





Abstract
Does the phenotype of women with normosmic congenital hypogonadotrophic hypogonadism (nCHH) and pituitary resistance to GnRH caused by biallelic mutations in the GnRH receptor (GNRHR) (nCHH/bi-GNRHR) differ from that of women with polycystic ovary syndrome (PCOS)?
Women with nCHH/bi-GNRHR have variable pubertal development but nearly all have primary amenorrhea and an exaggerated LH response to GnRH stimulation, similar to that seen in women with PCOS.
Women with nCHH/bi-GNRHR are very rare and their phenotype at diagnosis is not always adequately documented. The results of gonadotrophin stimulation by acute GnRH challenge test and ovarian features have not been directly compared between these patients and women with PCOS.
We describe the phenotypic spectrum at nCHH/bi-GNRHR diagnosis in a series of 12 women. Their reproductive characteristics and acute responses to GnRH were compared to those of 70 women with PCOS.
Patients and controls (healthy female volunteers aged over 18 years) were enrolled in a single French referral centre. Evaluation included clinical and hormonal studies, pelvic ultrasonography and GnRH challenge test. We also functionally characterized two missense GNRHR mutations found in two new consanguineous families.
Breast development was highly variable at nCHH/bi-GNRHR diagnosis, but only one patient had undeveloped breasts. Primary amenorrhea was present in all but two cases. In untreated nCHH/bi-GNRHR patients, uterine height (UH) correlated (P = 0.01) with the circulating estradiol level and was shorter than in 23 nulliparous post-pubertal age-matched controls (P < 0.0001) and than in 15 teenagers with PCOS under 20-years-old (P < 0.0001) in which PCOS was revealed by primary amenorrhea or primary–secondary amenorrhea. Unexpectedly, the stimulated LH peak response in nCHH/bi-GNRHR patients was variable, and often normal or exaggerated. Interestingly, the LH peak response was similar to that seen in the PCOS patients, but the latter women had significantly larger mean ovarian volume (P < 0.001) and uterine length (P < 0.001) and higher mean estradiol (P < 0.001), anti-Müllerian hormone (AMH) (P = 0.02) and inhibin-B (P < 0.001) levels.
In the two new consaguineous families, the affected nCHH/bi-GNRHR women carried the T269M or Y290F GNRHR missense mutation in the homozygous state. In vitro analysis of GnRHR showed complete or partial loss-of-function of the T269M and Y290F mutants compared to their wildtype counterpart.
The number of nCHH/bi-GNRHR patients reported here is small. As this disorder is very rare, an international study would be necessary to recruit a larger cohort and consolidate the phenotypic spectrum observed here.
In teenagers and young women with primary amenorrhea, significant breast and uterine development does not rule out CHH caused by biallelic GNRHR mutations. In rare patients with PCOS presenting with primary amenorrhea and a mild phenotype, the similar exaggerated pituitary LH responses to GnRH in PCOS and nCHH/bi-GNRHR patients could lead to diagnostic errors. This challenge test should therefore not be recommended. As indicated by consensus and guidelines, careful analysis of clinical presentation and measurements of testosterone circulating levels remain the basis of PCOS diagnosis. Also, analysis of ovarian volume, UH and of inhibin-B, AMH, estradiol and androgen circulating levels could help to distinguish between mild PCOS and nCHH/bi-GNRHR.
This study was supported by the French National Research Agency (ANR) grant ANR-09-GENO-017 KALGENOPATH, France; and by the Italian Ministry of Education, University and Research (MIUR) grant PRIN 2012227FLF_004, Italy. The authors declare no conflict of interest.
Introduction
GnRH triggers puberty and regulates reproduction in both genders by binding and activating the GnRH receptor (GnRHR) expressed on pituitary gonadotroph cells, which synthesize and secrete the gonadotrophins, LH and FSH (Millar et al., 2004; Flanagan and Manilall, 2017). The role of GNRHR loss-of-function mutations in the pathophysiology of isolated normosmic congenital hypogonadotropic hypogonadism (nCHH) was first reported in 1997 by our team (de Roux et al., 1997) and by Layman et al. (Layman et al., 1998). These mutations are now a well-established cause of nCHH in humans (Boehm et al., 2015; Francou et al., 2016). Familial forms of this genetic form of nCHH are usually autosomal recessive (Beranova et al., 2001; Brioude et al., 2010; Kim et al., 2010; Gianetti et al., 2012; Boehm et al., 2015; Francou et al., 2016; Maione et al., 2018), meaning that only subjects with biallelic mutations are affected, whereas heterozygotes are healthy carriers (Maione et al., 2018). Few female cases of nCHH due to biallelic GNRHR mutations have been reported but apart from some exceptions (de Roux et al., 1997, 1999; Layman et al., 1998; Beranova et al., 2001; Meysing et al., 2004; Kim et al., 2010; Gianetti et al., 2012; Hietamäki et al., 2017) their detailed clinical, neuroendocrine and hormonal characteristics are not always comprehensively described. The aim of this work was to describe the clinical characteristics, neuroendocrine and hormonal profiles, and uterine and ovarian morphology in a series of female patients with nCHH due to biallelic GNRHR mutations (nCHH/bi-GNRHR). In particular, we studied the response of pituitary gonadotrophins to GnRH stimulation in these patients, who have pituitary resistance to GnRH, by comparison with women of comparable age with polycystic ovary syndrome (PCOS) (Dewailly, 2016; Teede et al., 2018) revealed by primary, primo-secondary or secondary amenorrhea. Finally, we characterized in silico three additional mutations and, in vitro, the impact of two of these mutations present in the homozygous state in three patients belonging to two new consanguineous families.
Materials and Methods
Patients
We studied 12 women with nCHH/bi-GNRHR, three of whom have previously been partially described (de Roux et al., 1997, 1999). All 12 women were at least 17-years-old at the time of evaluation. At diagnosis, they all had low to low-normal estradiol (E2) levels, low, inappropriately normal or normal gonadotrophin levels (Table I), no other pituitary hormone deficiencies, normal serum prolactin levels and normal hypothalamo-pituitary MRI. None of these women had an eating disorder, and all had BMI values ≥20 kg/m2 (Table I). Their sense of smell and olfactory bulb structures were normal (Bry-Gauillard et al., 2017).
Clinical characteristics at diagnosis of women with normosmic congenital hypogonadotrophic hypogonadism carrying biallelic mutations in the GnRH receptor gene.
| Patients | Age | Height | BMI | Tanner | Menses | Uterine height | Ovarian volume | E2 | LH basal | LH peak | FSH basal | FSH peak | TT | ∆4-A | Allele 1/allele 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| years | m | kg/m2 | mm | (R/l), ml | pg/ml | IU/l | IU/l | IU/l | IU/l | ng/ml | ng/ml | ||||
| 1+ | 18 | 1.65 | 24.2 | B2/P3 | PA | 27 | NA / NA | 12 | 1.2 | 23 | 1.3 | 4.6 | 0.23 | 0.7 | Y290F/Y290F |
| 2+ | 22 | 1.67 | 24.7 | B3/P2 | PA | 32 | 0.9 / 1.0 | 20 | 1.5 | 28 | 0.8 | 3.4 | 0.20 | 0.9 | Y290F/Y290F |
| 3 | 21 | 1.64 | 21.9 | B2/P3 | PA | 27 | 3.3 / 2.8 | 9 | 0.4 | 10 | 0.8 | 3.2 | 0.24 | 1.2 | T269M/T269M |
| 4 | 19 | 1.74 | 20.5 | B3/P4 | Oligo | 64 | 5.1 / 6.9 | 34 | 4.6 | 42.5 | 5.1 | 8.9 | 0.55 | 2.3 | Q106R/V94A |
| 5 | 17 | 1.75 | 27.8 | B3/P4 | PA | 29 | 0.8 / 0.7 | 14 | 1.5 | 18 | 0.9 | 9.7 | 0.19 | 0.4 | Q106R/R139C |
| 6 | 27 | 1.64 | 23.4 | B2/P4 | PA | NA | 1.5 / 1.7 | 3 | 1.1 | 19 | 2.5 | 8.1 | 0.28 | 0.9 | Q106R/R262Q |
| 7+ | 18 | 1.71 | 23.0 | B5/P5 | PA | 51 | 2.2 / 1.5 | 22 | 5.9 | 81 | 7.4 | 19.5 | 0.21 | 1.2 | Q106R/R262Q |
| 8+ | 17 | 1.71 | 20.8 | B1/P4 | PA | 33 | 0.7 / 2.3 | 9 | 0.7 | 9 | 2.8 | 4.8 | 0.40 | 2.1 | Q106R/R262Q |
| 9 | 19 | 1.70 | 22.5 | B5/P5 | PA | 36 | 0.8 / 1.9 | 8 | 0.6 | NA | 0.7 | NA | 0.37 | 1.9 | Q106R/R139H |
| 10* | 37 | 1.65 | 24.0 | B5/P5 | PSA | 52 | 1.8 / 1.6 | 35 | 5.0 | 35 | 5.2 | 12.0 | 0.33 | 0.8 | Q106R/R262Q |
| 11+** | 16 | 1.70 | 22.4 | B3/P4 | PA | 39 | 1.7 / 1.8 | 23 | 2.3 | 10 | 2.7 | 5.5 | 0.17 | 1.1 | Q106R+S217R/R262Q |
| 12+** | 18 | 1.78 | 22.1 | B3/P5 | PA | 44 | 1.9 / 1.6 | 18 | 1.2 | 22 | 3.5 | 5.4 | 0.28 | 0.9 | Q106R+S217R/R262Q |
| Normal range++ | 17–33 | 1.59–1.70 | 21–26 | B4-B5 P4-P5 | Regular | 61–82 | 2.6–11 | 28–71+++ | 2.2–8+++ | 8.2–22+++ | 2.4–7.9+++ | 5.6–7.8+++ | 0.1–0.6+++ | 0.7–2.25 |
| Patients | Age | Height | BMI | Tanner | Menses | Uterine height | Ovarian volume | E2 | LH basal | LH peak | FSH basal | FSH peak | TT | ∆4-A | Allele 1/allele 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| years | m | kg/m2 | mm | (R/l), ml | pg/ml | IU/l | IU/l | IU/l | IU/l | ng/ml | ng/ml | ||||
| 1+ | 18 | 1.65 | 24.2 | B2/P3 | PA | 27 | NA / NA | 12 | 1.2 | 23 | 1.3 | 4.6 | 0.23 | 0.7 | Y290F/Y290F |
| 2+ | 22 | 1.67 | 24.7 | B3/P2 | PA | 32 | 0.9 / 1.0 | 20 | 1.5 | 28 | 0.8 | 3.4 | 0.20 | 0.9 | Y290F/Y290F |
| 3 | 21 | 1.64 | 21.9 | B2/P3 | PA | 27 | 3.3 / 2.8 | 9 | 0.4 | 10 | 0.8 | 3.2 | 0.24 | 1.2 | T269M/T269M |
| 4 | 19 | 1.74 | 20.5 | B3/P4 | Oligo | 64 | 5.1 / 6.9 | 34 | 4.6 | 42.5 | 5.1 | 8.9 | 0.55 | 2.3 | Q106R/V94A |
| 5 | 17 | 1.75 | 27.8 | B3/P4 | PA | 29 | 0.8 / 0.7 | 14 | 1.5 | 18 | 0.9 | 9.7 | 0.19 | 0.4 | Q106R/R139C |
| 6 | 27 | 1.64 | 23.4 | B2/P4 | PA | NA | 1.5 / 1.7 | 3 | 1.1 | 19 | 2.5 | 8.1 | 0.28 | 0.9 | Q106R/R262Q |
| 7+ | 18 | 1.71 | 23.0 | B5/P5 | PA | 51 | 2.2 / 1.5 | 22 | 5.9 | 81 | 7.4 | 19.5 | 0.21 | 1.2 | Q106R/R262Q |
| 8+ | 17 | 1.71 | 20.8 | B1/P4 | PA | 33 | 0.7 / 2.3 | 9 | 0.7 | 9 | 2.8 | 4.8 | 0.40 | 2.1 | Q106R/R262Q |
| 9 | 19 | 1.70 | 22.5 | B5/P5 | PA | 36 | 0.8 / 1.9 | 8 | 0.6 | NA | 0.7 | NA | 0.37 | 1.9 | Q106R/R139H |
| 10* | 37 | 1.65 | 24.0 | B5/P5 | PSA | 52 | 1.8 / 1.6 | 35 | 5.0 | 35 | 5.2 | 12.0 | 0.33 | 0.8 | Q106R/R262Q |
| 11+** | 16 | 1.70 | 22.4 | B3/P4 | PA | 39 | 1.7 / 1.8 | 23 | 2.3 | 10 | 2.7 | 5.5 | 0.17 | 1.1 | Q106R+S217R/R262Q |
| 12+** | 18 | 1.78 | 22.1 | B3/P5 | PA | 44 | 1.9 / 1.6 | 18 | 1.2 | 22 | 3.5 | 5.4 | 0.28 | 0.9 | Q106R+S217R/R262Q |
| Normal range++ | 17–33 | 1.59–1.70 | 21–26 | B4-B5 P4-P5 | Regular | 61–82 | 2.6–11 | 28–71+++ | 2.2–8+++ | 8.2–22+++ | 2.4–7.9+++ | 5.6–7.8+++ | 0.1–0.6+++ | 0.7–2.25 |
PA: primary amenorrhea; PSA: primo-secondary amenorrhea; Oligo: oligomenorrhea; TT: total testosterone; ∆4-A: delta-4-androstenedione; To convert estradiol (E2) in pg/ml to pmoles/l multiply by 3.671. To convert the testosterone values in ng/ml to nanomoles/l, multiply by 3.467 and to ng/dl multiply by 100. To convert ∆4-A values in ng/ml to nanomoles/l, multiply by 3.491. NA: not available.
Patient’s detailed genotypes (allele 1/allele 2; see also Fig. 1 and Supplementary Table SI for detailed genotypes); *in part previously reported (de Roux et al., 1997). ** in part previously reported (de Roux et al., 1999). +members of the same kindred. ++ from 21 healthy women; +++evaluated in early follicular phase.
Patients #1 to #9 belong to families #1 to #7 in Fig. 1.
Clinical characteristics at diagnosis of women with normosmic congenital hypogonadotrophic hypogonadism carrying biallelic mutations in the GnRH receptor gene.
| Patients | Age | Height | BMI | Tanner | Menses | Uterine height | Ovarian volume | E2 | LH basal | LH peak | FSH basal | FSH peak | TT | ∆4-A | Allele 1/allele 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| years | m | kg/m2 | mm | (R/l), ml | pg/ml | IU/l | IU/l | IU/l | IU/l | ng/ml | ng/ml | ||||
| 1+ | 18 | 1.65 | 24.2 | B2/P3 | PA | 27 | NA / NA | 12 | 1.2 | 23 | 1.3 | 4.6 | 0.23 | 0.7 | Y290F/Y290F |
| 2+ | 22 | 1.67 | 24.7 | B3/P2 | PA | 32 | 0.9 / 1.0 | 20 | 1.5 | 28 | 0.8 | 3.4 | 0.20 | 0.9 | Y290F/Y290F |
| 3 | 21 | 1.64 | 21.9 | B2/P3 | PA | 27 | 3.3 / 2.8 | 9 | 0.4 | 10 | 0.8 | 3.2 | 0.24 | 1.2 | T269M/T269M |
| 4 | 19 | 1.74 | 20.5 | B3/P4 | Oligo | 64 | 5.1 / 6.9 | 34 | 4.6 | 42.5 | 5.1 | 8.9 | 0.55 | 2.3 | Q106R/V94A |
| 5 | 17 | 1.75 | 27.8 | B3/P4 | PA | 29 | 0.8 / 0.7 | 14 | 1.5 | 18 | 0.9 | 9.7 | 0.19 | 0.4 | Q106R/R139C |
| 6 | 27 | 1.64 | 23.4 | B2/P4 | PA | NA | 1.5 / 1.7 | 3 | 1.1 | 19 | 2.5 | 8.1 | 0.28 | 0.9 | Q106R/R262Q |
| 7+ | 18 | 1.71 | 23.0 | B5/P5 | PA | 51 | 2.2 / 1.5 | 22 | 5.9 | 81 | 7.4 | 19.5 | 0.21 | 1.2 | Q106R/R262Q |
| 8+ | 17 | 1.71 | 20.8 | B1/P4 | PA | 33 | 0.7 / 2.3 | 9 | 0.7 | 9 | 2.8 | 4.8 | 0.40 | 2.1 | Q106R/R262Q |
| 9 | 19 | 1.70 | 22.5 | B5/P5 | PA | 36 | 0.8 / 1.9 | 8 | 0.6 | NA | 0.7 | NA | 0.37 | 1.9 | Q106R/R139H |
| 10* | 37 | 1.65 | 24.0 | B5/P5 | PSA | 52 | 1.8 / 1.6 | 35 | 5.0 | 35 | 5.2 | 12.0 | 0.33 | 0.8 | Q106R/R262Q |
| 11+** | 16 | 1.70 | 22.4 | B3/P4 | PA | 39 | 1.7 / 1.8 | 23 | 2.3 | 10 | 2.7 | 5.5 | 0.17 | 1.1 | Q106R+S217R/R262Q |
| 12+** | 18 | 1.78 | 22.1 | B3/P5 | PA | 44 | 1.9 / 1.6 | 18 | 1.2 | 22 | 3.5 | 5.4 | 0.28 | 0.9 | Q106R+S217R/R262Q |
| Normal range++ | 17–33 | 1.59–1.70 | 21–26 | B4-B5 P4-P5 | Regular | 61–82 | 2.6–11 | 28–71+++ | 2.2–8+++ | 8.2–22+++ | 2.4–7.9+++ | 5.6–7.8+++ | 0.1–0.6+++ | 0.7–2.25 |
| Patients | Age | Height | BMI | Tanner | Menses | Uterine height | Ovarian volume | E2 | LH basal | LH peak | FSH basal | FSH peak | TT | ∆4-A | Allele 1/allele 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| years | m | kg/m2 | mm | (R/l), ml | pg/ml | IU/l | IU/l | IU/l | IU/l | ng/ml | ng/ml | ||||
| 1+ | 18 | 1.65 | 24.2 | B2/P3 | PA | 27 | NA / NA | 12 | 1.2 | 23 | 1.3 | 4.6 | 0.23 | 0.7 | Y290F/Y290F |
| 2+ | 22 | 1.67 | 24.7 | B3/P2 | PA | 32 | 0.9 / 1.0 | 20 | 1.5 | 28 | 0.8 | 3.4 | 0.20 | 0.9 | Y290F/Y290F |
| 3 | 21 | 1.64 | 21.9 | B2/P3 | PA | 27 | 3.3 / 2.8 | 9 | 0.4 | 10 | 0.8 | 3.2 | 0.24 | 1.2 | T269M/T269M |
| 4 | 19 | 1.74 | 20.5 | B3/P4 | Oligo | 64 | 5.1 / 6.9 | 34 | 4.6 | 42.5 | 5.1 | 8.9 | 0.55 | 2.3 | Q106R/V94A |
| 5 | 17 | 1.75 | 27.8 | B3/P4 | PA | 29 | 0.8 / 0.7 | 14 | 1.5 | 18 | 0.9 | 9.7 | 0.19 | 0.4 | Q106R/R139C |
| 6 | 27 | 1.64 | 23.4 | B2/P4 | PA | NA | 1.5 / 1.7 | 3 | 1.1 | 19 | 2.5 | 8.1 | 0.28 | 0.9 | Q106R/R262Q |
| 7+ | 18 | 1.71 | 23.0 | B5/P5 | PA | 51 | 2.2 / 1.5 | 22 | 5.9 | 81 | 7.4 | 19.5 | 0.21 | 1.2 | Q106R/R262Q |
| 8+ | 17 | 1.71 | 20.8 | B1/P4 | PA | 33 | 0.7 / 2.3 | 9 | 0.7 | 9 | 2.8 | 4.8 | 0.40 | 2.1 | Q106R/R262Q |
| 9 | 19 | 1.70 | 22.5 | B5/P5 | PA | 36 | 0.8 / 1.9 | 8 | 0.6 | NA | 0.7 | NA | 0.37 | 1.9 | Q106R/R139H |
| 10* | 37 | 1.65 | 24.0 | B5/P5 | PSA | 52 | 1.8 / 1.6 | 35 | 5.0 | 35 | 5.2 | 12.0 | 0.33 | 0.8 | Q106R/R262Q |
| 11+** | 16 | 1.70 | 22.4 | B3/P4 | PA | 39 | 1.7 / 1.8 | 23 | 2.3 | 10 | 2.7 | 5.5 | 0.17 | 1.1 | Q106R+S217R/R262Q |
| 12+** | 18 | 1.78 | 22.1 | B3/P5 | PA | 44 | 1.9 / 1.6 | 18 | 1.2 | 22 | 3.5 | 5.4 | 0.28 | 0.9 | Q106R+S217R/R262Q |
| Normal range++ | 17–33 | 1.59–1.70 | 21–26 | B4-B5 P4-P5 | Regular | 61–82 | 2.6–11 | 28–71+++ | 2.2–8+++ | 8.2–22+++ | 2.4–7.9+++ | 5.6–7.8+++ | 0.1–0.6+++ | 0.7–2.25 |
PA: primary amenorrhea; PSA: primo-secondary amenorrhea; Oligo: oligomenorrhea; TT: total testosterone; ∆4-A: delta-4-androstenedione; To convert estradiol (E2) in pg/ml to pmoles/l multiply by 3.671. To convert the testosterone values in ng/ml to nanomoles/l, multiply by 3.467 and to ng/dl multiply by 100. To convert ∆4-A values in ng/ml to nanomoles/l, multiply by 3.491. NA: not available.
Patient’s detailed genotypes (allele 1/allele 2; see also Fig. 1 and Supplementary Table SI for detailed genotypes); *in part previously reported (de Roux et al., 1997). ** in part previously reported (de Roux et al., 1999). +members of the same kindred. ++ from 21 healthy women; +++evaluated in early follicular phase.
Patients #1 to #9 belong to families #1 to #7 in Fig. 1.
Twenty-two female patients with GnRH deficiency (GNRHD: hypothalamic CHH) (mean age 22 years, range: 17–29) served to compare the pituitary response to GnRH stimulation. In GNRHD, CHH was defined as reported elsewhere (Bry-Gauillard et al., 2017 and see Supplementary Data).
Seventy patients with PCOS (mean age 24 years, range 17–35) served to compare the GnRH response of women with nCHH/bi-GNRHR. PCOS was diagnosed according to the Rotterdam criteria, updated for the number of antral follicles (Dewailly, 2016; Teede et al., 2018). The mean serum AMH concentration in these women with PCOS was 65.7 ± 33.8 pmol/l (normal range in young women: 18–90), and the mean total circulating testosterone level was 0.79 ± 0.2 ng/ml (normal range in young women: 0.17–0.62).
As relevant comparators for uterine height (UH) measured in women with nCHH/bi-GNRHR, we selected 15 additional patients with PCOS. These patients were all under 20-years old (range: 16–19 years). Their condition was revealed by primary amenorrhea (n = 7) or primary–secondary amenorrhea (n = 8) (Rachmiel et al., 2008). Their BMI was between 22 and 29 kg/m2, their mean ovarian volume between 9 and 16 ml, and their mean antral follicular number/ovary between 18 and 29. Their mean (±SD) total circulating testosterone level was 0.71 ± 0.14 ng/ml.
Controls
Healthy female volunteers aged over 18 years were recruited through the press and enrolled after a thorough history-taking and clinical examination (Bry-Gauillard et al., 2017). All the healthy controls had a normal age of pubertal onset (12–13.6 years), regular spontaneous and ovulatory menstrual cycles (28–32 days) and normal BMI (see Supplementary Data).
Protocol
The GnRH test (100 microgrammes i.v.) was performed in 11 of the 21 controls between Day 3 and Day 6 after spontaneous menses onset. In 11 of the 12 nCHH/bi-GNRHR patients and in all 21 GNRHD women, the GnRH test was performed at an unspecified time (amenorrheic women) at diagnosis and before any prior pulsatile GnRH priming. In the PCOS group, the GnRH test was carried out 3–6 days after onset of bleeding provoked by non-antigonadotropic progestin administration (progesterone and retroprogesterone) or at a random period. In all controls and patients, serum LH and FSH levels were measured 15 min before and 0, 15, 30, 60 and 120 min after GnRH injection (time 0). Transvaginal pelvic ultrasound was performed in nCHH/bi-GNRHR patients by the same experienced ovarian ultrasonographer (Bry-Gauillard et al., 2017) using a Voluson E8 Expert device (BT13, 3D-4D; General Electric Systems, Velizy, France) equipped with a 6- to 12-MHz transvaginal transducer. Ultrasound measurements were made in real time, using a standardized protocol.
Hormone assays
Serum total estradiol (E2), total testosterone, AMH, inhibin-B, LH and FSH were measured with sensitive immunoassays as previously reported (Francou et al., 2016; Bry-Gauillard et al., 2017). The detection limits for E2 was 2 pg/ml (7.3 pmol/l).
Genetic analysis
In the 12 patients, genomic DNA extraction and the three coding exons and intron–exon junctions of GNRHR were sequenced as previously reported (de Roux et al., 1997; Tello et al., 2012; Francou et al., 2016). Genes encoding the GnRH receptor 1, kisspeptin 1, kisspeptin 1 receptor, tachykinin 3, tachykinin receptor 3 (Bouligand et al., 2009; Francou et al., 2016) and fibroblast growth factor receptor 1, prokineticin-2/prokineticin receptor-2, fibroblast growth factor-8, semaphorin 3 A, chromodomain helicase DNA-binding protein 7, WD repeat-containing protein-11, sex determining region Y box 10, FEZ family zinc finger 1, fibroblast growth factor 1 and interleukin 17 receptor D were also analyzed using methods published by us (Maione et al., 2018) and others (Supplementary Data). No additional rare variants of other genes involved in CHH/Kallmann syndrome (KS) were found in the 12 women with GNRHR mutations. Likewise, none of the women with GNRHD selected for the GnRH test carried GNRHR mutations, including in the heterozygous state.
Characterization of the V94A, T269M and Y290F GNRHR variants.
In silico analysis
The in silico characterization of V94A, T269M and Y290F rare variants, interspecies conservation analysis and sequence alignment were performed using Homologene on NCBI (http://www.ncbi.nlm.nih.gov/homologene) and Uniprot (The UniProt Consortium, http://www.uniprot.org/) databases. Protein function after selective amino acid substitution was predicted by means of SIFT (http://sift.jcvi.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), the PANTHER Coding SNP Analysis tool (http://www.pantherdb.org/tools/csnpScoreForm.jsp), Mutation Taster (http://www.mutationtaster.org/) and Alamut® (Interactive Biosoftware, Rouen, France).
In vitro functional analyses of T269m and Y290F GNRHR variants
In vitro functional analyses of the T269M and Y290F GNRHR transfected mutants were performed as previously reported (Tello et al., 2012 and Maione et al., 2013). Briefly, for radioligand binding assays, COS-7 cells (fibroblast-like cell line derived from monkey kidney) were transfected with wildtype or mutant GnRHR constructs, incubated with a radiolabeled GnRH analog (125I-[His5,D-Tyr6]-GnRH) alone or with graded concentrations of unlabeled GnRH and bound radioactivity was counted as previously described (Maione et al., 2013). IP accumulation was measured we previously reported (Maione et al., 2013). Briefly, COS-7 cells were transfected and incubated overnight with [3H]-myoinositol (Perkin Elmer, Boston, MA). Radiolabeled cells were stimulated with graded concentrations of GnRH. Total 3H-labeled IP were extracted from cell lysates and counted by liquid scintillation spectroscopy. The response to mitogen-activated protein kinase (MAPK) activation was tested in HEK293T by means of luciferase assay, as we prevously reported (Maione et al., 2013). Cells were transfected with the luc2P/SRE/Hygro plasmid (Promega, Madison, WI, USA) and the pMIR-REPORT™ beta-galactosidase vector (Applied Biosystems, Foster City, CA, USA) as a transfection efficiency housekeeper. Cells were then transfected and exposed to 10−8 M GnRH, full-serum medium or vehicle for 10 min before harvest. We used mouse anti-total and anti-phospho-p44/p42 ERK1/2 and monoclonal anti-actin antibodies at appropriate dilutions.
Statistical analyses
Hormonal parameters were compared by using a parametric t-test or the Mann–Whitney, Wilcoxon or Kolmogorov–Smirnov non-parametric test as appropriate. Statistical analyses were performed and graphics were produced using Prism software, version 5.0 f (GraphPad Software Inc., La Jolla, CA, USA).
Results
Pedigrees and clinical characteristics
Fig. 1 shows the pedigrees of the seven new families of patients with nCHH/bi-GNRHR. Patients belonging to Families #1, #2 and #3 had original genotypes, while patients in Families #4–#7 had recurrent mutations but different genotypes or previously reported genotypes in nCHH males (de Roux et al., 1997; Gianetti et al., 2012; Francou et al., 2016; Hietamäki et al., 2017) (see also Supplementary Table S1). Table I shows the main characteristics of the 12 nCHH/bi-GNRHR patients (see also Supplementary Figs S1 and S2). Patients #1 to #9 belong to Families #1–#7 in Fig. 1. Patients #10–#12 have been partially described elsewhere (de Roux et al., 1997, 1999). Ten patients (83%) had primary amenorrhea. Breast development at the time of diagnosis was variable: one patient had no breast development (B1), while the others had either partial (n = 8) or complete (n = 3) breast development.
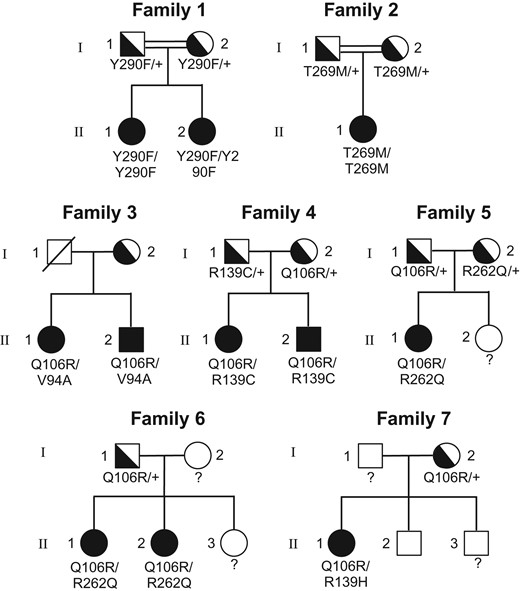
Pedigrees of the nine new women with normosmic congenital hypogonadotrophic hypogonadism carrying biallelic GNRHR mutations. GNRHR: GnRH receptor. Solid symbols indicate affected family members; semi-solid symbols indicate unaffected heterozygotes; open symbols indicate subjects with a normal genotype. Unknown genotypes are indicated by ‘?’. Circles indicate females and squares males. (see also Table I).
Pelvic imaging
Fig. 2 (Panel A) shows UH in nCHH/bi-GNRHR before any estrogen therapy. Compared to the 23 nulliparous healthy controls (UH: 72.0 ± 10.3 mm, mean±SD) and 15 teenagers or young women with PCOS revealed by primary amenorrhea or primo-secondary amenorrhea (UH: 67.0 ± 8.3 mm), the mean UH was significantly smaller in nCHH/bi-GNRHR women (37.0 ± 9.3 mm; respectively, P < 0.0001 and P < 0.001). Fig. 2 (Panel B) shows the positive correlation (r = 0.84; P = 0.001) between UH and the serum estradiol concentration at diagnosis, before any estrogen therapy. Mean (±SD) ovarian volume was 1.6 ± 0.7 ml (range 0.5–6) in nCHH/bi-GNRHR women at diagnosis and was significantly smaller than in healthy controls (5.6 ± 2.2 ml, range 2.6–11, P < 0.001) and those with PCOS (10.7 ± 3.2 ml, range 6.7–22.3 ml, P < 0.0001).
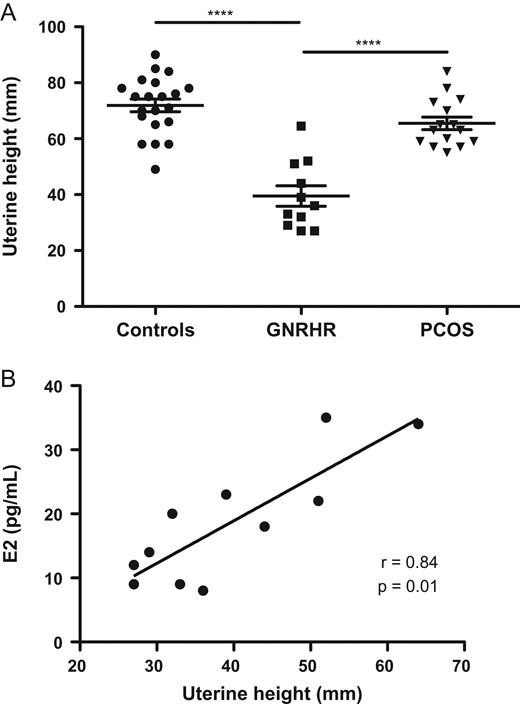
Uterine height in women in the study. A: Comparison of uterine height (UH) between 23 nulliparous healthy women in the early follicular phase (controls) in 11 normosmic congenital hypogonadotrophic hypogonadism (nCHH) patients carrying biallelic GNRHR mutations, and in 15 teenagers with polycystic ovary syndrome (PCOS) or young women in which the condition was revealed by primary amenorrhea or primary/secondary amenorrhea; UH was measured at diagnosis before any hormonal therapy; Horizontal line shows the mean ± SD error bars. B: correlation between UH and circulating estradiol (E2) levels in untreated nCHH patients carrying biallelic GNRHR mutations at diagnosis. All data are individual values. ***P < 0.001. Spearman’s rank correlation.
Hormonal evaluation
Fig. 3 shows the LH response to pituitary stimulation by GnRH. The LH peak was 14.9 ± 4.1 IU/l in healthy controls (Panel A) and 25.5 ± 21.2 IU/l in nCHH/bi-GNRHR women (Panel B), with highly variable individual responses (range 9.0–81.0 IU/l). In the women with CHH due to GnRH deficiency (GNRHD) (Panel C), the mean peak value was 4.8 ± 3.8 IU/l (range 0.2–14.0 IU/l), significantly lower than in the healthy controls and the women with nCHH/bi-GNRHR (Panel E). In patients with PCOS, the mean LH peak (37.5 ± 23.7 IU/l, Panels D and E) was not significantly different from that of nCHH/bi-GNRHR women. There was considerable overlap of the LH peaks between the patients with PCOS and nCHH/bi-GNRHR (Panels B, D and E). Fig. 3 also shows serum estradiol and inhibin-B levels in the four groups. Interestingly, mean±SD estradiol levels (58 ± 35 vs. 15.7 ± 10.0 pg/ml, P < 0.001) and inhibin-B levels (65 ± 36 vs. 13.0 ± 12.0 pg/ml, P < 0.001) were significantly higher in PCOS than in nCHH/bi-GNRHR patients. Serum total testosterone concentrations were lower in nCHH/bi-GNRHR (0.29 ± 0.1 ng/ml) than in PCOS women (0.79 ± 0.2 ng/ml, P = 0.0003). AMH levels were also lower in nCHH/bi-GNRHR (26.4 ± 12.3 pmol/l) than in PCOS women (61.1 ± 40.4 pmol/l, P = 0.02).
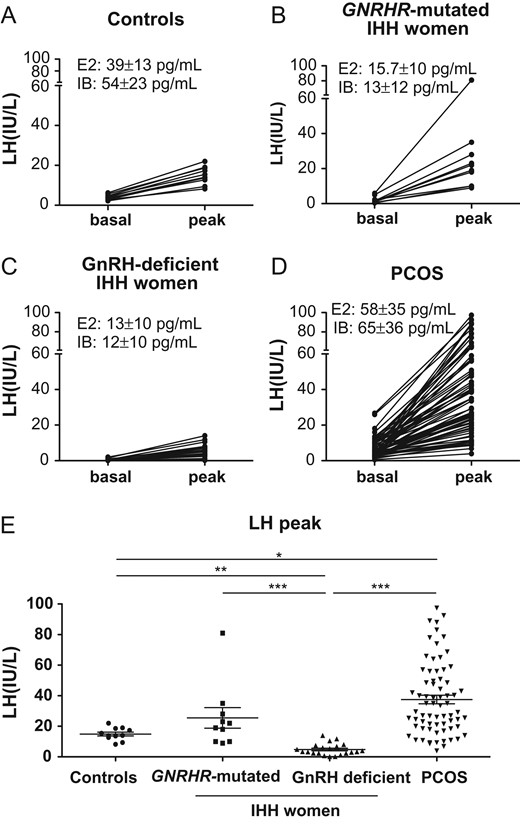
Basal and stimulated (peak) serum LH levels in response to a GnRH bolus in different female groups. A: Female healthy control in the early follicular phase (n = 11); B: Female nCHH patients carrying biallelic GNRHR mutations (n = 11); C: Female patients with CHH due to GnRH deficiency (n = 22: Kallmann syndrome n = 12, normosmic hypothalamic CHH n = 10); D: patients with PCOS (n = 70); E: Comparison of stimulated LH values (peaks) in healthy controls (n = 11), GNRHR-mutated patients (n = 11), GnRH-deficient CHH (n = 21) and PCOS patients (n = 70). Horizontal and error bars indicate mean ± SD. E2: serum estradiol levels (mean ± SD); IB: serum inhibin-B levels (mean ± SD). *P < 0.05, **P < 0.01, ***P < 0.001. Kolmogorov–Smirnov non-parametric test.
In silico mutation analysis
Fig. 4 shows the schematic structure of the GnRH receptor and the locations of the natural mutations found in the 12 patients described here (Panel A), some of which are recurrent (see also Table I). Natural mutations V94A, T269M and Y290F affect highly conserved amino acids (Panel B). The V94A variant is very rare, with an allelic frequency of 1.4 × 10−4 in the Exome Aggregation Consortium (ExAC) database. The T269M variant is also very rare (allelic frequency of 1.6 × 10−5 in ExAC). The Y290F variant was not found among 121,412 alleles analyzed in ExAC (Panel B, see below on the right). Prediction software packages (Min et al., 2016) concordantly indicated that the amino acid substitutions induced by these three mutations are predicted to affect the protein function and likely to be pathogenic.
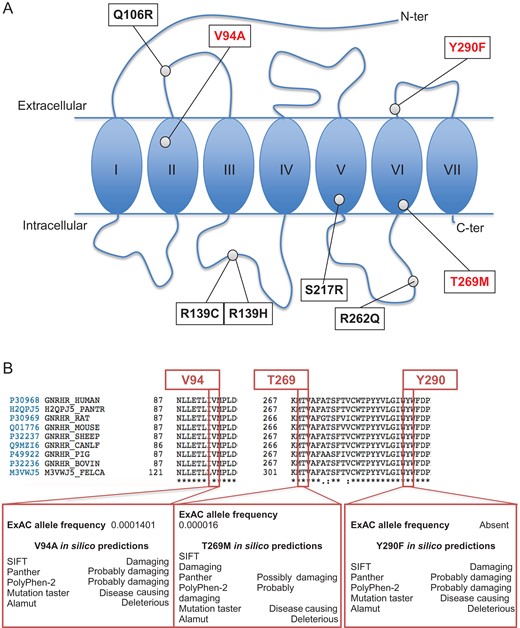
GnRHR mutations found in nCHH patients included in the study and in silico characterization of V94A, T269M and Y290F. A: Schematic representation of the GnRH receptor, with sites of mutations found in nine previously unreported nCHH female patients and in three previously reported cases (see Table I). In red are mutations analyzed in the present work, in black are previously reported and analyzed mutations. B: Characteristics of three mutations in three previously unreported families (Families #1–#3 in Fig. 1, and Subjects #1–#4 in Table I). Interspecies alignment of the V94A, T269 and Y290 amino acid residues, using the Uniprot consortium (available online at www.uniprot.org), showed that these three residues are conserved. Allelic frequency and in silico predictions of V94A, T269M and Y290F amino acid changes are reported in the lower inserts (for details see Materials and Methods and Results sections, and Supplementary Data).
Functional in vitro characterization of the T269M and Y290F GNRHR mutations
Fig. 5 shows the functional characteristics of the two natural mutations T269M and Y290F carried in the homozygous state in the two new families (Family 1 and Family 2, Fig. 1). These analyses demonstrate, respectively, complete or partial loss-of-function of the T269M and Y290F mutated receptors (details in Fig. 5, Panels A–D, and in Supplementary Data).
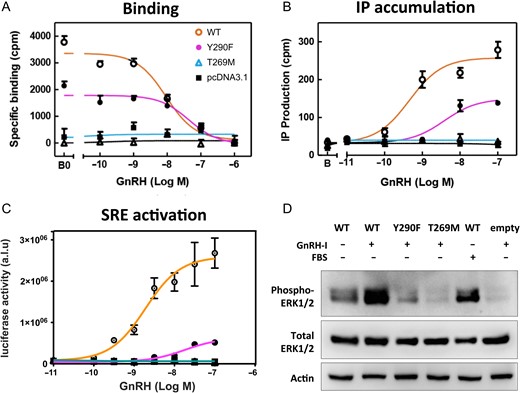
In vitro characterization of the Y290F and T269M GNRHR mutations. A: Competitive binding curves for the Y290F and T269M GNRHR mutants compared to the wildtype (WT) receptor in transiently transfected COS-7 cells (fibroblast-like cell line derived from monkey kidney). Cells were incubated with a 125I-labeled GnRH agonist in the presence of graded concentrations of unlabeled GnRH. Binding was reduced with the Y290F mutant and completely abolished with the T269M mutant. Data are means and range of a single representative experiment performed at least three times in duplicate. B: Inositol phosphate (IP) accumulation in transiently transfected COS-7 cells, showing impaired or abolished response with the Y290F and T269M GnRHR mutants. Data are means and range of a single representative experiment performed at least three times in duplicate. C: Serum responsive element (SRE)-coupled luciferase concentration-response curves in transiently transfected HEK293T cells (derived from human embryonic kidney). Luciferase activity (arbitrary units by luminometry; a.l.u.) are shown as a ratio of beta-galactosidase activity (optical density) used as internal control for transfection efficiency. SRE activity was reduced with the Y290F mutant and absent with the T269M mutant. This experiment was performed four times in triplicate. D: ERK1/2 phosphorylation by western blot in transiently transfected HEK293T cells. After transfection, cells were treated with 10 nM GnRH, 10% fetal bovine serum (FBS) or vehicle, for 10 min. ERK phosphorylation was reduced and absent with the Y290F and T269M mutants, respectively. Data are representative of three experiments. For more details see Materials and Methods and Results and Supplementary Data.
Discussion
The aim of this study was to further characterize the reproductive and endocrine phenotypes of women with CHH/bi-GNRHR. We found that the diagnosis was usually made during adolescence or early adulthood because of primary amenorrhea, as in women affected by others forms of nCHH or KS (Shaw et al., 2011; Bry-Gauillard et al., 2017). As also reported in a series of CHH female patients in which 12% have spontaneous menarche (Shaw et al., 2011), one patient had chronic oligomenorrhea and another had isolated bleeding (de Roux et al., 1997). The variable clinical features of the patients described here, also reported in women with normosmic CHH caused by biallelic GNRHR loss-of-function mutations (Beranova et al., 2001; Brioude et al., 2010; Kim et al., 2010; Gianetti et al., 2012) or KS (Shaw et al., 2011; Bry-Gauillard et al., 2017), seem to be related to differences in ovarian estradiol secretion, as suggested by the correlation between breast development and the serum estradiol concentration at diagnosis. Higher estradiol and milder phenotypes in nCHH/bi-GNRHR could be related to preserved intrinsic LH pulsatility, as reported by Beranova et al. (2001).
Similarly, we found that UH before treatment was less in these patients than in age-matched nulliparous healthy controls. Interestingly, UH also correlated with the circulating estradiol level, showing a relationship between the severity of estradiol deficiency and the degree of uterine hypoplasia. In agreement, previous studies including either CHH, or hypogonadotropic hypogonadism caused by thalassemia or panhypopituitarism, have described uterine hypoplasia in women with gonadotrophin deficiency and an increase of UH under estradiol or GnRH therapy (Stanhope et al. 1985; Cisternino et al.,1991; Tsilchorozidou and Conway, 2004; Tang et al. 2017).
A further aim of this study was to systematically evaluate the pituitary gonadotrophin response to GnRH stimulation in women with CHH/bi-GNRHR as a possible provocative diagnostic tool. Indeed, this pathological model allows one to study, in vivo, the impact of diminished GnRHR cell surface expression, binding and signaling on pituitary gonadotrophin secretion. In this series we found that these patients with pituitary resistance to GnRH in fact had, in line with few previous reports, highly variable LH responses to acute GnRH administration (Layman et al.,1998; de Roux et al., 1999; Beranova et al., 2001; Layman et al.,2001; Dewailly et al. 2002; Meysing et al., 2004; Hietamäki et al., 2017). These LH responses were significantly greater than that of FSH (Table I and Supplementary Figs S1 and S2). These in vivo data are not in line with an in vitro study performed by Meysing et al. where a mutated transfected GNRHR rresulted in a greater loss of LHß transcriptional stimulation than of FSHß (Meysing et al. 2004). It is therefore not always easy to reconcile functional mutant analyses performed in artificial in vitro systems and in vivo integrated responses (de Roux et al., 1999).
Unexpectedly, none of the nCHH/bi-GNRHR women reported here had lesser responses than the healthy controls. The high supraphysiologic dose of exogenous GnRH (100 μg bolus) could therefore overcome the receptor deficiency. Even more surprisingly, nearly half the CHH/bi-GNRHR patients had greater LH responses to GnRH than the controls. Interestingly, the patient with the strongest response to a GnRH bolus (Patient#7, Table I) had a mild phenotype associated with partial loss-of-function mutations. Women carrying biallelic GNRHR mutations with partial loss-of function affecting both alleles and a milder phenotype, have been shown to ovulate either spontaneously or in response to pulsatile GnRH administration via a portable pump (Seminara et al., 2000; Dewailly et al., 2002). Additional studies will be needed to evaluate whether the acute bolus response can predict or not the therapeutic efficacy of chronic pulsatile GnRH administration (Meysing et al. 2004; Abel et al., 2013; Beneduzzi et al., 2014).
In a diagnostic perspective, a normal or even an exaggerated LH response to GnRH does not rule out CHH due to biallelic GNRHR mutation. Our results also challenge the notion that a normal response to GnRH rules out a pituitary cause of CHH (Frohman, 1995). The GnRH provocative test cannot therefore be used for positive diagnosis of gonadotrophin deficiency in this genetic form of CHH. This finding conflicts with some textbooks, which state that the response to GnRH is strongly and constantly impaired in patients with CHH (Frohman, 1995). We should, however, also remember that some reports have shown a lack of pituitary LH response to acute GnRH in patients with CHH/bi-GNRHR (de Roux et al., 1999; Beranova et al., 2001; Costa et al., 2001). The variability of pituitary response to acute GnRH stimulation in CHH could also explain the lack of reliability of this challenge test to differentiate, in boys, CHH from constitutional delay of growth and puberty (Harrington and Palmert, 2012).
In this work we also compared the LH responses of CHH/bi-GNRHR women with those of women with CHH and hypothalamic GnRH deficiency. The two groups had similar serum levels of estradiol and inhibin-B indicating similar severity of the CHH phenotype. The LH peak in response to GnRH was, on average, lower in the GnRHD patients than in nCHH/bi-GNRHR women, with a very weak response in more than 80% of cases. This might suggest that pituitary gonadotrophic dysfunction could be, on average, more severe in this group. GnRHD as demonstrated in KS is associated with hypoplasia/atrophy of pituitary gonadotrophic cells (Kovacs and Sheehan, 1982), which could explain the very weak response to acute GnRH stimulation, possibly due to a drastic reduction in gonadotrophin synthesis and accumulation by gonadotroph cells that makes them incapable of releasing LH after a single stimulatory dose of GnRH (Santen, 1999). To differentiate the minority of patients with CHH/bi-GNRHR who respond weakly or not to acute GnRH stimulation from those with GnRHD, who in majority did not respond to this acute GnRH challenge test, priming by pulsatile GnRH administration for a few days or weeks has been propose as a useful tool (Bouligand et al., 2009; Abel et al., 2013). It must be recognized, however, that even with this more complex diagnostic/therapeutical tool, variable responses have been described in CHH patients with either pituitary abnormalities (Seminara et al., 2000; Gianetti et al., 2012) or hypothalamic GnRH deficiency (Sykiotis et al., 2010; Abel et al., 2013).
PCOS is the most common cause of anovulation, affecting more than 5% of women (Dewailly, 2016; Teede et al., 2018). PCOS diagnosis remains controversial with challenges defining individual components within the diagnostic criteria and significant clinical heterogeneity across the phenotypes (Teede et al., 2018) but hyperandrogenism and irregular cycles are the most consensual features. In a minority of patients the condition can be suspected in teenagers referred for primary amenorrhea without clear hyperandrogenism (Rachmiel et al., 2008). In this clinical context, PCOS diagnosis is difficult (Teede et al., 2018) and the differential diagnosis between nCHH/bi-GNRHR with mild reproductive phenotype and PCOS can be challenging (Caburet et al., 2017). The exaggerated LH response to GnRH in nCHH/bi-GNRHR women could lead to an erroneous diagnosis of PCOS in centers practicing this challenge test. Indeed, many studies have shown that an exaggerated LH response to GnRH is frequent in women with PCOS (Patel et al., 2004). We show here that the stimulated LH peak is very similar in these two patient groups, which leads us not to recommend this challenge test because of this lack of specificity. Moreover, we also show that, contrary to women with PCOS, nCHH/bi-GNRHR women have a significantly below-normal UH, and low ovarian volume similar to those reported in pre-pubertal girls with physiological gonadotrophin deficiency (Kelsey et al., 2013). In addition, we found that nCHH/bi-GNRHR women had markedly lower estradiol, inhibin-B, AMH and androgen levels than PCOS women. Therefore, differential diagnosis for PCOS definitively cannot be based on a GnRH test but must take into account the patient’s clinical history and presentation (i.e. hyperandrogenism and irregular cycles), as recommended by many guidelines (Teede et al., 2018) and also relying on both ovarian morphology and ovarian steroid/peptide hormone levels.
The majority of the nCHH/bi-GNRHR patients reported here were compound heterozygotes, as are most subjects reported in the literature (Beranova et al., 2001; Brioude et al., 2010; Kim et al., 2010; Beneduzzi et al., 2014; Francou et al., 2016; Maione et al., 2018). In the two new consanguineous families described here, the affected women carried mutations in the homozygous state, while their unaffected relatives carried them in the heterozygous state. Our results show that, in all these families, transmission also occurs in an autosomal recessive manner and in a monogenic context.
We found that the T269M mutation ablated both GnRH binding and signal transduction. Since the threonine269 residue is located close to the cytoplasmic end of transmembrane domain 6 of the wildtype GnRHR (Fig. 4A) and therefore unlikely to directly contact GnRH, these results suggest that the mutant protein may be poorly expressed in vitro. Threonine269 is conserved in GnRHRs of different species (Fig. 4B) and it is thought to form conserved interhelical contacts with other residues of the receptor that both stabilize GnRHR protein expression and regulate receptor activation in response to GnRH binding (Flanagan and Manilall, 2017). This mutation has recently been reported in an nCHH woman who is a simple heterozygote, together with a recurrent GNRHR mutation. However, in this report the two mutations were located on the same allele and were associated with a frequent GNRH1 variant (23%) (Zernov et al., 2016). In another recent publication, this mutation was also found in the heterozygous state in a man with CHH, in association with a KISS1R frameshift mutation in a context of probable oligogenism (Nair et al., 2016).
The other mutation in the homozygous state was Y290F. This variant was predicted to be damaging by all software used and we could demonstrate a partial loss-of-function in vitro. This mutation has not previously been reported in CHH patients and is not present in the ExAC database. It is therefore an extremely rare variant. However, using a site-directed mutagenesis approach we previously predicted (Coetsee et al., 2008) that Y290 was essential for binding interaction with Tyr5 of GnRH and demonstrated that binding affinity was impaired when mutating this residue to F, as in the patients, and showed that the aromatic moiety and the hydroxyl group in the GnRH ligand and the GnRHR are essential for high binding affinity. We also show here that the Y290F substitution also affects MAPK phosphorylation and serum responsive element-related signaling, indicating a partial loss-of-function in these two GnRHR major signaling pathways.
Conclusion
Breast and uterine development was variable in this series of women with nCHH/bi-GNRHR and correlated with their circulating estradiol level. Primary amenorrhea was largely present, making it a major sign of this genetic form of CHH. Unexpectedly, the LH response to GnRH stimulation was similar to normal subjects or exaggerated as in patients with PCOS. This could lead to a misdiagnosis but careful evaluation of ovarian volume, circulating AMH and inhibin-B could provide clues to differentiate the two conditions. Finally, we show in vitro the deleterious nature of the two natural mutations found in the homozygous state in two new consanguineous families.
Authors’ roles
Study design: L.M. and J.Y.
Execution: L.M., A.F., I.C.N., A.M., B.F., S.T., J.B. and C.A.F.
Analysis: L.M., A.F., B.D., C.A.F., J.B., P.E.M., R.P.M. and J.Y.
Manuscript drafting: L.M., C.A.F., R.P.M. and J.Y.
Critical discussion: A.F., B.F., A.G.M. and J.B.
Funding
The study was finded by National Research Agency (Agence Nationale de la Recherche (ANR)) grant ANR-09-GENO-017 KALGENOPATH, France; and by the Italian Ministry of Education, University and Research 89 (Ministero Italiano dell'Università e della Ricerca (MIUR)) grant Projects of Relevant National Interest (PRIN) 90 2012227FLF_004, Italy.
Conflict of interest
The authors declare no conflict of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}