reduced.
Infertility-related examination of affected individuals.
| Item1 | F1: II-5 | F2: II-1 | F3: II-1 | Normal value range | |
|---|---|---|---|---|---|
| Age2 | – | 29 | 35 | 40 | – |
| Endocrine detection | T | 4.8 | 5.7 | 2.85 | 2.5–8.4 ng/ml |
| FSH | 6.5 | 8.6 | 4.76 | 1.5–12.4 mIU/ml | |
| LH | 5.0 | 4.0 | 3.57 | 1.7–8.6 mIU/ml | |
| E2 | 38.0 | 42.0 | 27.06 | 7.6–42.6 pg/ml | |
| Testicular volume | Left | 11 | 12 | 12 | 10–15 ml |
| Right | 11 | 12 | 12 | ||
| Semen analysis3 | Sperm concentration | 6.7 | 0.5 | 1.28 | >15 × 106 (/ml) |
| Semen volume | 4.5 | 2.3 | 2.6 | >1.5 ml | |
| Progressive motility (%) | 0 | 0 | 11.1 | >32 (%) | |
| Karyotype | – | 46,XY | 46,XY | 46,XY | – |
| Y-Chromosome microdeletion | – | No deletion | No deletion | No deletion | – |
| Item1 | F1: II-5 | F2: II-1 | F3: II-1 | Normal value range | |
|---|---|---|---|---|---|
| Age2 | – | 29 | 35 | 40 | – |
| Endocrine detection | T | 4.8 | 5.7 | 2.85 | 2.5–8.4 ng/ml |
| FSH | 6.5 | 8.6 | 4.76 | 1.5–12.4 mIU/ml | |
| LH | 5.0 | 4.0 | 3.57 | 1.7–8.6 mIU/ml | |
| E2 | 38.0 | 42.0 | 27.06 | 7.6–42.6 pg/ml | |
| Testicular volume | Left | 11 | 12 | 12 | 10–15 ml |
| Right | 11 | 12 | 12 | ||
| Semen analysis3 | Sperm concentration | 6.7 | 0.5 | 1.28 | >15 × 106 (/ml) |
| Semen volume | 4.5 | 2.3 | 2.6 | >1.5 ml | |
| Progressive motility (%) | 0 | 0 | 11.1 | >32 (%) | |
| Karyotype | – | 46,XY | 46,XY | 46,XY | – |
| Y-Chromosome microdeletion | – | No deletion | No deletion | No deletion | – |
T, testosterone; FSH, follicle-stimulating hormone; LH, luteinizing hormone; E2, estradiol.
Infertility-related examinations were performed when the affected individuals (F1: II-5, F2: II-1, and F3: II-1) were 29, 35, and 40 years of age, respectively.
Semen parameters were evaluated according to the WHO Laboratory Manual for the Examination and Processing of Human Semen (First Edition, 2010) (Sanchez-Alvarez et al., 2012).
Infertility-related examination of affected individuals.
| Item1 | F1: II-5 | F2: II-1 | F3: II-1 | Normal value range | |
|---|---|---|---|---|---|
| Age2 | – | 29 | 35 | 40 | – |
| Endocrine detection | T | 4.8 | 5.7 | 2.85 | 2.5–8.4 ng/ml |
| FSH | 6.5 | 8.6 | 4.76 | 1.5–12.4 mIU/ml | |
| LH | 5.0 | 4.0 | 3.57 | 1.7–8.6 mIU/ml | |
| E2 | 38.0 | 42.0 | 27.06 | 7.6–42.6 pg/ml | |
| Testicular volume | Left | 11 | 12 | 12 | 10–15 ml |
| Right | 11 | 12 | 12 | ||
| Semen analysis3 | Sperm concentration | 6.7 | 0.5 | 1.28 | >15 × 106 (/ml) |
| Semen volume | 4.5 | 2.3 | 2.6 | >1.5 ml | |
| Progressive motility (%) | 0 | 0 | 11.1 | >32 (%) | |
| Karyotype | – | 46,XY | 46,XY | 46,XY | – |
| Y-Chromosome microdeletion | – | No deletion | No deletion | No deletion | – |
| Item1 | F1: II-5 | F2: II-1 | F3: II-1 | Normal value range | |
|---|---|---|---|---|---|
| Age2 | – | 29 | 35 | 40 | – |
| Endocrine detection | T | 4.8 | 5.7 | 2.85 | 2.5–8.4 ng/ml |
| FSH | 6.5 | 8.6 | 4.76 | 1.5–12.4 mIU/ml | |
| LH | 5.0 | 4.0 | 3.57 | 1.7–8.6 mIU/ml | |
| E2 | 38.0 | 42.0 | 27.06 | 7.6–42.6 pg/ml | |
| Testicular volume | Left | 11 | 12 | 12 | 10–15 ml |
| Right | 11 | 12 | 12 | ||
| Semen analysis3 | Sperm concentration | 6.7 | 0.5 | 1.28 | >15 × 106 (/ml) |
| Semen volume | 4.5 | 2.3 | 2.6 | >1.5 ml | |
| Progressive motility (%) | 0 | 0 | 11.1 | >32 (%) | |
| Karyotype | – | 46,XY | 46,XY | 46,XY | – |
| Y-Chromosome microdeletion | – | No deletion | No deletion | No deletion | – |
T, testosterone; FSH, follicle-stimulating hormone; LH, luteinizing hormone; E2, estradiol.
Infertility-related examinations were performed when the affected individuals (F1: II-5, F2: II-1, and F3: II-1) were 29, 35, and 40 years of age, respectively.
Semen parameters were evaluated according to the WHO Laboratory Manual for the Examination and Processing of Human Semen (First Edition, 2010) (Sanchez-Alvarez et al., 2012).
In Family 1, the couple (F1: II-5) accepted ICSI treatment in a local hospital (the medical records were lost) but had failed ICSI outcomes.
In Family 2, four ICSI cycles were implemented for the proband (F2: II-1) and his wife (25 years old), including two cycles using his sperm and two cycles using donor sperm (Supplementary Table S1). In the first ICSI attempt, 14 metaphase II oocytes were used for fertilization. The 2PN fertilization and eight-cell embryo rates were 42.8% (6/14) and 0% (0/14), respectively. Two 4BC embryos were transferred simultaneously, but implantation failed for an unknown reason. In the second ICSI cycle attempt, eight metaphase II oocytes were used for fertilization in cooperation with oocyte activation treatment, but only one developed into the eight-cell stage (12.5%). Two embryos were cryopreserved, and two cleavage embryos were chosen for the first transfer, but no implantation was achieved. The two cryopreserved embryos were successively used for blastocyst culture. Finally, two 4BC embryos were transplanted in two separate treatments: one resulted in a biochemical pregnancy and with the other, there was no implantation. After consecutive implantation failures, donor sperm were used in the following two ICSI cycles. Specifically, one cleavage embryo and one high-quality blastocyst were transplanted during the fourth ICSI cycle, and the affected individual’s wife became pregnant and delivered a healthy baby.
In Family 3, the proband (F3: II-1) and his wife received two ICSI cycles. During the first ICSI cycle (performed in local hospital), three transfers were carried out, but only one implanted then ended with miscarriage. During the second ICSI cycle, two cleavage embryos and one 4BC blastocyst were transferred in two separate treatments, but all failed to implant and no clinical pregnancy was achieved.
The three probands had abnormal aneuploid spermatozoa and highly uncondensed sperm nuclear chromatin
To investigate the sperm phenotypes of the three probands, H&E staining of the ejaculated sperm from F1: II-5, F2: II-1, and F3: II-1 was performed. This revealed a highly rates of sperm morphology abnormalities (97.4% for F1: II-5, 99.1% for F2: II-1, and 97.5% for F3: II-1) in both the sperm heads and flagella of all sperm samples compared to that of the normal control, suggesting an oligoasthenoteratozoospermia phenotype (Fig. 1A;Supplementary Fig. S1A). Specifically, some spermatozoa (29.2% for F1: II-5, 30.4% for F2: II-1, and 42.4% for F3: II-1) were large-headed in these probands (Fig. 1A;Supplementary Table S2). Spermatozoa with several flagella were also frequently observed (8.9% for F1: II-5, 8.7% for F2: II-1, and 7.5% for F3: II-1), which was strongly indicative of sperm polyploidy. Moreover, we observed numerous tailless unidentifiable cells and termed them as round cells, which were especially high in the semen sample of F1: II-5 (32.7%) (Supplementary Table S2). As frequent SPACA1 (an acrosome-related protein) expression were detected in these round cells, we speculate that most of these round cells might belong to immature spermatids (Supplementary Fig. S1B). Subsequent sperm-FISH analysis using two probe sets for chromosomes 3/8/12, and chromosomes 18/X/Y revealed a significantly higher rate of aneuploidy (such as 3/8,8/12,12; 8/12; 3/8/12,12,12,12; 18/XY; 18/YY; 18/XXY) in the spermatozoa from men with biallelic KCTD19 variants (Fig. 1B) than that from a normal control (79.6 ± 7.3% versus 0.15 ± 0.07% for chromosomes 3/8/12, 76.7 ± 10.5% versus 0.25 ± 0.2% for chromosomes 18/X/Y, P < 0.01; n > 100 spermatozoa for each individual in each experiment), implying a disorder in chromosome segregation during meiosis I and/or meiosis II. Moreover, these tailless cells and large-headed sperm were more efficiently stained with acidic aniline (98.0% for F1: II-5, 99.0% for F2: II-1, and 97.0% for F3: II-1, Fig. 1C), a reagent that specifically binds to the lysine-rich residues of sperm histones. This result indicates that the exchange of nuclear histones with protamine is inefficient, and the sperm chromatin is highly uncondensed in the probands, which could explain the large heads of the gametes. TEM analysis was performed to further examine the ultrastructural structure defects of KCTD19 mutant spermatozoa (Chatzimeletiou et al., 2022). The results revealed that the majority of the spermatozoa (84.4% for F3: II-1) showed less condensed chromatin with granular formation inside the nucleus (Fig. 1D). The malformed sperm head was wrapped with redundant cytoplasm and disorganized flagella components (Fig. 1D). Together, these observations indicated defective nuclear haploidy and chromatin compaction in these two probands.
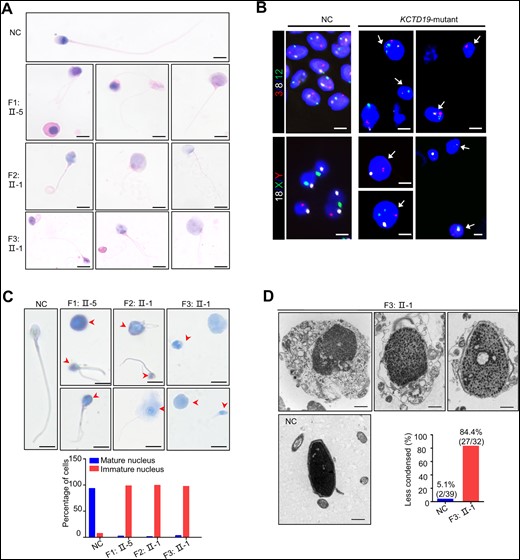
Sperm morphology for the normal control and patients with biallelic KCTD19 variants. (A) H&E staining analysis of spermatozoa from a fertile individual (normal control, NC) and the three probands with biallelic KCTD19 variants. Scale bars = 5 μm. (B) Fluorescence in situ hydriization (FISH) analysis using probe sets for chromosomes 3 (red)/8 (white)/12 (green) and chromosomes 18 (white)/X (green)/Y (red) in spermatozoa from the NC and three probands. Sperm nuclei stained with DAPI (blue). The sperm aneuploidy rates for chromosomes 3/8/12 and chromosomes 18/X/Y are 79.6% ± 7.3% and 76.7% ± 10.5%, respectively, in the probands, compared with 0.15% ± 0.07% and 0.25% ± 0.2%, respectively, in the NC. Representative FISH images of the probands are shown. The white arrows indicate aneuploid sperm. Scale bars = 5 μm. (C) Acidic aniline staining to detect the maturity of epididymal sperm nuclei of the NC and the three probands. Immature nuclei with increased histones are dyed blue or violet (arrowheads, up). The histograms (down) indicate the percentage of the mature and immature nucleus observed in males with biallelic KCTD19 variants. Scale bars = 5 μm. (D) Transmission electron microscopy analysis of spermatozoa from the NC and one proband (F3: II-1) with biallelic KCTD19 variants. The sperm heads from the proband showed abnormal morphology and were frequently embraced by residual cytoplasm containing unassembled flagellar components. Notably, the nuclear chromatin was granular and less condensed. The histograms indicate the percentage of the less condensed chromatin observed in man with biallelic KCTD19 variant. Scale bars = 1 μm.
Identification of homozygous KCTD19 variants in the three affected probands
WES was performed for 536 infertile males with idiopathic oligozoospermia to uncover the underlying genetic causes, including the three probands (F1: II-5, F2: II-1, and F3: II-1) with uncondensed sperm nuclear chromatin. For F1: II-5 from a consanguineous family, homozygous regions were further established using homozygosity analysis (Supplementary Fig. S2). After a stringent mutation screening strategy described in our previous studies (Tan et al., 2019; Tu et al., 2020), three homozygous missense variants in KCTD19 were identified in the three unrelated probands (Fig. 2A and Table 2). These KCTD19 variants were confirmed via Sanger sequencing. One homozygous KCTD19 missense variant (NM_001100915: c.G628A, p.E210K; M1) was identified in F1: II-1, F1: II-4, and F1: II-5. Two other homozygous KCTD19 missense variants, c.C893T:p.P298L (M2) and c.G2309A:p.G770D (M3), were identified in F2: II-1 and F3: II-1, respectively (Fig. 2A). We failed to verify the three variants in family members because the samples were unavailable. Specifically, the identified KCTD19 M1 and M2 variants were located at highly conserved sites in the BTB_POZ superfamily (Fig. 2B). All identified missense KCTD19 variants were absent or rare in public human databases, including the 1000 Genomes Project and gnomAD (v.2.1.1, 141 456 samples) and were predicted to be deleterious by multiple bioinformatics software (including SIFT, Polyphen2, Mutation Taster, and CADD) (Table 2). KCTD19 has been established as a meiosis-related gene based on evidence from a mouse model (Li et al., 2022), therefore, we speculate that the identified homozygous KCTD19 variants could be responsible for the observed infertility phenotypes.

Identification of three homozygous KCTD19 missense variants in three unrelated Chinese families. (A) Pedigrees of the three unrelated Han Chinese families. Solid symbols indicate the infertile males. Whole exome sequencing was performed for members indicated by the black arrows. Sanger sequencing chromatograms of KCTD19 variants are shown below the pedigrees and the red arrows indicate the variant positions. (B) Schematic representation of the protein domains of KCTD19. Variant positions were indicated. Sequence alignment shows the conservation of the mutated residues among different species. BTB, Broad-Complex, Tram track, and Bric-a-brac; POZ, poxvirus and zinc finger.
KCTD19 variants identified by WES and bioinformatic prediction.
| Sample ID | Gene | RefSeq ID | Location | Base change: Amino Acid Alteration | Type | Min depth | Function | dbSNP ID | 1000G | gnomAD | gnomAD East Asians | SIFT | Polyphen2 | Mutation taster | CADD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1: II-5 | KCTD19 | NM_001100915 | Chr16: 67337064 | c.G628A: p.E210K | Homozygous | 56 | Missense | NA | NA | NA | NA | D | D | D | 33 |
| F2: II-1 | KCTD19 | NM_001100915 | Chr16: 67333359 | c.C893T: p.P298L | Homozygous | 250 | Missense | NA | NA | 0.00001603 | NA | D | D | D | 33 |
| F3: II-1 | KCTD19 | NM_001100915 | Chr16: 67325650 | c.G2309A: p.G770D | Homozygous | 150 | Missense | NA | NA | NA | NA | D | D | D | 28 |
| Sample ID | Gene | RefSeq ID | Location | Base change: Amino Acid Alteration | Type | Min depth | Function | dbSNP ID | 1000G | gnomAD | gnomAD East Asians | SIFT | Polyphen2 | Mutation taster | CADD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1: II-5 | KCTD19 | NM_001100915 | Chr16: 67337064 | c.G628A: p.E210K | Homozygous | 56 | Missense | NA | NA | NA | NA | D | D | D | 33 |
| F2: II-1 | KCTD19 | NM_001100915 | Chr16: 67333359 | c.C893T: p.P298L | Homozygous | 250 | Missense | NA | NA | 0.00001603 | NA | D | D | D | 33 |
| F3: II-1 | KCTD19 | NM_001100915 | Chr16: 67325650 | c.G2309A: p.G770D | Homozygous | 150 | Missense | NA | NA | NA | NA | D | D | D | 28 |
The human genome assembly version GRCh37/hg19 was used in this study. The variant is considered deleterious when its CADD score >4. NA, not available; 1000G, 1000 Genomes project; D, damaging/disease causing.
KCTD19 variants identified by WES and bioinformatic prediction.
| Sample ID | Gene | RefSeq ID | Location | Base change: Amino Acid Alteration | Type | Min depth | Function | dbSNP ID | 1000G | gnomAD | gnomAD East Asians | SIFT | Polyphen2 | Mutation taster | CADD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1: II-5 | KCTD19 | NM_001100915 | Chr16: 67337064 | c.G628A: p.E210K | Homozygous | 56 | Missense | NA | NA | NA | NA | D | D | D | 33 |
| F2: II-1 | KCTD19 | NM_001100915 | Chr16: 67333359 | c.C893T: p.P298L | Homozygous | 250 | Missense | NA | NA | 0.00001603 | NA | D | D | D | 33 |
| F3: II-1 | KCTD19 | NM_001100915 | Chr16: 67325650 | c.G2309A: p.G770D | Homozygous | 150 | Missense | NA | NA | NA | NA | D | D | D | 28 |
| Sample ID | Gene | RefSeq ID | Location | Base change: Amino Acid Alteration | Type | Min depth | Function | dbSNP ID | 1000G | gnomAD | gnomAD East Asians | SIFT | Polyphen2 | Mutation taster | CADD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1: II-5 | KCTD19 | NM_001100915 | Chr16: 67337064 | c.G628A: p.E210K | Homozygous | 56 | Missense | NA | NA | NA | NA | D | D | D | 33 |
| F2: II-1 | KCTD19 | NM_001100915 | Chr16: 67333359 | c.C893T: p.P298L | Homozygous | 250 | Missense | NA | NA | 0.00001603 | NA | D | D | D | 33 |
| F3: II-1 | KCTD19 | NM_001100915 | Chr16: 67325650 | c.G2309A: p.G770D | Homozygous | 150 | Missense | NA | NA | NA | NA | D | D | D | 28 |
The human genome assembly version GRCh37/hg19 was used in this study. The variant is considered deleterious when its CADD score >4. NA, not available; 1000G, 1000 Genomes project; D, damaging/disease causing.
Mutant KCTD19 led to reduced protein stability and increased degradation via ubiquitination in vitro
KCTD19 has four BTB_POZ domains/superfamily members, which can mediate versatile protein–protein interactions that facilitate homodimerization or heterodimerization (Oura et al., 2021). The state of dimerization may be important for offering specificity to certain substrates while minimizing interactions with other unwanted proteins. To assess the impact of these three KCTD19 variants, we modeled the 3D structure of KCTD19 using the AlphaFold database. The c.G628A variant identified in F1: II-1, F1: II-4, and F1: II-5 is predicted to replace an acidic glutamic acid (E210) with a basic lysine (210K), which could disrupt one hydrogen bond between E210 and S209 within the α-helix of the first KCTD19 BTB_POZ superfamily (Fig. 3A). Therefore, this alteration could increase the instability of KCTD19 and result in its degradation. The c.C893T variant identified in F2: II-1 was predicted to replace an imino heterocyclic proline (P298) with an aliphatic leucine (298L) (Fig. 3B). P298 is at the connecting region of two helixes in the second KCTD19 BTB_POZ superfamily, and the heterocyclic property of proline could terminate the extension of the α-helix, which maintains a specific protein conformation. This alteration could change the spatial structure of KCTD19, leading to impaired protein stability or protein–protein interactions. The c.G2309A variant identified in F3: II-1 was predicted to replace a hydrophobic neutral glycine (G770) with a hydrophilic acidic aspartic acid (770D) (Fig. 3C), which might increase protein solubility and also result in protein instability.
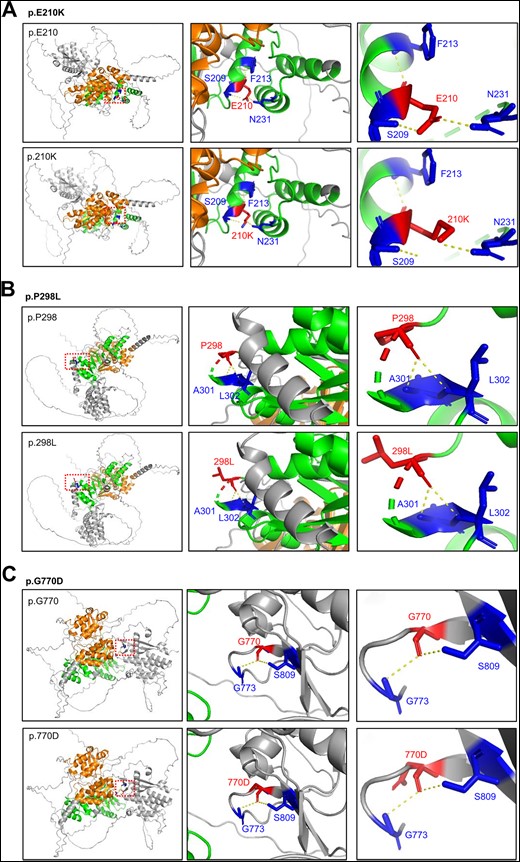
Three-dimensional (3D) structural model prediction of three KCTD19 variants. The 3D structural model of wild type KCTD19 was obtained from the AlphaFold Database (https://alphafold.ebi.ac.uk/) and was analyzed using the PyMol software. Modeling of p.E210K (A), p.P298L (B), and p.G770D (C) mutant KCTD19 proteins. Mutated residues and the corresponding residues connected through hydrogen bonding are shown in red and blue, respectively. The hydrogen bonds are denoted by the yellow dotted lines.
To experimentally evaluate the functional consequences of the three KCTD19 variants found in the five patients, we constructed WT and mutant KCTD19 plasmids, including c.G628A (p.E210K, M1), c.C893T (p.P298L, M2), c.G2309A (p.G770D, M3), and transfected them into HEK293T cells. Immunofluorescence (IF) analysis showed that both WT KCTD19 and KCTD19 mutants were mainly located in the cytoplasm of HEK293T cells (Supplementary Fig. S3). However, the fluorescence signal of these KCTD19 mutants was lower than that of the WT alleles (Supplementary Fig. S3), and the decreased expression levels of these three variants were further verified using western blot assays (Fig. 4A). To determine whether the reduced protein expression was caused by ubiquitination, we co-expressed KCTD19-3×Flag and Ubiquitin-HA plasmids into HEK293T cells. We found that KCTD19 mutants led to a highly increased Ub-KCTD19 conjugate (Fig. 4B), indicating that these three variants may accelerate KCTD19 degradation through the ubiquitination pathway in HEK293T cells. Taken together, these results reveal that the three homozygous variants of KCTD19 could have detrimental effects on the expression levels of KCTD19, thus disrupting its normal function during spermatogenesis.
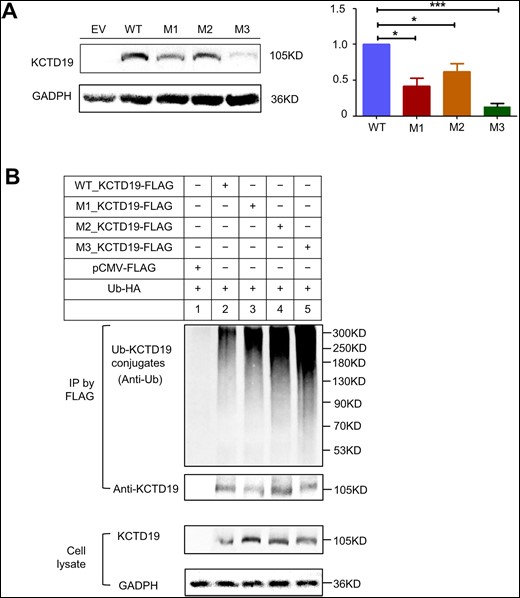
Impact of KCTD19 mutations on protein stability and protein degradation by ubiquitination in vitro. (A) Immunoblotting (IB) analysis revealing the effects of the mutations on KCTD19 protein expression levels in HEK293T cells transfected with wild type (WT) or mutant (M) plasmids. WT, M1, M2, and M3 represent plasmids encoding WT KCTD19-3×FLAG, p.E210K_KCTD19-3×FLAG, p.P298L_KCTD19-3×FLAG, and p.G770D_KCTD19-3×FLAG, respectively. The histograms below indicate the relative expression levels of KCTD19. The data are shown as means and standard errors of the mean. *P < 0.05, ***P < 0.001. (B) Detection of KCTD19 ubiquitination in HEK293T cells cotransfected with WT/mutant KCTD19-3×Flag and pCMV-Ubiquitin (Ub)-HA. Top: anti-Flag immunoprecipitation (IP) pellets immunoblotted with anti-Ub for Ub-KCTD19 conjugates (upper), or anti-KCTD19 to normalize the loading amounts (lower). Bottom: IB of KCTD19 protein levels in respective cell lysates, with GAPDH serving as a loading control.
Mutant KCTD19 impaired the nuclear colocalization between KCTD19 and ZFP541 in vitro
Recent studies have revealed that KCTD19 forms a complex with ZFP541 to ensure the progression of male meiosis prophase by regulating the transcriptional activity of meiotic genes (Oura et al., 2021; Li et al., 2022). We then analyzed the interaction between mutated KCTD19 and ZFP541 through co-IP experiments in co-transfected HEK293T cells. We found that the mutated KCTD19 still interacted with ZFP541; however, the ZFP541 interaction strength for KCTD19 p.E210K and p.P298L seemed to be weakened (Fig. 5A). Next, we studied the KCTD19-ZFP541 colocalization in HEK293T cells. We found that when cells were co-transfected with WT KCTD19 and ZFP541, nuclear KCTD19 expression was highly increased and showed strong colocalization with ZFP541 (38.7% ± 8.1%, n = 100 co-expressed cells in each experiment), implying that the KCTD19-ZFP541 complex may play a role in the nuclear localization of KCTD19 (Fig. 5B). However, the mutated KCTD19 (p.E210K, p.P298L, and p.G770D) exhibited significantly reduced nuclear colocalization with ZFP541 compared with WT KCTD19 (11.7% ± 6.0%, 9.3% ± 6.1%, and 12.0% ± 5.0%, respectively, P < 0.05, n = 100 co-expressed cells in each experiment, two-tailed t-test). Moreover, the cytoplasmic translocation of ZFP541 was elevated when ZFP541 was co-transfected with mutated KCTD19 (Fig. 5B). These results indicate that KCTD19 variants could disturb the normal nuclear localization of KCTD19 and ZFP541 mediated by the KCTD19-ZFP541 complex. Further investigation is needed to uncover the exact effects that these three KCTD19 variants exert during spermatogenesis.
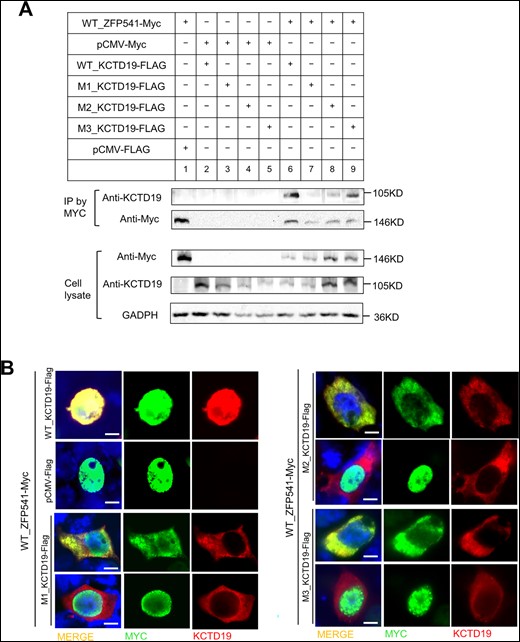
Impact of KCTD19 mutations on protein–protein interaction in vitro. (A) Co-immunoprecipitation assays of the effects of KCTD19 mutations on the binding of full-length KCTD19 to ZFP541 in co-transfected HEK293T cells. (B) Immunofluorescence assay for the expression of wild type (WT) or mutated KCTD19 and ZFP541 in cotransfected HEK293T cells through antibodies against MYC (green) and KCTD19 (red). Scale bars = 5 μm.
Discussion
In this study, we report three rare homozygous KCTD19 missense variants in five infertile males from three unrelated Chinese families. The three probands exhibited an oligoasthenoteratozoospermia phenotype, with the majority of spermatozoa presenting with uncondensed nuclear content and/or nuclear aneuploidy. These three KCTD19 variants reduced the abundance of KCTD19 and its nuclear colocalization with its functional partner, ZFP541, in HEK293T cells. Our observations, together with previously reported evidence in homozygous Kctd19 mutant mice (Oura et al., 2021), collectively support the critical role of KCTD19 in human male infertility with an oligoasthenoteratozoospermia phenotype.
In mice, KCTD19 is a key transcriptional regulator that is essential for mouse meiotic prophase progression (Horisawa-Takada et al., 2021). Kctd19 knockout male mice show an azoospermia phenotype due to spermatocyte meiosis metaphase I arrest, characterized by chromosome misalignment (Oura et al., 2021). However, in our study, patients harboring homozygous KCTD19 missense variants manifested oligozoospermia or severe oligozoospermia, but not azoospermia. Similar phenotypic differences varying from oligozoospermia to azoospermia have also been frequently observed in several mutations of meiotic genes, such as M1AP (Arango et al., 2013; Wyrwoll et al., 2020), RPL10L (Jiang et al., 2017; Tu et al., 2020), and SYCP1 (Nabi et al., 2022). Notably, our in vitro experiments using HEK293T cells indicated reduced protein expression of these KCTD19 variants due to accelerated ubiquitination-mediated degradation. KCTD19 is required for the progression of meiosis I prophase by cooperating with ZFP541 and other proteins in mice (Oura et al., 2021; Li et al., 2022). The KCTD19 M1 and M2 within the BTB_POZ superfamily might impair the nuclear colocalization of KCTD19 and ZFP541 by disrupting their interaction, indicating that the BTB_POZ superfamily may be important for the protein–protein interaction. Although KCTD19 M3 near the C-terminus is not located at critical domains, the related glycine at site 770 is highly conserved across species and M3 seemed to cause even worse protein degradation than M1 and M2, indicating a potential role of this region for maintaining protein stability. In addition, KCTD19 may fulfill its function through self-dimerization; thus, these variants may alternatively affect the activity of KCTD19 by disturbing its self-dimerization.
Moreover, our observations also suggest that these KCTD19 mutants retained partial function and resulted in partial arrest during meiotic I metaphase. A few germ cells seem to escape this meiotic metaphase I checkpoint and proceed to differentiate into mature spermatozoa, which might be the cause of the severe oligozoospermia. However, as KCTD19 was dysfunctional, a delay of metaphase entry occurred and chromosomes misaligned at the metaphase plate, leading to stochastic chromosome-segregation mistakes during meiosis I and II, producing mostly aneuploid spermatozoa. For example, successive segregation errors of homologues during meiosis I and sister chromatids during meiosis II can be reflected by trisomic/tetrasomic spermatozoa (such as spermatozoa showing 18/XXY or 3/8/12,12,12,12 in our study), while one segregation error in either meiosis I (detected as XY) or meiosis II (detected as XX or YY) might happen and produce disomic spermatozoa (Chatzimeletiou et al., 2011). Moreover, nullisomic spermatozoa (such as spermatozoa showing 18 or 8/12 in our study) could result from the non-disjunction at either the mitotic stages of diploid spermatogonia or the meiotic stages of spermatocytes (Mateizel et al., 2002). Another prominent feature of the surviving spermatozoa was the large sperm heads with highly uncondensed nuclear chromatin, as was demonstrated by TEM analysis. In concert with this, acidic aniline staining of sperm showed a high percentage of histones in these nuclei, suggesting the presence of greatly abnormal nucleosomic structures when KCTD19 was dysfunctional. During spermatogenesis, the sperm undergo a series of specific epigenomic modifications to ensure proper chromatin condensation (Gunes and Esteves, 2021). It is still unclear whether KCTD19 could participate in nuclear condensation during sperm maturation through regulating epigenetic process, and further investigations are needed.
Overall, in this study, we found that the sperm from patients with KCTD19 mutations presented a high rate of chromosome aneuploidy and defective chromatin compaction, exhibiting large sperm heads and flagellar malformations in appearance. To date, the large-head sperm phenotype has only been referred to for two genes in humans: aurora kinase C (AURKC) and nucleoporin-210-like (NUP210L) (Dieterich et al., 2007; Arafah et al., 2021). AURKC mutations have been considered to be responsible for most macrozoospermia cases, and the affected patients had large-headed multiflagellar polyploid spermatozoa caused by a failure in chromosome segregation and cytokinesis, with normal or low sperm counts (Dieterich et al., 2007). Multiflagellar spermatozoa were also frequently seen in KCTD19 mutant spermatozoa. Patients with NUP210L mutations exhibited abnormally large-headed spermatozoa with extremely uncondensed nuclear chromatin and absent acrosomes, possibly resulting from the failure to adequately replace core histones by protamines (Arafah et al., 2021). The observed sperm defects described here are different from other specific morphological defects, such as globozoospermia, acephalic spermatozoa, and multiple morphological anomalies of the flagella (Harbuz et al., 2011; Koscinski et al., 2011; Shang et al., 2018; Zhu et al., 2018; Wang et al., 2020; Tu et al., 2021), for which the disease-causing genes are mainly functionally related to spermiogenesis. Therefore, enlarged spermatozoa with an uncondensed nuclear content and/or an aneuploidy observed in patients with biallelic KCTD19, AURKC, or NUP210L variants may represent a common sperm defect phenotype, and genetic factors should be considered as the underlying etiology.
ICSI has become the most common fertilization method in patients with oligoasthenozoospermia. A previous study showed that patients with oligoasthenozoospermia resulting from meiotic abnormalities had comparable rates of fertilization, pregnancy, implantation, and miscarriage to oligoasthenozoospermia patients with normal meiosis (Aran et al., 2003). However, all ICSI treatments using spermatozoa from patients with biallelic KCTD19 variants failed. Remarkably, unsatisfactory ICSI outcomes have also been reported in patients with AURKC and NUP210L mutations (Ben Khelifa et al., 2011; Arafah et al., 2021). We retrospectively analyzed the ICSI outcome of a severe oligozoospermia patient with one homozygous mutation in RPL10L and found that his ICSI treatments also failed (Tu et al., 2020). Collectively, these findings reinforce the fact that ICSI should not be recommended in oligozoospermia patients with genetic pathogenesis of meiotic arrest. However, owing to the small sample size, we could not completely rule out other potential alterations in the patients’ genomes or epigenomes that may contribute to the impaired semen parameters and poor embryonic development (Gunes and Esteves, 2021). Further investigations in a larger cohort should be performed to evaluate the ICSI outcome of oligozoospermia caused by meiotic gene mutations. Such efforts would provide important clinical implications for genetic counseling and personalized reproductive treatment for affected families.
In conclusion, our study describes a specific oligoasthenoteratozoospermia phenotype characterized by large-headed spermatozoa with aberrant nuclear contents. Our genetic analyses revealed that this anomaly is likely caused by homozygous variants of KCTD19, a transcriptional regulator that is essential for meiotic prophase progression. Further investigations using Kctd19 knock-in mouse models may help elucidate the pathologic role of these three missense mutations and the molecular mechanisms underlying the function of KCTD19 in spermatogenesis.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
Acknowledgements
The authors thank all the affected individuals and their families for participating in and supporting this study.
Authors’ roles
Q.Z., Y.-Q.T., and C.T. conceived and designed the study. L.M., C.T., J.D., H.Z., and D.C. were involved in the patients care and evaluation. W.W., L.S., J.H., and D.Y. performed the experiments and analyzed the data. L.M. and C.T. supplied bioinformatics support. G.L. and G.L. participated in the project supervision. W.W. wrote the manuscript. Q.Z., Y.-Q.T., and C.T. critically reviewed the manuscript and all authors read and approved the final manuscript.
Funding
This work was supported by the National Key Research and Developmental Program of China (2022YFC2702604 to Y.-Q.T.), the National Natural Science Foundation of China (81971447 and 82171608 to Y.-Q.T., 82101961 to C.F.T.), a key grant from the Prevention and Treatment of Birth Defects from Hunan Province (2019SK1012 to Y.-Q.T.), a Hunan Provincial Grant for Innovative Province Construction (2019SK4012), and the China Postdoctoral Science Foundation (2022M721124 to W.L.W.).
Conflict of interest
The authors declare no conflicts of interest.
References
Author notes
Weili Wang and Lilan Su contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}