





Abstract
How much residual cryoprotectant remains in thawed/warmed ovarian tissues after slow freezing or vitrification?
After thawing/warming, at least 60 min of diffusion washing in media was necessary to significantly reduce the residual cryoprotectants in ovarian tissues frozen by slow freezing or vitrification.
Ovarian tissue cryopreservation (OTC) by slow freezing has been the conventional method; while the vitrification method has gained popularity for its practicality. The main concern about vitrification is how much potentially toxic residual cryoprotectant remains in the warmed tissues at the time of transplantation.
This was an animal study using the ovarian tissues from 20 bovine ovaries. The duration of this study was from 2018 to 2020.
Ovarian cortex tissues were prepared from 20 bovine ovaries and assigned randomly to groups of fresh (non-frozen) control, slow freezing with 1.5 M dimethyl sulfoxide (DMSO), 1.5 M 1,2-propanediol (PROH) and vitrification with 35% ethylene glycol (EG). The residual cryoprotectant concentrations in thawed/warmed tissues were measured by gas chromatography at the following time points: frozen (before thawing/warming), 0 min (immediately after thawing/warming), 30, 60 and 120 min after diffusion washing in media. Next, the ultrastructural changes of primordial follicles, granulosa cells, organelles and stromal cells in the ovarian tissues (1 mm × 1 mm × 1 mm) were examined in fresh (non-frozen) control, slow freezing with DMSO or PROH and vitrification with EG groups. Real-time quantitative PCR was carried out to examine the expressions of poly (ADP-ribose) polymerase-1 (PARP1), a DNA damage sensor and caspase-3 (CASP3), an apoptosis precursor, in thawed/warmed ovarian tissues that were washed for either 0 or 120 min and subsequently in tissues that were ex vivo cultured for 24 or 48 h. The same set of tissues were also used to analyze the protein expressions of gamma H2A histone family member X (γH2AX) for DNA double-strand breaks and activated caspase-3 (AC3) for apoptosis by immunohistochemistry.
The residual cryoprotectant concentrations decreased with the extension of diffusion washing time. After 60 min washing, the differences of residual cryoprotectant between DMSO, PROH and EG were negligible (P > 0.05). This washing did not affect the tissue integrity or significantly elevate the percentage of AC3 and γH2AX positive cells, indicating that tissues are safe and of good quality for transplantation.
N/A.
Since the study was performed with ovarian tissues from bovines, generalizability to humans may be limited. Potential changes in ovarian tissue beyond 120 min were not investigated.
This study addresses concerns about the cytotoxicity of EG in warmed ovarian tissues and could provide insights when devising a standard vitrification protocol for OTC.
The study was funded by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science to N.S.
Introduction
The survival rate of child, adolescent and young adult cancer patients continues to improve each year owing to the advancement in anticancer therapies in the past decade. However, anticancer therapies damage the ovaries which ultimately leads to premature ovarian failure and reduces the reproductive potential of young cancer patients (Gargus et al., 2018). The timely referral to consultations regarding the options to preserve fertility before undergoing gonadotoxic treatments is critical.
Several options for cryopreservation, namely oocytes, embryos and/or ovarian tissue are available to women whose fertility is threatened by oncological or non-oncological indications. Parameters, such as patient’s age, disease indications, marital status and ethical views, should be taken into consideration when choosing the most suitable fertility preservation strategy (Meirow and Nugent, 2001). Cryopreservation of mature oocytes and embryos are clinically established fertility preservation methods for reproductive women who have adequate time for ovarian stimulation and egg retrieval (Taylan and Oktay, 2017). Studies have shown superior fertilization and pregnancy rate in mature oocytes and embryos that are cryopreserved by vitrification (Kuwayama et al., 2005; Stehlik et al., 2005; Edgar and Gook, 2012; Anderson et al., 2020).
For pre-pubertal girls and women who require immediate start of cancer therapy, ovarian tissue cryopreservation (OTC) is the only viable option to preserve their fertility potential (Donnez and Dolmans, 2017; ASRM, 2019). OTC can be performed using slow freezing or vitrification. In 2004, Donnez et al. (2004) reported the first live birth after OTC by slow freezing and ovarian tissue transplantation (OTT). So far, the slow freezing method has resulted in over 200 live births after transplantation of frozen-thawed ovarian tissues (Dolmans et al., 2021). On the other hand, OTC by vitrification was developed later and so far, only five live births have resulted from transplantation of vitrified/warmed ovarian tissues. Vitrification is a rapid and simple technique which does not require expensive equipment. Keros et al. (2009) conducted a systematic comparison of different slow freezing and vitrification protocols using ovarian tissues obtained from the same patients. Their data revealed that follicle preservation was similar among the methods, but under electron microscopy the morphological integrity of ovarian stroma was better preserved after vitrification. In 2017, Shi et al. (2017) found no significant difference between slow freezing and vitrification in terms of proportion of the intact primordial follicles and its density in ovarian tissue, but vitrification was associated with significantly less DNA damage. The number of stromal cells was significantly higher after vitrification (Shi et al., 2017). Amorim et al. (2011) also compared the two methods and concluded that the optimal method to cryopreserve the ovarian tissue has yet to be established because most studies showed discrepant outcomes. Amorim’s group proposed that discrepant results were due to the differences in each study’s parameters such as animal species, sizes of ovarian tissue, cryoprotectants, equilibration time, carrier devices, thawing methods and evaluation methods (Amorim et al., 2011).
Given the limitations with using human ovarian tissues, we chose bovine ovaries as the animal model to investigate the residual cryoprotectant levels and its toxicity in thawed/warmed ovarian tissues. The bovine ovaries share profound similarities to those of humans in size, texture and composition, as well as comparable dynamics of follicle developments (Newton et al., 1999; Oktay and Karlikaya, 2000; Baerwald et al., 2009, 2012). Amorim et al. (2011) suggested that the preliminary tests done in the bovine model may provide valuable results that serve as basis to initiate the similar test using human tissues.
The use of cryoprotectants in the medium helps to prevent cell injuries due to ice crystal formation and hypertonicity during cryopreservation; however, they may cause cytotoxicity at high concentration. Indeed, many research groups have raised concerns about the residual cryoprotectant after vitrification and warming (Iwatani et al., 2006; Pegg, 2007; Yavin and Arav, 2007). Vitrification protocols call for high concentrations of cryoprotectants, which are potentially toxic to the human body, and the residual cryoprotectants have been reported to remain high in warmed ovarian tissues before transplantation (Best, 2015; Nakamura et al., 2017). Nakamura et al. (2017) investigated the residual cryoprotectant concentrations in ovarian tissues during and immediately after thawing/warming; however, in clinical practice, ovarian tissues are rarely transplanted immediately after thawing/warming. We hypothesized that if the frozen ovarian tissues are washed in culture media for some time after thawing/warming, the concentration of cryoprotectants would decrease by simple diffusion. Therefore, the present study aimed to address the concerns about residual cryoprotectants by evaluating the relationship between the washing time after thawing/warming and residual cryoprotectant concentrations. This study also compared apoptosis, DNA damage and ultrastructural changes in slow frozen and vitrified ovarian tissues.
Materials and methods
Ovarian tissue collection and preparation
The ovaries (n = 20) from 10 cows (mean age, 5 years) were resected in a local abattoir and delivered to the laboratory within 3 h after the animals were sacrificed for human consumption. The study design is outlined in Supplementary Fig. S1.
Ovarian tissue cryopreservation and thawing
The bovine ovarian cortex tissues were processed into square ovarian sheets (length × width × thickness: 10 mm × 10 mm × 1 mm) as described previously (Suzuki et al., 2012; Sugishita et al., 2018). On average 10 square ovarian sheets were harvested from each ovary. In total, 200 ovarian sheets were collected and assigned randomly to the following groups: slow freezing with dimethyl sulfoxide (DMSO; n = 60), slow freezing with 1,2-propanediol (PROH; n = 60), vitrification with ethylene glycol (EG; n = 60) or fresh (non-frozen) control (n = 20). Slow freezing was performed in cryotubes (1396-201S; Fukaekasei, Tokyo, Japan) and vitrification in open devices (Ova Cryo TypeM; Kitazato, Shizuoka, Japan). All tissues were maintained in liquid nitrogen for at least 1 week.
Slow freezing with 1.5 M DMSO or 1.5 M PROH
The slow freezing procedure was performed as described by Gosden et al. (1994) and Gook et al. (2017) with some modifications. For slow freezing with DMSO, the bovine ovarian sheets (n = 60) were loaded to cryotubes containing 1.5 ml of Leibovitz L-15 medium (Fujifilm Wako, Osaka, Japan) supplemented with 1.5 M DMSO (472301; Sigma Aldrich, MO, USA) and 10% v/v human serum albumin (HSA; LifeGlobal, Guilford, CT, USA). These cryotubes were then rotated on ice for 15 min before loading into the programed freezer (Kryo-Series III, Planner, Middlesex, UK). The freezing program was set to cool the tissues at a rate of 2°C/min to −7°C and then to hold them at −7°C for 10 min for seeding. The temperature was then lowered by −0.3 °C/min to −40°C and thereafter by −10°C/min to −140°C.
For slow freezing with PROH, the bovine ovarian sheets (n = 60) were loaded to cryotubes containing 1.5 ml of Leibovitz L-15 medium supplemented with 1.5 M PROH (398039; Sigma Aldrich, MO, US), 10% v/v HSA and 0.1 M sucrose EMPROVE® (EMD Millipore, Billerica, MA, USA). Similarly, these cryotubes were rotated on ice for 15 min before loading into the programmed freezer. The freezing program was set to cool the tissues at a rate of 2°C/min to −7°C and then to hold them at −7°C for 10 min for seeding. The temperature was then lowered by −0.3 °C/min to −30°C and thereafter by −50°C/min to −150°C. Finally, the cryotubes were stored in liquid nitrogen for at least 1 week (Supplementary Fig. S2A).
Thawing procedure after slow freezing
The thawing procedures were performed as described by Gook et al. (2017) with some modifications. The total thawing time was 21 min. First, the cryotubes were placed in a water bath at 90–95°C for 1 min until the ice had melted. Then, the tissues were transferred to 20 ml of four rehydration solutions at room temperature (25°C) for 5 min each to dilute the cryoprotectant gradually (A to D for slow freezing with DMSO, E to H for slow freezing with PROH). Rehydration A/E: Leibovitz L-15 medium supplemented with 1.0 M DMSO/PROH, 0.2 M sucrose and 10% v/v HSA. Rehydration B/F: Leibovitz L-15 medium supplemented with 0.5 M DMSO/PROH, 0.2 M sucrose and 10% v/v HSA. Rehydration C/G: Leibovitz L-15 medium supplemented with 0.5 M DMSO/PROH and 10% v/v HSA. Rehydration D/H: Leibovitz L-15 medium supplemented with 10% v/v HSA (Supplementary Fig. S2B).
Vitrification with 35% EG
Open vitrification was performed as described previously with slight modifications (Sugishita et al., 2018). In total, 60 pieces of bovine ovarian tissues were immersed in a series of vitrification solutions containing increasing EG concentrations. The vitrification solution was composed of HEPES-buffered 199 (M199-HEPES; Invitrogen, Carlsbad, CA, USA) with 20% (v/v) serum substitute supplement (SSS; Irvine Scientific, Santa Ana, CA, USA). Firstly, the tissues were immersed in 20 ml of the vitrification solution containing 1.61 mol/l EG (10% EG, Cryo 1) for 5 min, next 20 ml of 3.22 mol/l EG (20% EG, Cryo 2) for 5 min and lastly 20 ml of 5.64 mol/l EG (35% EG), 5% (w/v) polyvinylpyrrolidone (Sigma-Aldrich, St. Louis, Mo, USA) and 0.5 M sucrose EMPROVE® (Cryo 3) for 15 min. All steps were performed at room temperature (25°C). After 25 min, the tissues were loaded into the open ovarian storage device, immersed vertically into liquid nitrogen and stored in liquid nitrogen until further use. No crystallization was observed in any tissues during preparation (Supplementary Fig. S2A).
Warming procedure after vitrification
The total time for warming the vitrified ovarian tissue was 14 min. For warming, the open device was placed directly into the warming solution (100 ml of M199-HEPES with 20% v/v SSS and 0.8 M sucrose or Warm 1) pre-warmed to 37°C in the water bath for 1 min. Next, the tissues were transferred to 20 ml of M199 with 20% v/v SSS and 0.4 M sucrose (Warm 2) at room temperature (25°C) for 3 min. Lastly, the tissues were immersed in 20 ml of M199 with 20% v/v SSS (Warm 3 and 4) at room temperature for 5 min twice (Supplementary Fig. S2B).
Quantification of residual cryoprotectants by gas chromatography
Concentrations of DMSO, PROH and EG were quantified (n = 3 per group) by gas chromatography–mass spectrometry using GCMS-QP2010 Ultra (Shimadzu) with an Inert Cap Pure-WAX column (0.25 mm × 30 mm, 0.50 µm thickness; GL Science) by Shimadzu Techno-Research Inc. (Kyoto, Japan). The residual cryoprotectant concentrations in thawed/warmed tissues from each group were measured by gas chromatography at the following time points: frozen (before thawing/warming), 0 min (immediately after thawing/warming), 30, 60 and 120 min after diffusion washing in media. The diffusion washing media used was DMEM plus HEPES (Thermo Fisher, MA, USA) with 10% v/v HSA (Fuji Film Wako, Osaka, Japan), 1% v/v antibiotic/antimycotic (Anti-Anti; Fuji Film Wako, Osaka, Japan), 1% v/v insulin-transferrin-selenium (Fuji Film Wako, Osaka, Japan) and 0.2% v/v FSH (MSD, Osaka, Japan). The ovarian tissues (0.075–0.140 g) were mixed with methanol, homogenized and centrifuged at 9100g for 5 min then the supernatants were subsequently analyzed.
Deuterated DMSO (DMSO-d6), EG (EG-d6) and PROH (PG-d8) were used as internal standards. The column temperature was initially held at 80°C for 2 min, then raised at a rate of 5°C/min to 140°C and subsequently at a rate of 25°C/min to 240°C. The injector temperature was set at 240°C. We measured the amount of cryoprotectants in frozen ovarian tissues (before thawing/warming) and post-thawing/warming with diffusion wash for 0, 30, 60 or 120 min. The percentage of the residual cryoprotectant was obtained by dividing the weight of the cryoprotectant over the weight of ovarian tissue (including cryoprotectant).
Transmission electron microscopy
To examine the ultrastructural changes in follicles, granulosa and stromal cells in bovine ovarian tissues, we examined five small pieces (1 mm × 1 mm × 1 mm) from each of the four groups (slow frozen in DMSO or PROH, vitrified in EG and non-frozen control). First, the tissues were fixed in 2.5% v/v glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 1–7 days at 4°C. After rinsing in phosphate buffer, the samples were post-fixed with 1% v/v osmium tetroxide in phosphate buffer. Then, the tissues were dehydrated in an ethanol gradient of increasing concentration, immersed in propylene oxide for solvent substitution, embedded in EPON 812 resin and sectioned. Ultrathin sections (60–80 nm) were stained with uranyl acetate and lead citrate and then examined and photographed by transmission electron microscopy (JEM-1220; JEOL, Ltd., Tokyo, Japan).
Residual cryoprotectant cytotoxicity
To examine the effect cytotoxicity of residual cryoprotectants, we first compared DNA damage and apoptosis expressions in thawed/warmed tissues without diffusion washing (0 min) and with adequate washing (120 min). Next, to examine their suitability for transplantation we subjected these tissues (0 and 120 min) to 24 or 48 h of ex vivo culture at 37°C under a 5% CO2 atmosphere. The ex vivo culture period partially represented the avascular period of the tissue after transplantation. The thawed/warmed ovarian tissues were ex vivo cultured in dishes with cell culture inserts. The culture dishes were from Falcon, 351008, NY, USA and the Millicell cell culture inserts were from Merck Millipore, PICM0RG50, MA, USA. The culture media used was DMEM/F12 containing 10% v/v HSA (Fuji Film Wako, Osaka, Japan), 0.05 mg/ml ascorbic acid, 1% v/v antibiotic/antimycotic solution (Invitrogen) and 0.3 IU/ml FSH (Kawamura et al., 2013). All experimental groups were analyzed for apoptosis and DNA damage by quantitative PCR (qPCR) and immunohistochemistry.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from the ovarian tissue from respective experimental groups (n = 9 per group) by RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The RNA concentration and purity were determined by NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Complementary DNA was synthesized from 150 ng RNA, which was first treated with AccRT Genomic DNA Removal Kit (APB Applied Biological Materials Inc., Vancouver, Canada). After DNase inactivation, the reverse transcription was performed in a final volume of 10 μl with PrimeScript RT Master Mix (TaKaRa, Shiga, Japan) and reverse transcribed at 37°C for 15 min and 85°C for 5 s. The cDNA samples were stored at −30°C until use. Before quantitative real-time PCR, synthesized cDNA was diluted five times with EASY Dilution (TaKaRa, Shiga, Japan). Then, qPCR was performed in StepOnePlus Real-Time PCR System with StepOne Software v2.3 (Thermo Fisher Scientific, Waltham, MA, USA) by using SYBR Green-based PCR assay and TB Green Fast qPCR Mix (TaKaRa, Shiga, Japan). The amplification condition was set as follows: 95°C for 30 s and 40 cycles of 95°C for 5 s and 60°C for 10 s. After the amplification cycles, the melt-curve analyses were performed at 95°C for 15 s, 60°C for 1 min and 95°C for 15 s. For each tissue, qPCR was performed in triplicate.
The mRNA expression levels of caspase-3 (CASP3), an apoptosis precursor and poly (ADP-ribose) polymerase-1 (PARP1), a first responder molecule for single-strand DNA damage, were investigated. The expression level of CASP3 and PARP1 were assessed as a ratio to the level of β-actin, the housekeeping gene that was assumed to remain constant across the experimental groups. The primer sequences and sizes of the amplified fragments are shown in Supplementary Table SI.
Immunohistochemistry
Immunohistochemistry with activated caspase-3 (AC3) antibody (1:500, WKAF835, Active, Rabbit-Poly; R&D Systems, Inc., MN, USA) or gamma H2A histone family member X (γH2AX) antibody (1:500, FN-IHC0059, Rabbit-Poly; Bethyl Laboratories, Inc. TX, USA) with second antibody (1:500, anti-IgG, Rabbit, Goat-Poly, HRP; Bethyl Laboratories, Inc. TX, USA) were used to detect apoptosis and DNA double-strand breaks, respectively. To evaluate the toxicity of residual cryoprotectants (DMSO, PROH and EG) in tissues after transplantation, we first compared AC3 and γH2AX positive cell ratios in thawed/warmed tissues that were washed for 0 or 120 min, next in tissues that were further subjected to 24 or 48 h of ex vivo culturing (n = 3 per group).
To prepare tissue sections for immunohistochemistry, the ovarian tissues were fixed in formalin, then serial dehydrated in graded ethanol before embedding in paraffin. The paraffin-embedded tissue blocks were serially cut into 4-μm sections. Immunohistochemistry staining was performed by an automated machine (HISTOSTAINER; Nichirei, Tokyo, Japan) (Supplementary Fig. S3). For quantification, the AC3 or γH2AX positive cells were counted in 10 randomly selected locations across the tissue section and presented as ratios to the total number of cells by Patholoscope software (Mitani, Fukui, Japan). The Patholoscope software analyzed the immunopositivity with the parameters set in advance for AC3 and γH2AX. In the analyzed location, the yellow circles indicated immuno-positive cells; while the purple circle indicated immuno-negative cells (Supplementary Fig. S3). The parameters were set as follows: cell size, 13 pixels; staining concentration, 17; extraction of immuno-negative cells, 27; extraction of immuno-positive cells, 12; measurement area, automation, 75; deletion of small particles, 24; and deletion of elongated particles, 68. In the negative control, the primary antibody was omitted in each immunohistochemistry staining.
Statistical evaluation
The gas chromatography was compared by general linear model and the qPCR and immunohistochemistry data were compared by Mann–Whitney test with SPSS version 24.0 software (SPSS Inc., USA). A P-value <0.05 was considered statistically significant.
Results
Concentration of residual cryoprotectants assessed by gas chromatography
The residual concentrations of DMSO, PROH and EG in thawed/warmed ovarian tissues (n = 3 per group) were measured by gas chromatography at the following time points: pre-thawing/warming (frozen), post-thawing/warming with 0, 30, 60 and 120 min of diffusion washing in media. The residual concentrations of DMSO, PROH and EG decreased with the extension of diffusion washing time (Fig. 1, Supplementary Table SII).
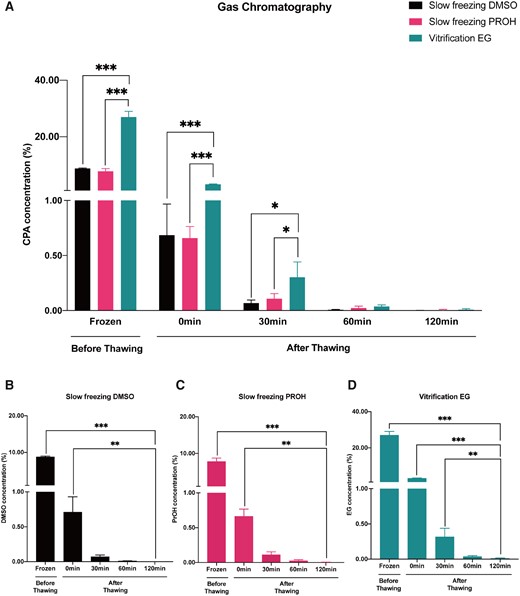
The cryoprotectant concentrations in percentage measured by gas chromatography. (A) Residual dimethyl sulfoxide (DMSO), 1,2-propanediol (PROH) and ethylene glycol (EG) amount reduced to a comparable level after 60 min of washing. (B–D) The residual DMSO, PROH and EG concentrations decreased with extension of diffusion washing time. Histograms represent mean concentrations with standard errors expressed as cryoprotectant mass/tissue mass (including cryoprotectant) percent. General linear model analysis was used for the statistical analysis (***P < 0.001; **P < 0.01; *P < 0.05).
Since vitrification protocol required a higher concentration of cryoprotectant, the residual EG concentration remained significantly higher than DMSO and PROH in slow frozen tissues at frozen and after thawing/warming with 0 and 30 min of diffusion washing (pre-thawing and 0 min post-thawing, DMSO versus EG, P = 0; PROH versus EG, P = 0; 30 min post-thawing, DMSO versus EG, P = 0.022; PROH versus EG, P = 0.049). However, after 60 min of diffusion washing, all three residual cryoprotectants reduced to a comparable level with no significant difference (DMSO versus PROH; DMSO versus EG; PROH versus EG, P > 0.05). When the diffusion washing time was extended to 120 min, the residual cryoprotectant concentrations reduced to minimal (DMSO versus PROH; DMSO versus EG; PROH versus EG, P > 0.05; Fig. 1A).
Next, we replotted the data to facilitate comparison of the efficiency of diffusion washing to reduce the residual cryoprotectant to minimum (Fig. 1B–D). In slow freezing with DMSO, the concentration at frozen (pre-thawing versus 120 min, P = 0) and 0 min (0 min versus 120 min, P = 0.003) were significantly higher. However, when diffusion washing time was implemented for 30 min, the residual DMSO concentration was comparable to that after 60 and 120 min of diffusion washing (30 min versus 120 min, 60 min versus 120 min, P > 0.05) (Fig. 1B). Similarly, the residual PROH concentration in slow frozen tissues was significantly higher at frozen and 0 min (pre-thawing versus 120 min, P = 0; 0 min versus 120 min, P = 0.005). It also took 30 min of diffusion washing after thawing to reduce the residual PROH to a comparable level with 60 and 120 min of diffusion washing (30 min versus 120 min, 60 min versus 120 min, P > 0.05; Fig. 1C). In the vitrification EG group, however, it was not until 60 min after the diffusion washing that the residual EG concentration became comparable with that of 120 min (60 versus 120 min, P > 0.05). The residual EG at frozen, 0 and 30 min remained significantly higher than that of 120 min (pre-thawing versus 120 min, P = 0; 0 min 120 min, P = 0; 30 min versus 120 min, P = 0.003; Fig. 1D).
Histological evaluation by transmission electron microscope
To compare the ultrastructural changes in ovarian tissues, the transmission electron microscope examination was performed in fresh (non-frozen) control and thawed/warmed samples (DMSO, PROH and EG; n = 5 from each group). Specifically, we evaluated the morphology of primordial follicles, granulosa cells (GC), organelles and stromal cells (Supplementary Fig. S4).
The fresh control tissues showed normal morphology. The primordial follicle showed a layer of GC that adhered closely to the oocyte (Supplementary Fig. S4A). The gap junction (Gj) and compact collagen fibers could be seen around the GC (Supplementary Fig. S4E). In the cytoplasm, the stromal tissue was intact and organelles such as mitochondria (M), rough endoplasmic reticulum (rER) and stromal cells (SC) were well defined (Supplementary Fig. S4I, M).
In the slow freezing group with DMSO, the morphology of primordial follicles and GCs was comparable to that in fresh control (Supplementary Fig. S4B, F). A rich population of mitochondria was observed inside the follicles, some with uniform cristae, but for some the cristae appeared swollen (*). Abundant ribosomes (R) were also observed (Supplementary Fig. S4J). Stromal cells maintained their morphology outside the follicles and the stromal tissues appeared intact (Supplementary Fig. S4N).
In the slow freezing group with PROH, the morphology of primordial follicles and GCs were maintained (Supplementary Fig. S4C, G). A lysosome, which emerges when cell organelles degenerate, next to the GC was observed (Supplementary Fig. S4G). Some mitochondria in the follicle showed swelling and the cristae structure was lost (Supplementary Fig. S4K). Stromal tissues with compact collagen fibers were maintained and the stromal cells appeared normal (Supplementary Fig. S4O).
In the vitrification group with EG, the morphology of primordial follicles and GCs was similar to that in fresh control (Supplementary Fig. S4D, H). Some follicles contained fat droplets (Supplementary Fig. S4D). Rough endoplasmic reticulum and ribosomes were seen in the follicles. Some mitochondrial swelling was observed (Supplementary Fig. S4L), while the stromal matrix consisted of rich collagen fibers and stromal cells were well preserved (Supplementary Fig. S4P).
Cytotoxicity of residual cryoprotectant in ovarian tissues
Next, we quantified mRNA expression of PARP1 and CASP3 by qPCR, and immune-positive cell ratios of γH2AX and AC3 by immunohistochemistry in thawed/warmed ovarian tissues with high (0 min) and minimum (120 min) concentration of residual cryoprotectant. After sufficient diffusion washing of 120 min, the DNA damage sensor PARP1 mRNA expression was up-regulated in slow frozen tissues with DMSO and vitrified tissues with EG (DMSO 0 min versus 120 min, P = 0; EG 0 min versus 120 min, P = 0), while it stayed comparable for slow frozen tissues with PROH (PROH 0 min versus 120 min, P > 0.05) (Fig. 2A; Supplementary Table SIII).
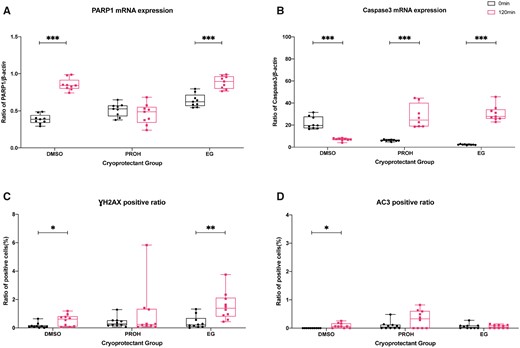
Quantification of DNA damage and apoptosis expression by real-time quantitative PCR and immunostaining in the thawed/warmed ovarian tissues with high and minimum residual cryoprotectant. Boxplots with all points showing min to max of poly (ADP-ribose) polymerase-1 (PARP1) and caspase 3 (CASP3) mRNA expression with respect to β-actin (A, B) and gamma H2A histone family member X (γH2AX) and activated caspase-3 (AC3) immune-positive cell ratios (C, D) in thawed/warmed ovarian tissues with high (0 min) and minimum (120 min) concentration of residual cryoprotectant. The groups were compared by Mann–Whitney test (***P < 0.001; **P < 0.01; *P < 0.05). DMSO, dimethyl sulfoxide; EG, ethylene glycol; PROH, 1,2-propanediol.
In the evaluation of the cell apoptosis precursor expression level, CASP3 level was increased after 120 min of diffusion washing in PROH-slow frozen (P = 0) and EG-vitrified tissues (P = 0) compared to their respective samples at 0 min. On the other hand, DMSO-slow frozen showed significantly lower CASP3 expression after 120 min washing (P = 0; Fig. 2B; Supplementary Table SIII).
To demonstrate the presence of DNA double-strand breaks and activated apoptosis cells, γH2AX- and AC3-positive cells were quantified by immunohistochemistry and compared between high (0 min) and minimum (120 min) residual cryoprotectant in each group (Fig. 2C and D; Supplementary Figs. S5 and S6; Supplementary Table SIV). After 120 min diffusion washing, the γH2AX-positive cell ratio was elevated in DMSO-slow frozen (P = 0.043) and EG-vitrified tissues (P = 0.002); while it stayed comparable in PROH-slow frozen tissues (P > 0.05; Fig. 2C). In AC3 immunostaining, after 120 min of diffusion washing the AC3-positive cell ratio was increased in DMSO-slow frozen tissues (P = 0.043), but it stayed comparable in PROH-slow frozen and EG-vitrified tissues (P > 0.05; Fig. 2D). Despite some elevations were observed in the γH2AX- and AC3-positive cell ratios after diffusion washing, the overall positive cell ratios remained low and within 2% of the total cells.
Cytotoxicity of residual cryoprotectant in ex vivo culture
To determine the suitability of thawed/warmed ovarian tissues for transplantation, we examined the effect of cytotoxicity of residual cryoprotectants (0 versus 120 min) in ovarian tissues after 24 or 48 h of ex vivo culture. The ex vivo culture period partially represented the avascular period of the tissue after transplantation. Again, we quantified the PARP1 and CASP3 mRNA expression by qPCR (Fig. 3; Supplementary Table SV), and γH2AX- and AC3-positive cell ratios by immunohistochemistry (Fig. 4; Supplementary Figs. S7, S8, S9 and S10; Supplementary Table SVI). In mRNA evaluation after 24 h of ex vivo culture, PARP1 and CASP3 consistently increased in tissues with minimal residual cryoprotectant (0 versus 120 min after 24 h ex vivo: DMSO, P = 0.006 for PARP1, P = 0 for CASP3; PROH, P = 0 for PARP1, P = 0.004 for CASP3; EG, P = 0 for PARP1 and CASP3) (Fig. 3A and B). After 48 h of ex vivo culture, PARP1 and CASP3 expressions were amplified further in slow frozen tissues with DMSO and PROH (0 versus 120 min after 48 h ex vivo: DMSO, P = 0.011 for PARP1, P = 0 for CASP3; PROH, P = 0 for CASP3; Fig. 3C and D). Interestingly, vitrified/warmed tissues with minimum EG showed lower CASP3 level compared to tissues with high residual EG after 48 h of ex vivo culture.
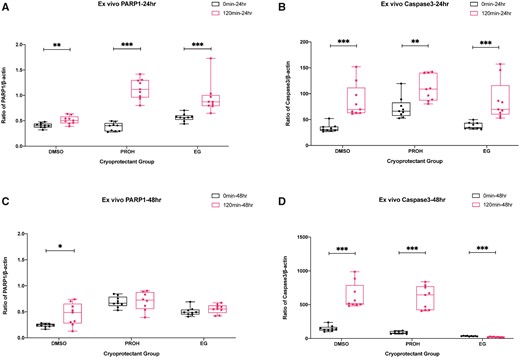
Quantification of PARP1 and CASP3 mRNA levels after 24 and 48 h ex vivo culture of the thawed/warmed ovarian tissues with high and minimum residual cryoprotectant. Boxplots with all points showing min to max of poly (ADP-ribose) polymerase-1 (PARP1) and caspase 3 (CASP3) mRNA expression with respect to β-actin. The effect of residual cryoprotectant (0 versus 120 min) was examined after 24 h of ex vivo culture (A, B) and 48 h of ex vivo culture (C, D). The groups were compared by Mann–Whitney test (***P < 0.001; **P < 0.01; *P < 0.05). DMSO, dimethyl sulfoxide; EG, ethylene glycol; PROH, 1,2-propanediol.
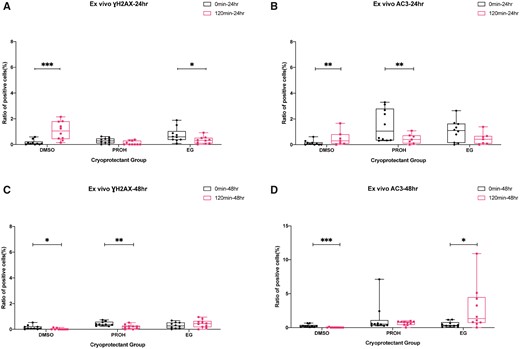
Immunostaining of γH2AX and AC3 after 24 and 48 h ex vivo culture of the thawed/warmed ovarian tissues with high and minimum residual cryoprotectant. Boxplots with all points showing min to max of gamma H2A histone family member X (γH2AX) and activated caspase-3 (AC3) immune-positive cell ratios. The effect of residual cryoprotectant (0 versus 120 min) was examined after 24 h of ex vivo culture (A, B) and 48 h of ex vivo culture (C, D). The groups were compared by Mann–Whitney test (***P < 0.001; **P < 0.01; *P < 0.05). DMSO, dimethyl sulfoxide; EG, ethylene glycol; PROH, 1,2-propanediol.
The immunohistochemistry staining showed that after 24 h of ex vivo culture, the γH2AX and AC3 positive cell ratios were significantly higher in slow frozen tissues with minimum residual DMSO (0 versus 120 min after 24 h ex vivo: DMSO, P = 0 for γH2AX, P = 0.004 for AC3; Fig. 4A and B). After reducing the cryoprotectant to minimum (120 min washing), the PROH slow frozen tissues and EG vitrified tissues showed lower AC3 positive cell ratios (P = 0.005) and γH2AX positive cell ratios (P = 0.035), respectively (Fig. 4A and B). Inconsistent trends were observed after 48 h of ex vivo culture (Fig. 4C and D). Slow frozen tissues with minimum residual DMSO and PROH showed lower γH2AX positive cell ratios (DMSO, P = 0.023; PROH, P = 0.004). A lower AC3 positive ratio was observed in slow frozen tissues with minimum residual DMSO (DMSO, P = 0), while higher AC3 positive cell ratio was observed in vitrified tissues with minimum residual EG (EG, P = 0.019) after 48 h of ex vivo culture (Fig. 4C and D).
Discussion
In 2017, Nakamura et al. (2017) conducted a gas chromatography study to measure the residual EG and DMSO concentrations in ovarian tissues during thawing and warming steps following slow freezing and vitrification, respectively. Nakamura et al. found the residual cryoprotectant concentrations remained high in vitrified/warmed samples compared to that in slow frozen/thawed samples and they expressed concerns over the cytotoxicity of residual cryoprotectants to human body after OTT. In regard to these findings, it should be pointed out the broad time discrepancies at which the cryoprotectant concentrations were measured (Supplementary Fig. S11). For example, the DMSO and EG concentrations were measured 1, 6 and 16 min after immersion in the warming solution for vitrified tissues; whereas for slow frozen tissues, the DMSO concentration was measured 47 and 67 min after immersion in the thawing solution. In addition, Nakamura et al. lengthened the slow freezing protocol described by Isachenso et al. to 65 min instead of 46.5 min; on the other hand, they shortened the vitrification protocol described by Kagawa et al. to 16 min instead of 26 min (Kagawa et al., 2009; Isachenko et al., 2012; Supplementary Fig. S11).
To fairly compare the residual cryoprotectant concentrations following different OTC methods, the time required to complete the thawing and warming and the time points at which the cryoprotectant concentrations were measured should be similar. In this study, the total time required for thawing following slow freezing and warming following vitrification was similar; that is 21 and 14 min, respectively. In addition, the residual cryoprotectant concentrations were measured at the same time points: pre-thawing/warming (frozen) and post-thawing/warming with 0, 30, 60 or 120 min of diffusion washing. We hypothesized that washing time and residual cryoprotectant concentration is inversely related so when the thawed/warmed ovarian tissue is immersed in the washing media, the osmotic and concentration gradient move the cryoprotective agent out in exchange for water until close to equilibrium state is reached. The gas chromatography confirmed the residual DMSO, PROH and EG concentrations decreased with the extension of diffusion washing time. After 60 min of washing, the residual cryoprotectant concentration reduced significantly regardless of the cryopreservation method. When the washing was extended to 120 min, the residual cryoprotectant concentration reduced to minimum. It took 30 min of washing to minimize the residual DMSO and PROH in slow frozen tissues; however, it took 60 min of washing to minimize the residual EG in vitrified tissues. Since vitrification relies on the use of high cryoprotectant concentration to prevent the ice formation during freezing and warming, it is within reason that higher residual EG concentration was detected in vitrified tissues before warming and longer diffusion washing time was required to reach equilibrium. These findings suggested the vitrified/warmed ovarian tissues should undergo at least 60 min of diffusion washing in culture media before transplantation.
The ultrastructural comparisons of fresh (non-frozen) and thawed/warmed ovarian tissues by transmission electron microscope investigated the morphology of primordial follicles, GCs, stromal cells and organelles such as mitochondria, ribosomes, and lysosomes. The Gjs were tightly maintained with all three cryoprotectants. Some swellings and degenerated mitochondria were observed in the DMSO group, indicating damages due to freezing. In the PROH group, we did not see a marked decrease or disappearance of cell organelles. On the other hand, lysosomes were observed in follicles and mitochondrial degeneration was present near the stromal cells. Thus, some areas in tissues slow frozen by PROH might have been damaged. The EG group had some mitochondria with partial degeneration; however, the damage was no worse than that in the DMSO and PROH groups. Moreover, the stromal cells were aligned and rich in collagen fibers, indicating the integrity of stromal cells. Several studies have been done to demonstrate that the toxic effect of these permeable cryoprotectants was mainly during the exposure period (Rodrigues et al., 2004a,b). The ovarian tissues’ exposure period to cryoprotectants was 15 on ice and 20 min at 25°C for slow freezing and vitrification protocol, respectively (as illustrated in Supplementary Fig. S2). According to the results of these studies, the exposure periods in our protocols would not likely cause toxicity due to the cryoprotectants. Furthermore, Celestino et al. (2008) showed that DMSO and EG at 1.5 M did not decrease the percentage of viable follicles in bovine ovarian fragments when compared to control; whereas the deleterious decrease in viable follicles was significant when no cryoprotectant or cryoprotectant concentration at 3 M was used. Although PROH at 1.5 M significantly reduced the viable preantral follicle counts according to Celestino et al., the exposure time and temperature (20 min at 20°C) applied was longer and higher compared to those in our protocol (15 min on ice) (Celestino et al., 2008). In agreement with our observations, Keros et al. (2009) also found no apparent morphological change in follicles with either a slow-freezing or vitrification method and the good morphology with rich collagen fibers was observed in stromal cells. Shi et al. conducted a meta-analysis and found no morphological difference between slow-freezing and vitrification methods. However, less DNA damage was seen with the vitrification method than slow-freezing method and stromal cell count was higher after vitrification (Shi et al., 2017). Therefore, our findings of no marked damage among the three groups seemed consistent with these reports.
The tissue integrity demonstrated by transmission electron microscopy may not represent the functionality. To provide additional evidence on whether the residual cryoprotectant affected the tissue viability, we examined the level of DNA damage and apoptotic mRNA and protein expression in thawed/warmed ovarian tissues. The thawed/warmed ovarian tissues with high (0 min) and minimal residual cryoprotectant (120 min) were compared in their PARP1 and CASP3 mRNA expression levels and γH2AX- and AC3-positive cells ratios. PARP1, the DNA damage sensor, was lowly expressed at 0 min, then the expression increased in DMSO-slow frozen and EG-vitrified tissues after 120 min washing; while it remained similar for PROH-slow frozen tissues. The delayed increase in PARP1 expression may suggest the transcriptional time delay in response to DNA damages caused by cytotoxicity of cryoprotectants at high concentration. Similarly, the immunohistochemistry showed a delayed increase in γH2AX-positive cell ratios in DMSO-slow frozen and EG-vitrified tissues after 120 min of washing. As an early DNA damage sensor, PARP1 mRNA expression with respect to B-Actin was under 1%. In addition, the protein expression of γH2AX in response to accumulation of DNA damage was under 2% of total cell population, suggesting both OTC methods provided good viability that the gene repair mechanisms were not activated.
Apoptosis evaluations in tissues with and without diffusion washing were also performed to determine the cryoprotectant cytotoxicity. Overall, the CASP3 expression was more pronounced than PARP1 and the delayed increase in CASP3 was observed in PROH-slow frozen and EG-vitrified tissues. Interestingly, in DMSO-slow frozen tissues the CASP3 expression decreased after 120 min of washing. The cytotoxicity of cryoprotectant may have more impact on apoptosis than DNA damage; however, the immunohistochemistry showed low number of AC3-positive cells in both slow frozen and vitrified tissues before and after washing. The low ratios of AC3-positive cells observed may suggest that only a small percentage of apoptosis precursors (CASP3) were activated. Or the translational changes such as protein synthesis and post-translational modifications which reflect the elevated CASP3 may take longer than 120 min. Taken together, we demonstrated that the adverse effects of residual cryoprotectant cytotoxicity could be reduced in tissues when implementing at least 60 min of diffusion washing after thawing/warming.
Otala et al. (2002) reported that apoptosis was initiated in ovaries that were culture for 24 h. Indeed, the qPCR results showed PARP1 and CASP3 mRNA expressions increased profoundly in tissues after 24 h of ex vivo culture in tissues with minimum residual cryoprotectants (120 min). We speculate the culture damages such as tissue hypoxia and reactive oxygen species productions (Silva et al., 2018) may be the major contributors for accelerating the DNA damages and apoptosis in ex vivo cultures rather than the cytotoxicity of residual cryoprotectants. Inconsistencies were observed in immunostaining results. Further studies are required to evaluate the optimal culture time before OTT. In addition, hormonal studies such as estradiol and anti-mullerian hormone could also be investigated to evaluate the post-cryopreservation tissue survival.
Conclusion
In conclusion, the findings from the present study demonstrated the importance to introduce a minimum of 60 min washing step following thawing/warming of cryopreserved ovarian tissues to minimize the cryoprotectant toxicity. In addition, the molecular evaluations and ultrastructural observations supported that vitrification is an effective alternative to slow freezing in cryopreservation of ovarian tissues.
Data availability
The data underlying this article will be shared upon reasonable request to the corresponding author.
Acknowledgements
We thank M. Sekiguchi and M. Kashiwagi-Iwano for technical assistance.
Authors’ roles
Y.S. and N.S. designed the study and wrote the manuscript. Y.S., L.M., Y.S.-T., S.N., S.F., A.U., A.T.-O., J.M.-I., H.S., Y.N. and M.K. conducted the studies. Y.S., L.M., Y.S.-T., S.N. and T.I.-K. conducted the statistics. Y.S. L.M., Y.S.-T., S.N., K.E., J.K. and N.S. participated in discussions and manuscript editing.
Funding
This work was supported by the Grant-in-Aid for Scientific Research (B) (17H04342 to N.S.).
Conflict of interest
The authors declare no conflict of interest.
References
Author notes
Yodo Sugishita, Lingbo Meng should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}