





Abstract
Is next generation sequencing (NGS) capable of detecting smaller sub-chromosomal rearrangements in human embryos than the manufacturer’s quoted resolution suggests?
NGS was able to detect unbalanced chromosome segments smaller than the manufacturer’s resolution.
Array Comparative Genomic Hybridization (array-CGH) has been the gold standard platform used for PGD of chromosome rearrangements. NGS is a viable alternative to array-CGH for PGD of chromosome arrangements given that the manufacturer’s guidelines quote a resolution of ≥20 Mb. However, as many patients carry a chromosome rearrangement <20 Mb, the detection limits of NGS warrant further investigation.
This study involved a retrospective assessment of stored DNA samples from embryos that had previously been diagnosed as unbalanced by array-CGH as part of routine PGD in two separate IVF clinics between November 2013 and April 2017. SurePlex whole genome amplification (WGA) products derived from DNA extracted from an embryo biopsy sample known to carry an unbalanced form of a chromosome rearrangement were subjected to a specific NGS workflow (VeriSeq PGS). The results from the two technologies were compared for each sample.
WGA products from 200 embryos known to carry unbalanced rearrangements were sequenced and analysed. These embryos had been created by 75 patients known to carry a chromosome rearrangement (68 reciprocal translocations, 3 pericentric inversions, 1 paracentric inversion, 2 insertions and 1 dual reciprocal and inversion). Each sample was assessed for the size of the segmental gain/loss (Mb), copy number for each segment and chromosome, segregation pattern, the number of bins in the analysis software used and concordance with array-CGH results.
A total of 294 unbalanced chromosome segments were assessed. NGS was capable of detecting 285/294 (97%) unbalanced segments previously identified using array-CGH. The final PGD diagnosis was concordant for 200/200 (100%) embryos. In total, 44/75 (59%) patients contained an unbalanced chromosome segment below the quoted 20 Mb manufacturer’s stated resolution. Of these, 35/44 (80%) patients had segments that were able to be detected using NGS, whilst maintaining clinical outcome concordance.
Our study subset did not include any rearrangements involving the Y chromosome. NGS has less available bins per chromosome compared to the array-CGH platform used, thus it remains possible that chromosome rearrangements predicted to be small but still detectable by array-CGH may not be feasible for testing using NGS. This should be considered when undertaking a theoretical feasibility assessment for detecting the chromosome rearrangement in question. Only one specific workflow for WGA and NGS was investigated in this study.
This study has shown that NGS is available for the detection of unbalanced chromosome rearrangements ≥10 Mb.
Part sponsorship of the VeriSeq PGS kits used was provided by Illumina. The remainder of the kits were provided by two commercial IVF clinics. None of the authors has any conflicting interests to declare.
N/A.
Introduction
Approximately 3% of the human population carry a balanced chromosome rearrangement (Phelan et al., 1996). Chromosome rearrangement carriers are considered ‘balanced’ because all their genetic information is present, however they are at increased risk of producing chromosomally unbalanced gametes (Gardner, 2012). There are different categories of chromosome rearrangements that include reciprocal translocations, inversions (paracentric and pericentric) and insertions to name a few, all of which arise from different mechanisms.
One such sub-group of chromosomal rearrangements, reciprocal translocations, are carried by ~0.2% of the population (Jacobs et al., 1992). For a reciprocal translocation, a two-way exchange of material takes place between two chromosomes. The translocation chromosomes pair to form a quadrivalent at the pachytene stage of meiosis. Only embryos with a normal chromosomal complement or balanced rearrangement are capable of forming a healthy ongoing pregnancy (Supplementary Fig. S1A). Another sub-group of chromosomal rearrangements are inversions. Approximately 0.12–0.7% of the population carry a pericentric inversion and 0.1–0.5% of the population carry a paracentric inversion (Van Dyke et al., 1983; Pettenati et al., 1995). Inversions occur following a two-break event on one chromosome, whereby the segment rotates 180°, reinserts itself and the breaks unite. While paracentric inversions are generally innocuous (Gardner, 2012), pericentric inversions result in the formation of an inversion loop during meiosis and, if recombination occurs within the inverted segment, four possible gametes are possible (Supplementary Fig. S1B). Insertions are the most uncommon form of chromosome rearrangement. Approximately 1 in 80 000 of the population are carriers of insertions (Van Hemel and Eussen, 2000). Insertions involve three breaks on two chromosomes. The first two breaks occur on the same chromosome, with the interstitial segment released and inserted into a third break on a different chromosome. Following fertilization, created gametes can result in four possible segregation types; two with the correct amount of genetic material (balanced insertion carriers) and two without (unbalanced). These unbalanced combinations contain either a deletion or duplication of the insertion segment (Supplementary Fig. S1C). In rare cases, a quadrivalent may form leading to recombinant chromosomes (Gardner, 2012).
PGD offers an alternative to invasive prenatal diagnosis for couples who carry a chromosome rearrangement. PGD occurs as part of IVF treatment prior to the transfer and implantation of an embryo, providing the opportunity to distinguish normal and/or balanced embryos (which have the potential to result in a successful ongoing pregnancy) from unbalanced embryos (which may result in implantation failure, miscarriage or the birth of a chromosomally unbalanced child). However, it must be noted that embryos that carry a normal chromosomal complement and embryos that carry balanced forms of rearrangements may not be differentiated during the PGD process. Initially Array Comparative Genomic Hybridization (array-CGH) (24sure+, Illumina) was used for the detection of unbalanced chromosome rearrangements, with a quoted resolution of 10 Mb. However, since this platform was made obsolete, the validation of alternate technology/platforms for the detection of unbalanced chromosome rearrangements in preimplantation embryos was required. Many of these alternate platforms have a minimum resolution that is greater than that of array-CGH. For example, Illumina quote a resolution of ≥20 megabases (Mb) for SurePlex whole genome amplification (WGA) products when using the VeriSeq PGS protocol (as per manufacturer guidelines). While unbalanced chromosome rearrangements involving segments ≥20 Mb are therefore capable of being detected by this next generation sequencing (NGS) protocol, smaller rearrangements (<20 Mb) require further validation to define the limit of detection prior to NGS being offered as a viable alternative to array-CGH.
The primary aim of this study was to assess the concordance between array-CGH and NGS for the detection of unbalanced rearrangements in blastocyst biopsy samples and to define the limit of detection for NGS. Other factors assessed were the specific chromosomes involved in the rearrangement, the size of the segment/s involved, the specific chromosome breakpoints involved, the number of bins available (i.e. normalized read count for each region of the genome) and whether all unbalanced forms of the chromosome rearrangement could be detected.
Materials and Methods
Assessment of individual chromosome data
To determine the precise size and the number of bins available for each chromosome, a whole genomic male DNA (60 pg) control (Promega #G1471) was sequenced using the VeriSeq PGS kit—MiSeq (Illumina, USA), as described below. The chromosome length and number of bins for each chromosome (including chromosome arms) was determined using the ‘Data Table’ report produced by the Bluefuse Multi software. The ‘Data Table’ lists all available bins and the corresponding location of the chromosome band along with the human genome region. Using this information, the available bins per chromosome arm were counted. Chromosome length was calculated by subtracting the genomic position of the last available data point from the first and converting to Mb. A secondary sizing measure was also performed using the highlight and draw function of the software which automatically calculates the width of the selected region in Mb.
Study design and study groups
This study involved a retrospective analysis of 200 embryos, from 75 patients who were known to carry a chromosome rearrangement. Patients had presented to one of two clinic sites, Monash IVF (Clayton, Australia) and Repromed (Dulwich, Australia) between November 2013 and April 2017. The study entailed the use of stored frozen WGA DNA products derived from embryos known to carry a chromosome rearrangement that had previously been diagnosed as unbalanced using array-CGH (24sure+, Illumina).
WGA products were generated following blastocyst biopsies conducted at the two clinics. Samples were processed and analysed using a standard NGS protocol platform (VeriSeq PGS—MiSeq, Illumina). The results of the two PGD technologies were compared. This study was approved by the Repromed Scientific Advisory Committee. Financial support for the study was provided in part by Illumina in the form of provision of VeriSeq NGS kits. The remainder of kits and staff time were provided by the Monash IVF Group.
PGD feasibility assessment
Prior to commencing an IVF–PGD cycle, all couples undergo mandatory genetic counselling as per National Health and Medical Research Council guidelines (NHMRC, 2017). To determine if PGD was a feasible testing option, a theoretical assessment of the chromosome rearrangement breakpoints was performed to determine if the array-CGH/NGS platform had adequate resolution to distinguish the normal/balanced embryos from all unbalanced embryos. For array-CGH, the chromosome rearrangement breakpoints, as reported in the patient’s karyotype, were entered into the 24Sure+ reciprocal translocation detection tool (BlueGnome, Illumina). This tool determines the probability of detection which is calculated based on the position of the chromosome breakpoints and the size of the translocated fragments. If the overall probability of detection was >0.98 (manufacturer’s recommendation), PGD using array-CGH was considered to be feasible.
The feasibility of detection for a chromosome rearrangement by NGS was also determined using the 24Sure+ reciprocal translocation detection tool to calculate the size of the chromosomal segments. Feasibility was also assessed by ‘mapping’ the co-ordinates of the chromosome rearrangement using the Bluefuse Multi Software V4.4 (Illumina) to determine the size of the chromosome fragments and the number of bins available in each fragment. In order to avoid a potential PGD misdiagnosis, it is important to assess each karyotype individually and ascertain if all unbalanced modes of segregation can be distinguished from the normal/balanced modes of segregation. For analysis of the individual karyotypes, based on the chromosome breakpoints, each reciprocal translocation was classified into four segments (two per chromosome), each inversion was classified into three segments (inverted segment, material distal to inverted segment, material proximal to inverted segment) and each insertion was classified into three segments (inserted segment, material distal to inserted segment, material proximal to inserted segment). It was then determined whether each potentially unbalanced segment was large enough (i.e. ≥20 Mb) to be detected using NGS. For example, for a reciprocal translocation a minimum of three out of the four possible chromosome segments need to be of sufficient size to ensure detection. If only two chromosome segments are able to be detected, unbalanced adjacent-1 embryos will be misdiagnosed as normal/balanced. Offering PGD with less than three detectable segments for reciprocal translocations carries an inherent risk of misdiagnosis, as adjacent-1 segregation is often the most common form of malsegregation that can give rise to the birth of an abnormal child (Midro et al., 2000; Gardner, 2012; Beyer and Willats, 2017). NGS was considered a feasible testing platform for the chromosome rearrangement in question if the enough segments were >20 Mb. In cases involving segments <20 Mb, the feasibility of NGS was further assessed by processing unbalanced DNA (e.g. from embryos previously diagnosed as unbalanced by array-CGH or from an unbalanced product of conception) on the NGS platform. If the imbalance was detected on NGS and NGS was capable of distinguishing all unbalanced forms of segregation from the normal/balanced forms, NGS could be offered for PGD testing in future cycles.
Whole genome amplification
Following blastocyst biopsy on Day 5/6 post-oocyte collection (Munne and Wells, 2017), each biopsy sample was washed through a series of 20 μl 1xPBS drops (Cell Signalling Technologies, USA) before being transferred to a sterile 0.2 μl PCR tube containing 2.5 μl of 1xPBS. WGA was performed using the SurePlex DNA Amplification System as per the manufacturer’s instructions (Illumina).
Array-CGH
WGA products (as well as male SureRef reference DNA (#PR-40-415205-PK, Illumina)) were fluorescently labelled with Cy3 or Cy5 fluorophores using the Fluorescent Labelling System (Illumina) and competitively hybridized to 24Sure+ arrays (Illumina). Arrays were washed and then scanned using an Agilent C DNA microarray scanner (using two colour scan settings at 10 μm resolution). WGA, labelling, hybridization, array washing and array scanning were all performed using the manufacturer’s protocol and reagents. Samples were analysed using BlueFuse Multi Software V4.1–4.4 (Illumina), with analysis performed as per the manufacturer’s recommendations. Samples were considered unbalanced if a copy number change ≥40% (calculated based on X chromosome separation level) was observed within the location of the chromosome rearrangement (as indicated by the patient’s karyotype). Samples were considered normal/balanced if the copy number of each segment deviated <40%. Samples displaying a copy number change (gain/loss) in chromosomes not involved in the rearrangement were considered aneuploid if a gain/loss of ≥80% was detected (calculated based on X chromosome separation level).
Reanalysis using NGS
Archived embryonic DNA samples (previously amplified using SurePlex) were retrieved from long term storage and subjected to NGS using the VeriSeq PGS kit—MiSeq (Illumina). Tagmentation, sample barcoding, parallel sequencing and alignment were all performed using the manufacturer’s protocol and reagents. Samples were analysed using BlueFuse Multi Software V4.4 (Illumina), with analysis performed as per the manufacturer’s recommendations. Samples were considered aneuploid if a copy number change ≥70% was detected or mosaic if a copy number change ≥30% and <70% was detected. Samples with a copy number change ranging from ≥20 to <30% were also reported as mosaic if the overall dynamic range for the sample was within 10% and all the QC measures were within the acceptable range as per manufacturer’s guidelines. Any copy number deviations <20% were deemed to have a normal or balanced chromosome complement. These diagnostic thresholds were set previously based on in-house validation studies using set ratios of euploid and aneuploid DNA mixtures to mimic mosaic samples (data not shown). The final diagnosis also took into account the data quality, read depth, dynamic range/noise, the presence of ramping artefacts or step changes, and any recorded issues with biopsy or cell-to-tube transfer (e.g. the presence of lysed cells).
Limitations
A limitation of our study was that the data subset did not include any chromosome rearrangements involving the Y chromosome. All other chromosomes were represented at least once in the assessed rearrangements.
Another limitation of this study is the difference in detection capabilities between array-CGH and NGS technologies. NGS has been reported to be superior to array-CGH for the detection of chromosomal mosaicism (Munne et al., 2016; Spinella et al., 2018), however, NGS also has less available bins per chromosome compared to the array-CGH platform used. At the time of this study, embryos diagnosed as normal/balanced for the chromosome rearrangement but mosaic for other chromosome/s were diagnosed as ‘mosaic’ and not recommended for transfer.
Both array-CGH and NGS have differences for the detection of chromosome rearrangements involving acrocentric chromosomes (i.e. chromosomes 13, 14, 15, 21 and 22). Acrocentric chromosomes have a p-arm that consists of centromeric heterochromatin (band p11), satellite stalk (band p12) and satellite material (band p13) (Gardner, 2012). As the satellite DNA of all the acrocentric chromosomes contains significant amounts of repetitive DNA sequences (usually clinically insignificant), there are little or no data points available for these chromosomes (Table I). For example, the p-arm of chromosome 22 is not able to be detected by NGS; however, it is able to be detected by array-CGH due to extra coverage of the BAC clones in this region.
Detailed breakdown of each chromosome including the chromosome arm sizes and the number of NGS bins available on each arm.
| Chromosome | p-arm | q-arm | ||||
|---|---|---|---|---|---|---|
| # | Size (Mb) | # Bins | Size (Mb) | # Bins | Size (Mb) | # Bins |
| 1 | 248.2 | 196 | 119.3 | 105 | 115.7 | 91 |
| 2 | 242.0 | 210.0 | 92.7 | 80 | 149.3 | 130 |
| 3 | 197.1 | 175.0 | 88.7 | 81 | 105.7 | 94 |
| 4 | 190.3 | 168.0 | 47.8 | 43 | 142.5 | 125 |
| 5 | 180.0 | 157.0 | 47.3 | 41 | 132.2 | 116 |
| 6 | 170.3 | 150.0 | 60.2 | 52 | 110.1 | 98 |
| 7 | 158.2 | 133.0 | 56.4 | 50 | 101.8 | 83 |
| 8 | 145.1 | 127.0 | 42.3 | 37 | 102.7 | 90 |
| 9 | 140.0 | 97.0 | 40.2 | 34 | 99.8 | 63 |
| 10 | 134.5 | 114.0 | 37.6 | 33 | 97.0 | 81 |
| 11 | 134.2 | 116.0 | 49.9 | 44 | 84.3 | 72 |
| 12 | 132.7 | 115.0 | 32.9 | 29 | 99.8 | 86 |
| 13 | 94.7 | 86.0 | No data* | 94.7 | 86 | |
| 14 | 87.1 | 78.0 | No data* | 94.4 | 78 | |
| 15 | 80.1 | 68.0 | No data* | 80.1 | 68 | |
| 16 | 89.1 | 65.0 | 32.6 | 25 | 56.6 | 40 |
| 17 | 80.4 | 65.0 | 25.3 | 18 | 55.0 | 47 |
| 18 | 77.0 | 67.0 | 13.7 | 12 | 63.3 | 55 |
| 19 | 58.1 | 45.0 | 25.5 | 19 | 32.6 | 26 |
| 20 | 62.1 | 53.0 | 24.1 | 22 | 38.0 | 31 |
| 21 | 35.3 | 30.0 | 3.5 | 1 | 31.7 | 29 |
| 22 | 33.6 | 28.0 | No data* | 33.6 | 28 | |
| X | 149.7 | 123.0 | 56.3 | 47 | 93.4 | 76 |
| Y | 37.5 | 11.0 | 7.0 | 3 | 30.5 | 8 |
| Chromosome | p-arm | q-arm | ||||
|---|---|---|---|---|---|---|
| # | Size (Mb) | # Bins | Size (Mb) | # Bins | Size (Mb) | # Bins |
| 1 | 248.2 | 196 | 119.3 | 105 | 115.7 | 91 |
| 2 | 242.0 | 210.0 | 92.7 | 80 | 149.3 | 130 |
| 3 | 197.1 | 175.0 | 88.7 | 81 | 105.7 | 94 |
| 4 | 190.3 | 168.0 | 47.8 | 43 | 142.5 | 125 |
| 5 | 180.0 | 157.0 | 47.3 | 41 | 132.2 | 116 |
| 6 | 170.3 | 150.0 | 60.2 | 52 | 110.1 | 98 |
| 7 | 158.2 | 133.0 | 56.4 | 50 | 101.8 | 83 |
| 8 | 145.1 | 127.0 | 42.3 | 37 | 102.7 | 90 |
| 9 | 140.0 | 97.0 | 40.2 | 34 | 99.8 | 63 |
| 10 | 134.5 | 114.0 | 37.6 | 33 | 97.0 | 81 |
| 11 | 134.2 | 116.0 | 49.9 | 44 | 84.3 | 72 |
| 12 | 132.7 | 115.0 | 32.9 | 29 | 99.8 | 86 |
| 13 | 94.7 | 86.0 | No data* | 94.7 | 86 | |
| 14 | 87.1 | 78.0 | No data* | 94.4 | 78 | |
| 15 | 80.1 | 68.0 | No data* | 80.1 | 68 | |
| 16 | 89.1 | 65.0 | 32.6 | 25 | 56.6 | 40 |
| 17 | 80.4 | 65.0 | 25.3 | 18 | 55.0 | 47 |
| 18 | 77.0 | 67.0 | 13.7 | 12 | 63.3 | 55 |
| 19 | 58.1 | 45.0 | 25.5 | 19 | 32.6 | 26 |
| 20 | 62.1 | 53.0 | 24.1 | 22 | 38.0 | 31 |
| 21 | 35.3 | 30.0 | 3.5 | 1 | 31.7 | 29 |
| 22 | 33.6 | 28.0 | No data* | 33.6 | 28 | |
| X | 149.7 | 123.0 | 56.3 | 47 | 93.4 | 76 |
| Y | 37.5 | 11.0 | 7.0 | 3 | 30.5 | 8 |
*There is no data on the number of bins available for the acrocentric chromosomes 13, 14, 15 and 22.
Detailed breakdown of each chromosome including the chromosome arm sizes and the number of NGS bins available on each arm.
| Chromosome | p-arm | q-arm | ||||
|---|---|---|---|---|---|---|
| # | Size (Mb) | # Bins | Size (Mb) | # Bins | Size (Mb) | # Bins |
| 1 | 248.2 | 196 | 119.3 | 105 | 115.7 | 91 |
| 2 | 242.0 | 210.0 | 92.7 | 80 | 149.3 | 130 |
| 3 | 197.1 | 175.0 | 88.7 | 81 | 105.7 | 94 |
| 4 | 190.3 | 168.0 | 47.8 | 43 | 142.5 | 125 |
| 5 | 180.0 | 157.0 | 47.3 | 41 | 132.2 | 116 |
| 6 | 170.3 | 150.0 | 60.2 | 52 | 110.1 | 98 |
| 7 | 158.2 | 133.0 | 56.4 | 50 | 101.8 | 83 |
| 8 | 145.1 | 127.0 | 42.3 | 37 | 102.7 | 90 |
| 9 | 140.0 | 97.0 | 40.2 | 34 | 99.8 | 63 |
| 10 | 134.5 | 114.0 | 37.6 | 33 | 97.0 | 81 |
| 11 | 134.2 | 116.0 | 49.9 | 44 | 84.3 | 72 |
| 12 | 132.7 | 115.0 | 32.9 | 29 | 99.8 | 86 |
| 13 | 94.7 | 86.0 | No data* | 94.7 | 86 | |
| 14 | 87.1 | 78.0 | No data* | 94.4 | 78 | |
| 15 | 80.1 | 68.0 | No data* | 80.1 | 68 | |
| 16 | 89.1 | 65.0 | 32.6 | 25 | 56.6 | 40 |
| 17 | 80.4 | 65.0 | 25.3 | 18 | 55.0 | 47 |
| 18 | 77.0 | 67.0 | 13.7 | 12 | 63.3 | 55 |
| 19 | 58.1 | 45.0 | 25.5 | 19 | 32.6 | 26 |
| 20 | 62.1 | 53.0 | 24.1 | 22 | 38.0 | 31 |
| 21 | 35.3 | 30.0 | 3.5 | 1 | 31.7 | 29 |
| 22 | 33.6 | 28.0 | No data* | 33.6 | 28 | |
| X | 149.7 | 123.0 | 56.3 | 47 | 93.4 | 76 |
| Y | 37.5 | 11.0 | 7.0 | 3 | 30.5 | 8 |
| Chromosome | p-arm | q-arm | ||||
|---|---|---|---|---|---|---|
| # | Size (Mb) | # Bins | Size (Mb) | # Bins | Size (Mb) | # Bins |
| 1 | 248.2 | 196 | 119.3 | 105 | 115.7 | 91 |
| 2 | 242.0 | 210.0 | 92.7 | 80 | 149.3 | 130 |
| 3 | 197.1 | 175.0 | 88.7 | 81 | 105.7 | 94 |
| 4 | 190.3 | 168.0 | 47.8 | 43 | 142.5 | 125 |
| 5 | 180.0 | 157.0 | 47.3 | 41 | 132.2 | 116 |
| 6 | 170.3 | 150.0 | 60.2 | 52 | 110.1 | 98 |
| 7 | 158.2 | 133.0 | 56.4 | 50 | 101.8 | 83 |
| 8 | 145.1 | 127.0 | 42.3 | 37 | 102.7 | 90 |
| 9 | 140.0 | 97.0 | 40.2 | 34 | 99.8 | 63 |
| 10 | 134.5 | 114.0 | 37.6 | 33 | 97.0 | 81 |
| 11 | 134.2 | 116.0 | 49.9 | 44 | 84.3 | 72 |
| 12 | 132.7 | 115.0 | 32.9 | 29 | 99.8 | 86 |
| 13 | 94.7 | 86.0 | No data* | 94.7 | 86 | |
| 14 | 87.1 | 78.0 | No data* | 94.4 | 78 | |
| 15 | 80.1 | 68.0 | No data* | 80.1 | 68 | |
| 16 | 89.1 | 65.0 | 32.6 | 25 | 56.6 | 40 |
| 17 | 80.4 | 65.0 | 25.3 | 18 | 55.0 | 47 |
| 18 | 77.0 | 67.0 | 13.7 | 12 | 63.3 | 55 |
| 19 | 58.1 | 45.0 | 25.5 | 19 | 32.6 | 26 |
| 20 | 62.1 | 53.0 | 24.1 | 22 | 38.0 | 31 |
| 21 | 35.3 | 30.0 | 3.5 | 1 | 31.7 | 29 |
| 22 | 33.6 | 28.0 | No data* | 33.6 | 28 | |
| X | 149.7 | 123.0 | 56.3 | 47 | 93.4 | 76 |
| Y | 37.5 | 11.0 | 7.0 | 3 | 30.5 | 8 |
*There is no data on the number of bins available for the acrocentric chromosomes 13, 14, 15 and 22.
Another limitation to consider when determining if a chromosome rearrangement is feasible for testing is the resolution of the G-banded karyotype. Low-resolution karyotypes may make it difficult to determine if the region of consideration has sufficient bin coverage. Supplementary Fig. S2 illustrates the impact of the differing resolutions of two karyotypes for a chromosome (2;9) translocation carrier.
One final limitation is that only one specific workflow for WGA and NGS was investigated in this study and therefore the findings of this study can only be applied to the VeriSeq NGS platform and not other NGS workflows available in the market.
Results
Individual chromosome analysis
Comprehensive analysis of a male 60 pg Genomic DNA sample was used to determine the NGS resolution for each chromosome. Analysis of this control sample, which had QC metrics well above the optimal recommended thresholds (765213 reads after filtering), was performed to measure the size of each chromosome rearrangement (Mb) and the number of bins (refer to Table I). The p-arm regions of acrocentric chromosomes have no data points and the p-arm regions of chromosomes 19, 20 and 21 have very few bins.
Clinic data summary
For the purpose of this study, from November 2013 to April 2017 Monash IVF Group performed 150 PGD cycles for 102 patients requesting PGD for a chromosome rearrangement. A complete list of the chromosome rearrangements investigated in this study can be found in the Supplementary Table SI.
A total of 413 embryos were biopsied on Day 5/6 and tested using array-CGH. Array-CGH results indicated that 200/413 (48%) embryos carried an unbalanced form of the familial chromosome rearrangement, 125/413 (30%) embryos were normal/balanced with no additional chromosome abnormalities detected and 74/413 (18%) embryos were normal/balanced for the chromosome rearrangement but aneuploid/mosaic for another chromosome unrelated to the chromosome rearrangement. Overall, 73% (75/102) patients who underwent PGD had at least one unbalanced embryo. The types of chromosome rearrangements that resulted in unbalanced embryos included reciprocal translocations (68 patients), pericentric inversions (3 patients), paracentric inversion (1 patient), insertions (2 patients) and a single case of both a reciprocal translocation and a pericentric inversion. The array-CGH cycle information is summarized in Table II.
Patient numbers and array-CGH result information.
| Chromosome rearrangement | Total | |||||
|---|---|---|---|---|---|---|
| Insertion | Paracentric inversion | Pericentric inversion | Reciprocal translocation | Reciprocal translocation + inversion | ||
| Number of PGD cycles | 8 | 1 | 3 | 137 | 1 | 150 |
| Number of unique chromosome rearrangements | 3 | 1 | 8 | 86 | 1 | 99 |
| Number of patients | 3 | 1 | 8 | 89 | 1 | 102 |
| Number of embryos biopsied | 7 | 1 | 20 | 384 | 1 | 413 |
| Number of normal/ balanced embryos (%) | 0 (0) | 0 (0) | 12 (60) | 113 (29) | 0 (0) | 125 (30) |
| Number of unbalanced embryos (%) | 4 (57) | 1 (100) | 4 (20) | 190 (49) | 1 (100) | 200 (48) |
| Number of aneuploid embryos (%)a | 3 (43) | 0 (0) | 4 (20) | 67 (17) | 0 (0) | 74 (18) |
| Number of inconclusive embryos (%)b | 0 (0) | 0 (0) | 0 (0) | 10 (3) | 0 (0) | 10 (2) |
| Number of no result embryos (%)c | 0 (0) | 0 (0) | 0 (0) | 4 (1) | 0 (0) | 4 (1) |
| Chromosome rearrangement | Total | |||||
|---|---|---|---|---|---|---|
| Insertion | Paracentric inversion | Pericentric inversion | Reciprocal translocation | Reciprocal translocation + inversion | ||
| Number of PGD cycles | 8 | 1 | 3 | 137 | 1 | 150 |
| Number of unique chromosome rearrangements | 3 | 1 | 8 | 86 | 1 | 99 |
| Number of patients | 3 | 1 | 8 | 89 | 1 | 102 |
| Number of embryos biopsied | 7 | 1 | 20 | 384 | 1 | 413 |
| Number of normal/ balanced embryos (%) | 0 (0) | 0 (0) | 12 (60) | 113 (29) | 0 (0) | 125 (30) |
| Number of unbalanced embryos (%) | 4 (57) | 1 (100) | 4 (20) | 190 (49) | 1 (100) | 200 (48) |
| Number of aneuploid embryos (%)a | 3 (43) | 0 (0) | 4 (20) | 67 (17) | 0 (0) | 74 (18) |
| Number of inconclusive embryos (%)b | 0 (0) | 0 (0) | 0 (0) | 10 (3) | 0 (0) | 10 (2) |
| Number of no result embryos (%)c | 0 (0) | 0 (0) | 0 (0) | 4 (1) | 0 (0) | 4 (1) |
aEmbryos normal/balanced for chromosome rearrangement and aneuploid/mosaic for chromosome/s unrelated to the chromosome rearrangement.
bAn embryo was diagnosed as inconclusive if there was poor amplification from the biopsied cells or if the data was of poor quality, making it difficult to obtain a conclusive result.
cAn embryo was diagnosed as no result if there was no DNA detected after WGA.
Patient numbers and array-CGH result information.
| Chromosome rearrangement | Total | |||||
|---|---|---|---|---|---|---|
| Insertion | Paracentric inversion | Pericentric inversion | Reciprocal translocation | Reciprocal translocation + inversion | ||
| Number of PGD cycles | 8 | 1 | 3 | 137 | 1 | 150 |
| Number of unique chromosome rearrangements | 3 | 1 | 8 | 86 | 1 | 99 |
| Number of patients | 3 | 1 | 8 | 89 | 1 | 102 |
| Number of embryos biopsied | 7 | 1 | 20 | 384 | 1 | 413 |
| Number of normal/ balanced embryos (%) | 0 (0) | 0 (0) | 12 (60) | 113 (29) | 0 (0) | 125 (30) |
| Number of unbalanced embryos (%) | 4 (57) | 1 (100) | 4 (20) | 190 (49) | 1 (100) | 200 (48) |
| Number of aneuploid embryos (%)a | 3 (43) | 0 (0) | 4 (20) | 67 (17) | 0 (0) | 74 (18) |
| Number of inconclusive embryos (%)b | 0 (0) | 0 (0) | 0 (0) | 10 (3) | 0 (0) | 10 (2) |
| Number of no result embryos (%)c | 0 (0) | 0 (0) | 0 (0) | 4 (1) | 0 (0) | 4 (1) |
| Chromosome rearrangement | Total | |||||
|---|---|---|---|---|---|---|
| Insertion | Paracentric inversion | Pericentric inversion | Reciprocal translocation | Reciprocal translocation + inversion | ||
| Number of PGD cycles | 8 | 1 | 3 | 137 | 1 | 150 |
| Number of unique chromosome rearrangements | 3 | 1 | 8 | 86 | 1 | 99 |
| Number of patients | 3 | 1 | 8 | 89 | 1 | 102 |
| Number of embryos biopsied | 7 | 1 | 20 | 384 | 1 | 413 |
| Number of normal/ balanced embryos (%) | 0 (0) | 0 (0) | 12 (60) | 113 (29) | 0 (0) | 125 (30) |
| Number of unbalanced embryos (%) | 4 (57) | 1 (100) | 4 (20) | 190 (49) | 1 (100) | 200 (48) |
| Number of aneuploid embryos (%)a | 3 (43) | 0 (0) | 4 (20) | 67 (17) | 0 (0) | 74 (18) |
| Number of inconclusive embryos (%)b | 0 (0) | 0 (0) | 0 (0) | 10 (3) | 0 (0) | 10 (2) |
| Number of no result embryos (%)c | 0 (0) | 0 (0) | 0 (0) | 4 (1) | 0 (0) | 4 (1) |
aEmbryos normal/balanced for chromosome rearrangement and aneuploid/mosaic for chromosome/s unrelated to the chromosome rearrangement.
bAn embryo was diagnosed as inconclusive if there was poor amplification from the biopsied cells or if the data was of poor quality, making it difficult to obtain a conclusive result.
cAn embryo was diagnosed as no result if there was no DNA detected after WGA.
Unbalanced segmental size analysis
A total of 294 different chromosome segments were reanalysed using NGS (272 reciprocal translocation segments, 12 inversion segments, 6 insertion segments, 4 reciprocal translocation and inversion combined segments). The 73% percent (75/102) patients had at least one embryo that was diagnosed as unbalanced using array-CGH. Of these patients, 31/75 (41%) carried a chromosome rearrangement in which all segments were ≥20 Mb. The remaining 44/75 (59%) patients carried a chromosome rearrangement in which at least one chromosome segment was <20 Mb. Of the patients with at least one chromosome segment <20 Mb, 21/44 (49%) had segments between 10 and 20 Mb that contained a minimum of 10 bins and were successfully detected by NGS. The remaining 23/44 (52%) had segments <10 Mb or segments that contained <10 bins, of which detection was variable (refer to Table II).
Array-CGH versus NGS concordance
Overall there were 15/75 chromosome rearrangements or 16/294 (5%) chromosome segments (note that one rearrangement had two segments not concordant) that contained breakpoints that were not detected and/or were not concordant with their original array-CGH results (Table III). A total of 9/16 segments could not be detected by NGS, of which six were not concordant with their array-CGH result; three presented as either a full gain or loss of the whole chromosome and three had no change in the copy number observed for the chromosome (Fig. 1, Chromosome 2 example). The other 7/16 (44%) chromosome segments were detected, albeit presented as a mosaic copy number change (±20–50%). All of the chromosome segments that were not able to be detected by NGS were <10 Mb and/or contained <10 bins. Interestingly, there were five karyotypes in the data set that involved a common chromosome 22 breakpoint (22q11.2). The 22pter–22q11.2 segment was not able to be detected by NGS but was able to be detected by array-CGH (Fig. 2). However, it should be noted that regardless of these differences, the clinical calls on the embryos were 100% concordant between the two platforms. Although one segment was not able to be detected, the other segments were and therefore all unbalanced outcomes were able to be differentiated from the normal/balanced outcomes resulting in an unbalanced diagnosis.
Comparison of discordant chromosome rearrangement results.
| Karyotype | Array-CGH result | Detected by NGS | Clinical call between technologies |
|---|---|---|---|
| 46,XX,t(2;10)(q37.3;q24.3) | Deletion of 2q37.3–2qter | No | Concordant |
| Duplication of 10q24.3–10qter | Yes | ||
| 46,XX,t(2;21)(p11.2;q22.3) | Duplication of 2 pt–2p11.2 | Yes | Concordant |
| Trisomy 21 | Yes* | ||
| 46,XY,t(3;5)(q12;p15.31) | Deletion of 3q12–3 qter | Yes | Concordant |
| Duplication of 5pter–5p15.31 | Detected as mosaic (level 2.38) | ||
| 46,XX,t(3;6)(q21;p25) | Deletion of 3q21–3qter | Yes | Concordant |
| Duplication of 6pter–6p25 | Detected as mosaic (level 2.32) | ||
| Duplication of 3pter–3q2 | Yes | Concordant | |
| Deletion of 6p25–6qter | Detected as monosomy 6 | ||
| 46,XX,t(3;13)(q21;q34) | Duplication of 3q22.1–3qt | Yes | Concordant |
| 13q segment not detected | No | ||
| 46,XY,t(3;19)(p26.1;q13.41) | Deletion of 3p26.1–3pter | No | Concordant |
| Duplication of 19q13.41–19qter | Yes | ||
| 46,XX,t(4;15)(q25;q26.3) | Duplication of 4q25–4qter | Yes | Concordant |
| Deletion of 15q26.3–15qter | Detected as mosaic (level 1.45) | ||
| 46,XX,t(6;13)(p12.2;q33.2) | Deletion of 6pter–6p12.2 | Yes | Concordant |
| Duplication of 13q33.2–13qter | Detected as mosaic (level 2.25) | ||
| 46,XX,t(9;13)(q21.3;q12.1) | Duplication of 9pter–9q21.3 | Yes | Concordant |
| 13q segment not detected | No | ||
| 46,XX,t(9;21)(q31.2;q11.2) | Duplication of 9pter–pq31.2 | Yes | Concordant |
| Deletion of 21pter–21q11.2 | Detected as mosaic (level 1.44) | ||
| 46.XY,t(9;22)(q22.3;q11.2) | Deletion of 9q22.3–9qter | Yes | Concordant |
| Duplication of 22q11.2–22qter | Detected as mosaic trisomy 22 (level 2.42) | ||
| 46,XY,t(11;22)(q23.3;q11.2) | Deletion of 11q23.3–11qter | Yes | Concordant |
| Duplication of 22q11.2–22qter | Detected as trisomy 22 | ||
| Deletion of 11q23.3–11qter | Yes | Concordant | |
| Deletion of 22pter–22q11.2 | No | ||
| 46,XY,t(16;18)(q24.3;p11.1) | Duplication of 16pter–q24.3 | Detected as trisomy 16 | Concordant |
| Deletion of 18p11.1–18qter | Yes | ||
| 46,XX,t(16;18)(q24;p11.2) | Deletion of 16q24–16qter | Detected as mosaic (level 1.50) | Concordant |
| Duplication of 18pter–18p11.2 | Yes | ||
| 46,XY,t(16;19)(p11.2;p13.3) | Duplication of 16pter–16p11.2 | Yes | Concordant |
| Trisomy 19 | Yes* |
| Karyotype | Array-CGH result | Detected by NGS | Clinical call between technologies |
|---|---|---|---|
| 46,XX,t(2;10)(q37.3;q24.3) | Deletion of 2q37.3–2qter | No | Concordant |
| Duplication of 10q24.3–10qter | Yes | ||
| 46,XX,t(2;21)(p11.2;q22.3) | Duplication of 2 pt–2p11.2 | Yes | Concordant |
| Trisomy 21 | Yes* | ||
| 46,XY,t(3;5)(q12;p15.31) | Deletion of 3q12–3 qter | Yes | Concordant |
| Duplication of 5pter–5p15.31 | Detected as mosaic (level 2.38) | ||
| 46,XX,t(3;6)(q21;p25) | Deletion of 3q21–3qter | Yes | Concordant |
| Duplication of 6pter–6p25 | Detected as mosaic (level 2.32) | ||
| Duplication of 3pter–3q2 | Yes | Concordant | |
| Deletion of 6p25–6qter | Detected as monosomy 6 | ||
| 46,XX,t(3;13)(q21;q34) | Duplication of 3q22.1–3qt | Yes | Concordant |
| 13q segment not detected | No | ||
| 46,XY,t(3;19)(p26.1;q13.41) | Deletion of 3p26.1–3pter | No | Concordant |
| Duplication of 19q13.41–19qter | Yes | ||
| 46,XX,t(4;15)(q25;q26.3) | Duplication of 4q25–4qter | Yes | Concordant |
| Deletion of 15q26.3–15qter | Detected as mosaic (level 1.45) | ||
| 46,XX,t(6;13)(p12.2;q33.2) | Deletion of 6pter–6p12.2 | Yes | Concordant |
| Duplication of 13q33.2–13qter | Detected as mosaic (level 2.25) | ||
| 46,XX,t(9;13)(q21.3;q12.1) | Duplication of 9pter–9q21.3 | Yes | Concordant |
| 13q segment not detected | No | ||
| 46,XX,t(9;21)(q31.2;q11.2) | Duplication of 9pter–pq31.2 | Yes | Concordant |
| Deletion of 21pter–21q11.2 | Detected as mosaic (level 1.44) | ||
| 46.XY,t(9;22)(q22.3;q11.2) | Deletion of 9q22.3–9qter | Yes | Concordant |
| Duplication of 22q11.2–22qter | Detected as mosaic trisomy 22 (level 2.42) | ||
| 46,XY,t(11;22)(q23.3;q11.2) | Deletion of 11q23.3–11qter | Yes | Concordant |
| Duplication of 22q11.2–22qter | Detected as trisomy 22 | ||
| Deletion of 11q23.3–11qter | Yes | Concordant | |
| Deletion of 22pter–22q11.2 | No | ||
| 46,XY,t(16;18)(q24.3;p11.1) | Duplication of 16pter–q24.3 | Detected as trisomy 16 | Concordant |
| Deletion of 18p11.1–18qter | Yes | ||
| 46,XX,t(16;18)(q24;p11.2) | Deletion of 16q24–16qter | Detected as mosaic (level 1.50) | Concordant |
| Duplication of 18pter–18p11.2 | Yes | ||
| 46,XY,t(16;19)(p11.2;p13.3) | Duplication of 16pter–16p11.2 | Yes | Concordant |
| Trisomy 19 | Yes* |
*Concordant with the array-CGH for full chromosome change, but did not detect segment.
Comparison of discordant chromosome rearrangement results.
| Karyotype | Array-CGH result | Detected by NGS | Clinical call between technologies |
|---|---|---|---|
| 46,XX,t(2;10)(q37.3;q24.3) | Deletion of 2q37.3–2qter | No | Concordant |
| Duplication of 10q24.3–10qter | Yes | ||
| 46,XX,t(2;21)(p11.2;q22.3) | Duplication of 2 pt–2p11.2 | Yes | Concordant |
| Trisomy 21 | Yes* | ||
| 46,XY,t(3;5)(q12;p15.31) | Deletion of 3q12–3 qter | Yes | Concordant |
| Duplication of 5pter–5p15.31 | Detected as mosaic (level 2.38) | ||
| 46,XX,t(3;6)(q21;p25) | Deletion of 3q21–3qter | Yes | Concordant |
| Duplication of 6pter–6p25 | Detected as mosaic (level 2.32) | ||
| Duplication of 3pter–3q2 | Yes | Concordant | |
| Deletion of 6p25–6qter | Detected as monosomy 6 | ||
| 46,XX,t(3;13)(q21;q34) | Duplication of 3q22.1–3qt | Yes | Concordant |
| 13q segment not detected | No | ||
| 46,XY,t(3;19)(p26.1;q13.41) | Deletion of 3p26.1–3pter | No | Concordant |
| Duplication of 19q13.41–19qter | Yes | ||
| 46,XX,t(4;15)(q25;q26.3) | Duplication of 4q25–4qter | Yes | Concordant |
| Deletion of 15q26.3–15qter | Detected as mosaic (level 1.45) | ||
| 46,XX,t(6;13)(p12.2;q33.2) | Deletion of 6pter–6p12.2 | Yes | Concordant |
| Duplication of 13q33.2–13qter | Detected as mosaic (level 2.25) | ||
| 46,XX,t(9;13)(q21.3;q12.1) | Duplication of 9pter–9q21.3 | Yes | Concordant |
| 13q segment not detected | No | ||
| 46,XX,t(9;21)(q31.2;q11.2) | Duplication of 9pter–pq31.2 | Yes | Concordant |
| Deletion of 21pter–21q11.2 | Detected as mosaic (level 1.44) | ||
| 46.XY,t(9;22)(q22.3;q11.2) | Deletion of 9q22.3–9qter | Yes | Concordant |
| Duplication of 22q11.2–22qter | Detected as mosaic trisomy 22 (level 2.42) | ||
| 46,XY,t(11;22)(q23.3;q11.2) | Deletion of 11q23.3–11qter | Yes | Concordant |
| Duplication of 22q11.2–22qter | Detected as trisomy 22 | ||
| Deletion of 11q23.3–11qter | Yes | Concordant | |
| Deletion of 22pter–22q11.2 | No | ||
| 46,XY,t(16;18)(q24.3;p11.1) | Duplication of 16pter–q24.3 | Detected as trisomy 16 | Concordant |
| Deletion of 18p11.1–18qter | Yes | ||
| 46,XX,t(16;18)(q24;p11.2) | Deletion of 16q24–16qter | Detected as mosaic (level 1.50) | Concordant |
| Duplication of 18pter–18p11.2 | Yes | ||
| 46,XY,t(16;19)(p11.2;p13.3) | Duplication of 16pter–16p11.2 | Yes | Concordant |
| Trisomy 19 | Yes* |
| Karyotype | Array-CGH result | Detected by NGS | Clinical call between technologies |
|---|---|---|---|
| 46,XX,t(2;10)(q37.3;q24.3) | Deletion of 2q37.3–2qter | No | Concordant |
| Duplication of 10q24.3–10qter | Yes | ||
| 46,XX,t(2;21)(p11.2;q22.3) | Duplication of 2 pt–2p11.2 | Yes | Concordant |
| Trisomy 21 | Yes* | ||
| 46,XY,t(3;5)(q12;p15.31) | Deletion of 3q12–3 qter | Yes | Concordant |
| Duplication of 5pter–5p15.31 | Detected as mosaic (level 2.38) | ||
| 46,XX,t(3;6)(q21;p25) | Deletion of 3q21–3qter | Yes | Concordant |
| Duplication of 6pter–6p25 | Detected as mosaic (level 2.32) | ||
| Duplication of 3pter–3q2 | Yes | Concordant | |
| Deletion of 6p25–6qter | Detected as monosomy 6 | ||
| 46,XX,t(3;13)(q21;q34) | Duplication of 3q22.1–3qt | Yes | Concordant |
| 13q segment not detected | No | ||
| 46,XY,t(3;19)(p26.1;q13.41) | Deletion of 3p26.1–3pter | No | Concordant |
| Duplication of 19q13.41–19qter | Yes | ||
| 46,XX,t(4;15)(q25;q26.3) | Duplication of 4q25–4qter | Yes | Concordant |
| Deletion of 15q26.3–15qter | Detected as mosaic (level 1.45) | ||
| 46,XX,t(6;13)(p12.2;q33.2) | Deletion of 6pter–6p12.2 | Yes | Concordant |
| Duplication of 13q33.2–13qter | Detected as mosaic (level 2.25) | ||
| 46,XX,t(9;13)(q21.3;q12.1) | Duplication of 9pter–9q21.3 | Yes | Concordant |
| 13q segment not detected | No | ||
| 46,XX,t(9;21)(q31.2;q11.2) | Duplication of 9pter–pq31.2 | Yes | Concordant |
| Deletion of 21pter–21q11.2 | Detected as mosaic (level 1.44) | ||
| 46.XY,t(9;22)(q22.3;q11.2) | Deletion of 9q22.3–9qter | Yes | Concordant |
| Duplication of 22q11.2–22qter | Detected as mosaic trisomy 22 (level 2.42) | ||
| 46,XY,t(11;22)(q23.3;q11.2) | Deletion of 11q23.3–11qter | Yes | Concordant |
| Duplication of 22q11.2–22qter | Detected as trisomy 22 | ||
| Deletion of 11q23.3–11qter | Yes | Concordant | |
| Deletion of 22pter–22q11.2 | No | ||
| 46,XY,t(16;18)(q24.3;p11.1) | Duplication of 16pter–q24.3 | Detected as trisomy 16 | Concordant |
| Deletion of 18p11.1–18qter | Yes | ||
| 46,XX,t(16;18)(q24;p11.2) | Deletion of 16q24–16qter | Detected as mosaic (level 1.50) | Concordant |
| Duplication of 18pter–18p11.2 | Yes | ||
| 46,XY,t(16;19)(p11.2;p13.3) | Duplication of 16pter–16p11.2 | Yes | Concordant |
| Trisomy 19 | Yes* |
*Concordant with the array-CGH for full chromosome change, but did not detect segment.

Example of discordant Array Comparative Genomic Hybridization (array-CGH) versus next generation sequencing (NGS) results on chromosome 2. The array-CGH profile shows a deletion of 2q37.3–2qter (top chart). The NGS profile does not show this deletion of 2q37.3–2qter (Bottom chart).
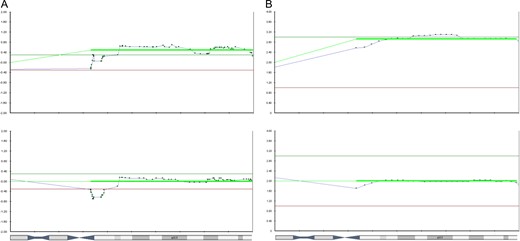
Discordant chromosome 22 results comparing the Array Comparative Genomic Hybridization (array-CGH) and next generation sequencing (NGS) copy number variation (CNV) charts from an embryo from a 46,XY,t(11;22)(q23.3;q11.2) translocation carrier. (A) Shows the array-CGH profile identified a 22q11.2–22qter duplication and a 22pter–22q11.2 deletion. (B) Shows the NGS profile identified trisomy 22 only.
Effect of data quality for detection of segments <10 Mb
An assessment of individual data quality was performed to determine if the processing run quality could affect the ability to detect segments <10 Mb in size. Figure 3 demonstrates this with an example from a 46,XX,t(2;9)(p24;q33) translocation carrier. It was determined that QA/QC measures are important for the detection of segmental gains/losses that are <20 Mb, whereby when an optimum number of reads can be achieved per sample (~500 000) a small segmental gain/loss can be detected that might not be detected with less optimal read number (~250 000).
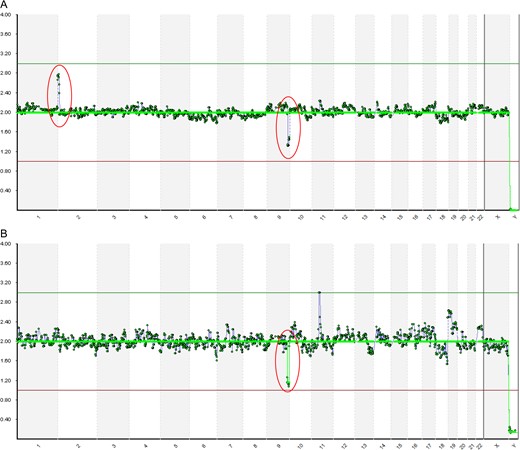
An example of how poor data quality can impact the detection of unbalanced chromosome segments for embryos from a 46,XX,t(2;9)(p24;q33) translocation carrier using next generation sequencing (NGS). The same SurePlex product is shown in both A and B. All quality control (QC) measures were above the acceptable level recommended by the manufacturer. (A) Shows the sample from a run with high data quality which resulted in the detection of the chromosome 2 duplication (2pter–2p25.1). (B) Shows the same sample from a run with suboptimal data quality (the number of reads passing filter were just above 250 000, which was well below the optimal 500 000 reads) which resulted in lack of detection of the chromosome 2 duplication.
Discussion
For many years, array-CGH has been the gold standard of testing for unbalanced chromosome rearrangements in PGD patients. The obsolescence of the 24sure+ array-CGH platform sparked a renewed interest in the validation of other testing platforms to enable continued testing for patients with a chromosome rearrangement. Given the broad use of NGS technology in most PGD clinics (Treff and Zimmerman, 2017), including our own, it was logical to transfer these patients to a NGS platform. This study compared the results of embryos unbalanced for a chromosome rearrangement between two testing platforms (array-CGH and NGS), to determine if NGS was a suitable testing alternative to offer chromosome rearrangements patients.
This study included an assessment of 99 unique chromosome rearrangements from 102 patients who presented to two IVF clinics. A total of 294 unbalanced chromosome segments were assessed by NGS and their results compared to their original array-CGH results. Overall, NGS had a 97% concordance rate with array-CGH for the detection of segmental imbalance in PGD embryos. However, when examining the results based on clinical significance (i.e. would the NGS results have changed the embryos clinical outcome), NGS has a 100% concordance rate for all of the 102 unbalanced embryos tested. In certain instances, the clinical outcome for that embryo would not change, for example a full gain/loss of a chromosome was detected by NGS instead of the duplication/deletion from the specific breakpoint. It is critical to recognize that this result is individualized to the embryo and that the inability to detect the specific breakpoints may result in all forms of malsegregation not being detected and therefore risk possible PGD misdiagnosis.
There are limited reports on the clinical usage of NGS for PGD for embryo screening for carriers of chromosome rearrangements. (Zhang et al., 2016) published a similar study looking at the ability of an in-house NGS platform ‘CNV-Seq’ to accurately detect segmental aneuploidy from known chromosome rearrangement patients previously identified using array-CGH. Karyotypes from prenatal screening matched the preimplantation diagnosis for all patients in the cohort. The CNV-Seq used in the study was quoted to have a higher resolution, detecting segmental aneuploidy as low as 0.8 Mb, compared to the VeriSeq platform used in our study. (Bono et al., 2015) performed a retrospective study comparing the results of unbalanced chromosome rearrangements in embryos previously diagnosed using array-CGH with the Ion torrent PGM sequencer (Life Technologies). The Ion torrent was reported to detect a 5 Mb fragment, however, it did not detect a previously identified 2.8 Mb fragment detected by array-CGH. To the best of our knowledge, there are no published studies directly comparing the results of unbalanced chromosome rearrangements in embryos between array-CGH and the NGS platform (VeriSeq) used in this study.
While it is encouraging that there is such a high level of concordance between array-CGH and NGS, including other NGS platforms, it is important to note that there are key differences between the two testing platforms. The resolution of detection varies for both technologies. NGS has been designed for calling whole chromosome abnormalities and even though the VeriSeq manufacturer states the limit of detection to be 20 Mb, other groups have reportedly validated the technology for the detection of de novo deletion and/or duplication segmental aneuploidies <20 Mb (Vera-Rodriguez et al., 2016; Lai et al., 2017). Array-CGH has been reported to detect segmental aneuploidies 5–6 Mb in size (Colls et al., 2012; Munne, 2012), while other NGS platforms have been reported to detect segmental aneuploidies as small as ~3 Mb in size (Fiorentino et al., 2014). The current study has similarly validated NGS for the detection of segmental aneuploidies below 10 Mb, however, we caution that the ability to detect a segmental aneuploidy of <10 Mb is not guaranteed and multiple other factors must be taken into account when using the technology for an inherited segmental change. Such factors include the location of the aneuploidy (the number of sequence bins in the region) and the quality of the data obtained. The 59% (44/75) patients in this study contained an unbalanced chromosome segment below the manufacturer’s guidelines, of which 35/44 (80%) were able to be detected by NGS. The remaining 20% of segments not detected were all below 10 Mb. This highlights that the resolution of each platform is a significant limitation when undertaking PGD for chromosome rearrangements in embryos.
NGS is more sensitive for the detection of chromosome mosaicism than array-CGH (Munne et al., 2016; Spinella et al., 2018) and the incidence of mosaicism may be a confounding factor when determining if a particular karyotype is feasible for PGD using NGS. Discordant results were observed with copy number variations of chromosome segments being detected as less than full gains/losses (ie at a mosaic level) at an incidence of 2.4% when the segment was below 10 Mb. The exact reasoning for this is unknown, however it could be speculated that when a segment gain/loss is <10 Mb, or if the DNA sample is of poor quality, that there is a reduced number and/or loss of reads available in those regions resulting in a mosaic call, or may be an artefact of the smoothing algorithm used by the calling software. The presence of segmental imbalances appearing to be mosaic may confound the interpretation of NGS data, which means that identification of the segmental imbalance may not be repeatable across all clinical testing sites.
A karyotype that has a small segmental gain/loss that may just be assessed as detectable by array-CGH testing using the probability of detection tool provided by the manufacturer may no longer be feasible using NGS due to the reduced number of bins available. This is a limitation of the NGS technology that should be taken into consideration when doing a theoretical feasibility test for each specific chromosome rearrangement. Therefore, the probability of detection tool provided for the 24Sure+ platform should not be solely relied upon for NGS to predict the likelihood of detection (although it can be a useful adjunct for predicting minimal sizes of gains and losses). Laboratories should instead define their own limits based on location of the chromosome rearrangement in conjunction with the number of bins in the specific region of interest.
Another significant limitation is the size of the specific chromosome segments involved in the rearrangement. Here we demonstrate that patients, who present with chromosome rearrangements with segments >10 Mb or contain a minimum of 10 bins, can undertake an IVF-PGD cycle using NGS. If segments are <10 Mb or containing <10 bins, further investigation should be performed to determine if testing is feasible. Further investigation could include validating the test on DNA from a previous pregnancy that carried an unbalanced form of the rearrangement or if similar gains/losses at breakpoints have been consistently detected in the same region.
Another key consideration when using VeriSeq NGS for the detection of unbalanced chromosome rearrangements is if an acrocentric chromosome is involved. Due to the fact that the p-arms of acrocentric chromosomes contain significant amounts of repetitive DNA sequences, there are little or no data points available for the p-arms of these chromosomes. Therefore, depending on the specific karyotype, there may not be enough bins in each segment to distinguish all of the possible embryo outcomes and therefore determine feasibility. Furthermore, the resolution of the banded karyotype provided, including variation between karyotypes of the same patient, may affect the ability to ascertain what the correct chromosome breakpoints are and if a specific segment contains a sufficient number of bins. In this study, Supplementary Fig. S2 shows an example from a specific patient case of the detrimental effect that two different karyotype resolutions can have on determining feasibility. It would be ideal to have a karyotype resolution of 800 bands, however, if only a low-resolution karyotype is available, clinics should seek repeat karyotypes with a higher resolution to ensure that prediction of detection is as accurate as possible.
The quality of an individual VeriSeq run will affect the analysis of PGD results. For example, it was observed in this study that when the number of filtered reads for an individual sample was less than the manufacturer’s ‘optimal’ recommendation (i.e. 500 000), but greater than the manufacturer’s ‘acceptable’ recommendation (i.e. 250 000), this resulted in the inability to detect a segment below <10 Mb. However, if the manufacturer’s optimal recommended number of reads was achieved or exceeded, detection was possible. This clearly demonstrated that run quality can impact the analysis of NGS results and poorer quality runs made the identification of chromosome segments <20 Mb difficult, increasing the risk of these segments not being detected if VeriSeq run quality is not optimal. If run quality is at or exceeds optimal measures as recommended by the manufacturer, the risk of a possible PGD misdiagnosis is reduced. Therefore, laboratories may need to reassess what the acceptable run metrics are for VeriSeq when it is used to assess the presence of unbalanced chromosome rearrangements compared to whole chromosome copy number changes.
In order for NGS to be used for PGD on embryos from carriers of a chromosome rearrangement, each patient’s karyotype should be assessed on a case-by-case basis prior to the commencement of a stimulated IVF cycle. There are limitations, as discussed above, with using NGS for chromosome rearrangement carriers which should be taken into consideration when investigating if NGS will be able to distinguish all the unbalanced embryo outcomes from the normal/balanced embryo outcomes for a specific karyotype, and therefore determine if PGD testing is feasible. For a patient karyotype to be considered feasible for PGD using VeriSeq NGS, the recommendations as determined by the results of this study are as follows:
– For reciprocal translocation patients: a minimum of three (out of four) chromosome segments must be >10 Mb and/or contain >10 bins, to ensure all modes of malsegregation are able to be detected.
– For inversion and insertion carriers: the same sizing limits apply as for reciprocal translocations, however, a minimum of one out of the two distal segments must be detected for inversions, and the inserted segment must be detected for insertions.
– If a karyotype does not meet the above points and DNA from an unbalanced product is available, feasibility testing can be performed to determine if detection of the smallest and/or limiting segment is possible.
– The run QC metrics should be optimal as per the manufacturer’s guidelines (as opposed to the minimum acceptable limits) when determining if an embryo sample is normal/balanced.
This study identified that the effective resolution is half (ie 10 Mb) that of the manufacturer’s recommended resolution of 20 Mb. However, the ability of detection is not guaranteed when a chromosome rearrangement contains segments below 10 Mb or in chromosome regions containing <10 bins. Therefore, in these circumstances, PGD should only be offered if a positive control (i.e. DNA from an unbalanced product of conception) exists to confirm detection of the chromosome segments below these resolutions.
Acknowledgements
The authors would like to thank all the embryology staff at Monash IVF and Repromed for performing the embryo biopsy procedures and also the genetics staff for their genetic analysis and contributions towards this paper.
Authors’ roles
C.C.: Study concept and design, data acquisition, study execution, analysis, article drafting, critical discussion. C.B.: Study concept and design, study execution, analysis, article drafting, critical discussion. D.B.: Analysis, article drafting, critical discussion. T.F.: Analysis, article review, interpretation of data, critical discussion. J.L.: Articlet review, interpretation of data, critical discussion. E.W.: Article review, interpretation of data, critical discussion. D.Z.-F.: Article review, interpretation of data, critical discussion. J.M.: Study oversight, article review, critical discussion.
Funding
Part sponsorship of the VeriSeq PGS kits used was provided by Illumina. The remainder of the kits were provided by two commercial IVF clinics.
Conflict of interest
None.
References
Author notes
C. Cuman and C.E. Beyer authors consider that the first two authors should be regarded as joint first authors.

{kind=link}
{kind=link}
{kind=link}