





Abstract
Platycodon grandiflorus (Jacq.) A. DC, known for its saponin content, can potentially prevent and treat cerebrovascular diseases and COVID-19. Triterpenoid saponin biosynthesis in plants is enhanced by methyl jasmonate (MeJA) application. However, the underlying molecular mechanisms of MeJA-induced saponin biosynthesis remain unknown in P. grandiflorus. In the current study, exogenous application of 100 μmol/l MeJA was identified to be optimal for promoting saponin accumulation. RNA sequencing analysis demonstrated the PgbHLH28 gene as a key regulatory factor responding to MeJA during saponin accumulation. Overexpression of PgbHLH28 in P. grandiflorus increased saponin content, while silencing of PgbHLH28 significantly inhibited saponin synthesis, suggesting that PgbHLH28 acts as a positive regulator of saponin biosynthesis. Yeast one-hybrid and dual luciferase assays demonstrated that PgbHLH28 directly bound to the promoters of PgHMGR2 and PgDXS2 to activate gene expression. PgHMGR2 and PgDXS2 transformation promoted saponin accumulation, while silencing of these genes inhibited saponin biosynthesis. This study determined that MeJA promoted saponin accumulation in P. grandiflorus by inducing PgbHLH28 gene expression and activating downstream genes (PgHMGR2 and PgDXS2) involved in saponin biosynthesis. In conclusion, a complex regulatory network governing saponin biosynthesis following MeJA treatment was elucidated, offering a theoretical foundation for enhancing saponin content and biosynthesis efficacy in P. grandiflorus.
1. Introduction
Platycodon grandiflorus (Jacq.) A. DC. is a perennial herbaceous plant [1]. Its roots are rich in saponins, the major active components and a key indicator for breeding. Saponins possess pharmacological properties that are effective against chronic inflammatory diseases such as asthma and tuberculosis [2]. As a traditional herbal medicine in the pharmacopeias of China, Korea and Japan [3], the role of P. grandiflorus and other traditional Chinese herbs in the treatment of diseases, particularly COVID-19, has been gaining interest, leading to an expanding range of applications and a significant increase in demand [4, 5]. However, several challenges in P. grandiflorus’s cultivation process include the lack of superior varieties and cultivation measures. Therefore, a complete understanding of the transcriptional regulatory mechanisms of saponin biosynthesis, combined with corresponding cultivation measures, will provide effective solutions for developing cultivars with high-efficiency saponins.
The triterpenoid saponins in P. grandiflorus belong to the oleanane type, which are synthesized primarily in the mevalonic acid (MVA) pathway [6]. Farnesyl pyrophosphate (farnesyl-PP) is synthesized from isopentenyl diphosphate (IPP) and then it undergoes catalysis via squalene synthase (SS), squalene epoxidase (SE), farnesyl-diphosphate synthase (FPPS), oxidosqualene cyclase (OSC), and other major structural genes to form the saponin skeleton [6–8]. Finally, various saponin monomers are formed via hydroxylation, oxidation, and glycosylation catalyzed by cytochrome p450 monooxygenases (CYP450) and glycosyltransferases (GTs) [3, 9]. To date, understanding is primarily limited to the functional genes (i.e. SS, SE, and DS) in saponin biosynthesis. However, the regulatory networks controlling saponin biosynthesis are still unclear and require further elucidation [1, 10, 11].
Methyl jasmonate (MeJA) is a jasmonic acid derivative and is an important regulator of plant growth and secondary metabolite biosynthesis [12]. The exogenous application of MeJA can upregulate enzyme expression involved in terpenoid biosynthesis (i.e. SS, SE, β-AS, and HMGR) to promote the accumulation of terpenoid compounds [7]. Along with the direct regulation of terpenoid biosynthetic structural gene expression, MeJA also enhances saponin compound accumulation via induction of transcription factors (TFs), including bHLH, AP2/ERF, bZIP, and WRKY [13, 14], thereby regulating downstream structural genes. The bHLH TF plays a vital role in synthesizing monoterpenes, sesquiterpenes, and diterpenes in response to MeJA. In Arabidopsis thaliana, the MYC2 TF binds to the promoters of TPS11 and TPS21 genes, facilitating sesquiterpene formation [15]. In diterpenoid biosynthesis, the bHLH TF DPF in rice binds to the cis-acting element (N-box) located in CPS2 and CYP99A2 promoter regions, consequently enhancing the synthesis of diterpenoid toxins [16]. Along with participating in monoterpene and diterpene biosynthesis, bHLH TFs also play important regulatory roles in triterpenoid and tetraterpenoid biosynthesis. In cucumber, bitter leaf (Bl) and bitter fruit (Bt) belong to the bHLH TF family that participates in synthesizing a triterpenoid compound, cucurbitacin C [17]. Furthermore, bHLH TFs regulating triterpenoid biosynthesis have also been identified in plants such as Panax notoginseng [18], Panax ginseng [19], and Betula platyphylla [20]. Overexpression of the bHLH TF PIF5 in A. thaliana T87 suspension culture cells increased the content of tetraterpenoid compounds, such as carotenoids [21].
Exogenous MeJA application can increase the saponin content in the roots of P. grandiflorus. However, since the mechanisms of transcriptional regulation are unknown and require further elucidation, a regulatory network of MeJA-induced saponin biosynthesis was characterized using transcriptome data. The PgbHLH28 gene was identified as a key regulatory factor in MeJA-induced saponin biosynthesis. It binds to the promoter regions of saponin biosynthetic genes (PgDXS2 and PgHMGR2) activating their expression levels and promoting saponin accumulation. These findings are of great significance in understanding the role of MeJA in regulating saponin accumulation via transcriptional regulation. This new understanding provides guidelines for increasing saponin content, enhancing biosynthesis efficacy, and facilitating molecular breeding of P. grandiflorus containing high saponin.
3. Results
3.1. Optimal MeJA concentration for saponin content induction in P. grandiflorus
MeJA functions as an inducer and signaling molecule in saponin biosynthesis [22]. In order to investigate the effect of exogenously applied MeJA on saponin accumulation, we designed four different concentrations of MeJA (10 μmol/l, 50 μmol/l, 100 μmol/l, and 200 μmol/l) to treat the roots of P. grandiflorus. After MeJA treatment, the degree of root browning increased with the concentration of MeJA (Fig. 1A). Saponin accumulation after treatments with four different concentrations MeJA was compared. The results demonstrated that MeJA could promote saponin accumulation, and 100 μmol/l of MeJA was the optimal concentration for promoting saponin accumulation (Fig. 1B). Therefore, 100 μmol/l of MeJA was selected as the optimal treatment concentration for further experiments. Soluble sugars and starch, acting as substrates in the glycolysis, directly impact the biosynthesis and accumulation of saponins via pathways such as MVA and 2-C-methyl-D-erythritol-4-phosphate (MEP). Under the optimal MeJA concentration treatment, soluble sugar and starch content decreased compared to the control (Fig. 1C). Furthermore, the levels of intermediate products (i.e. β-amyrin and oleanolic acid) in saponin biosynthesis were significantly enhanced (Fig. 1C). In addition to the upstream substrates and intermediate products, the two saponin monomers (platycodin D and platycoside E) were also induced by MeJA (Fig. 1C). These results demonstrated that exogenous application of MeJA could promote saponin accumulation, and 100 μmol/l of MeJA was the optimal concentration for promoting saponin accumulation.
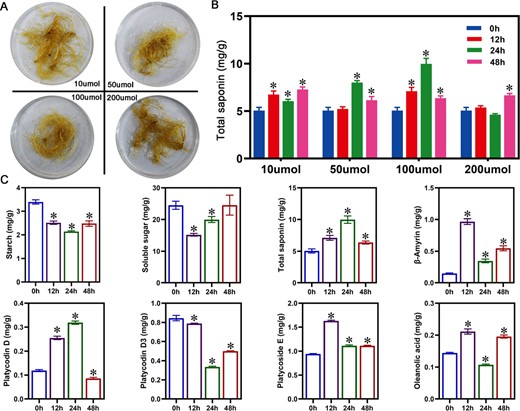
Changes in saponin content, saponin monomers, and intermediate synthesized products at different concentrations of MeJA. (A) Morphology of P. grandiflorus roots at four MeJA concentrations at 72 h. (B) Total saponin content at four MeJA concentrations. (C) Soluble sugars, starch, three saponin monomers, and intermediate products (β-amyrin and oleanolic acid) in P. grandiflorus roots treated with 100 μmol/l of MeJA.
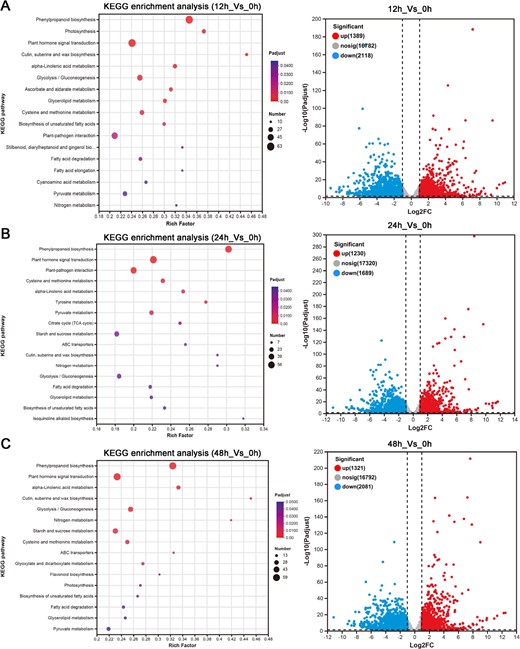
Volcanograms and KEGG enrichment analysis of DEGs in P. grandiflorus roots under 100 μmol/l MeJA treatment. (A) 12h_Vs_0h. (B) 24h_Vs_0h. (C) 48h_Vs_0h.
3.2. DEG and enrichment analyses
To investigate the molecular mechanism of MeJA-induced saponin biosynthesis in P. grandiflorus, transcriptome sequencing was performed on P. grandiflorus root samples treated with 100 μM MeJA at 0, 12, 24, and 48 h. A total of 96.37 Gb clean data were obtained. Alignment of the clean data with the P. grandiflorus genome produced alignment rates ranging from 94.34%–94.91% (Supplementary Table S1). DEG analysis identified a total of 5251 DEGs (1389 up and 2118 downregulated) in the 12h_Vs_0h comparison, 3133 DEGs (1230 up and 1689 downregulated) in the 24h_Vs_0h comparison, and 3401 DEGs (1321 up and 2081 downregulated) in the 48h_Vs_0h comparison. According to the KEGG enrichment results, the alpha-linolenic acid metabolism involved in MeJA biosynthesis, as well as the glycolysis/gluconeogenesis, starch and sucrose metabolism, and ABC transporters pathways involved in saponin biosynthesis and transport, were significantly enriched after MeJA treatment. Furthermore, plant hormone signal transduction pathways also showed significant enrichment (Fig. 2A–C). These results demonstrated that MeJA, as a hormone-signaling molecule, was found to induce triterpenoid saponin-related pathway gene expression, thereby promoting the accumulation of saponins in the roots of P. grandiflorus.
3.3. Genetic basis of P. grandiflorus saponin biosynthesis under MeJA treatment
Investigating the genetic foundation of saponin biosynthesis in P. grandiflorus under MeJA treatment yielded 7590 DEGs (average TPM > 1, CV > 0.1). In order to elucidate the regulatory mechanisms of saponin biosynthesis under MeJA treatment, the DEG co-expression networks were examined using the WGCNA package. Based on the expression patterns, all the DEGs were divided into 22 modules (Fig. 3A; Supplementary Table S2). The correlation analysis between the 22 modules and metabolites demonstrated a strong correlation between MEblue, MEturquoise, MEred, MEroyalblue, and MEgreen modules and β-amyrin, oleanolic acid, starch, and three individual saponin moieties. These results showed that the genes in these five modules were primarily involved in MeJA-induced saponin biosynthesis (Fig. 3B).
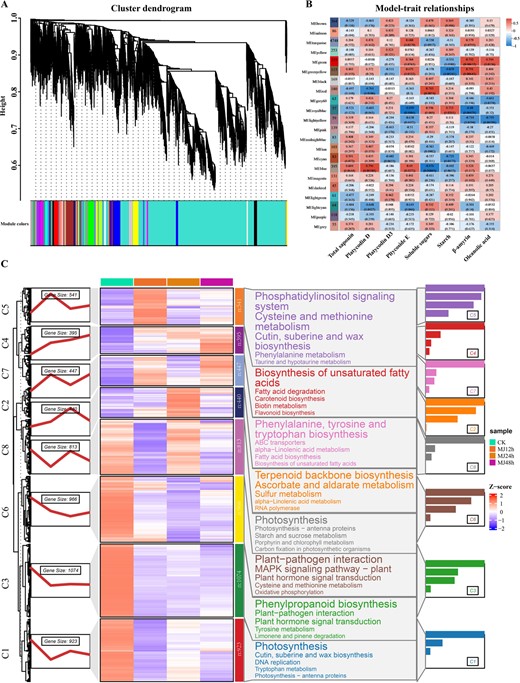
Transcriptome and metabolite correlation analysis of P. grandiflorus under MeJA treatment. (A) Dendrogram of the co-expression modules during saponin accumulation under MeJA treatment. The tree branches contain 22 modules, labeled with various colors. (B) Heat map displaying saponin correlation module. Different colors of each column denote specific modules. Each row corresponds to a metabolite. The positive relationship between the model and the content of various metabolites is indicated in red, and the negative correlation is in blue. (C) Temporal expression and KEGG enrichment of eight gene clusters.
To further understand the MeJA response and saponin biosynthesis regulation of the five modules, these genes were subdivided into eight clusters using Mfuzz based on their expression pattern similarities (Fig. 3C; Supplementary Table S3). KEGG enrichment analysis of the eight clusters demonstrated that Cluster 2 exhibited significant enrichment in alpha-linolenic acid metabolism, the key biosynthetic pathway for MeJA biosynthesis. Cluster 2 was also enriched in the terpenoid backbone biosynthesis pathway, the major pathway for synthesizing of triterpenoid saponins, monoterpenes, and sesquiterpenes. These results demonstrated that the 440 genes within Cluster 2 were the core genes involved in MeJA-induced saponin biosynthesis.
3.4. Generation of saponin biosynthesis regulatory networks under MeJA treatment
By analyzing the genetic basis of P. grandiflorus saponin biosynthesis under MeJA treatment, Cluster 2 was considered the main module in response to MeJA regulation of saponin synthesis. KEGG pathway analysis showed that Cluster 2 included alpha-linolenic acid metabolism and terpenoid backbone biosynthesis pathways, which are crucial for MeJA and terpenoid biosynthesis, respectively. Expression analysis of structural genes within alpha-linolenic acid and terpenoid backbone biosynthesis pathways showed MeJA could upregulate these genes (Fig. 4A–B; Supplementary Table S4). In order to generate a regulatory network for MeJA-induced saponin biosynthesis, eight structural genes (including PgGGPS2, PgUGTPg21, PgUGT85A4, PgUGT90A1, PgUGTPg26, PgHMGCR1, PgHMGR2, and PgDXS2) involved in saponin biosynthesis were identified, exhibiting highly correlated relationship with saponin metabolite content in Cluster 2 (Fig. 4C; Supplementary Table S5). At the same time, a total of 19 TFs (including bHLH, AP2/ERF, and Dof) were identified, which may promote saponin accumulation via regulating structural genes involved in saponin biosynthesis (Fig. 4D; Supplementary Table S5). The regulatory network of saponin accumulation induced by MeJA was generated by calculating the transcription accumulation pattern of TFs and structural genes related to saponin biosynthesis, as well as the potential binding affinity of TFs to structural gene promoters (Fig. 4D; Supplementary Table S5–S6). Network analysis identified that PgbHLH28 was highly correlated with the structural genes involved in saponin biosynthesis (i.e. PgGGPS2, PgHMGR2, PgHMGR1, and PgDXS2), which was identified as the most promising candidate for further investigation.
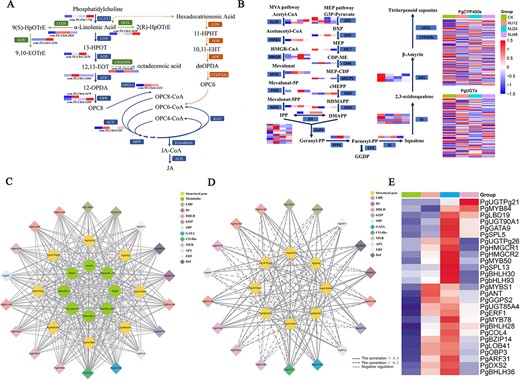
The regulatory network of saponin in P. grandiflorus. (A) Metabolic pathway for MeJA. (B) Metabolic pathway for saponin. (C) Green circles denote metabolites (including soluble sugar, starch, total saponin, β-amyrin, platycodin D, platycodin D3, platycoside E, and oleanolic acid). Yellow circles denote structure genes. Diamonds, each with a distinct color, represent various families of TFs identified in Cluster 2. (D) The regulatory network between structural genes and TFs of saponin biosynthesis under MeJA. (E) Heatmap of TF and structural gene expression involved in saponin biosynthesis in Cluster 2.
3.5. PgbHLH28 regulates saponin accumulation in P. grandiflorus
Through regulatory network analysis, the PgbHLH28 gene was identified as the key regulator in response to MeJA-induced saponin accumulation in P. grandiflorus. In order to verify the function of the PgbHLH28 gene, both overexpression and RNA interference vectors of the PgbHLH28 gene were constructed to demonstrate its function in P. grandiflorus (Fig. 5E). Transgenic overexpression plants (Fig. 5A–B, D), overexpression transgenic hairy roots (Supplemental Material Fig. S1A–B), and RNA-silencing transgenic hairy roots (Fig. 5C–D) were generated via Agrobacterium-mediated transformation. β-amyrin, oleanolic acid, and total saponin levels were significantly increased in PgbHLH28-overexpressing plants and decreased in silenced lines compared to the wild-type (WT) plants (Fig. 5H; Supplemental Material Fig. S1C). The relative expression of PgbHLH28 and other structural genes, such as PgDXS2 and PgHMGR2, in the network involved in saponin synthesis was significantly upregulated in the PgbHLH28-overexpressing plants and downregulated in RNA-silencing transgenic hairy roots compared to WT (Fig. 5F–G; Supplemental Material Fig. S1D).
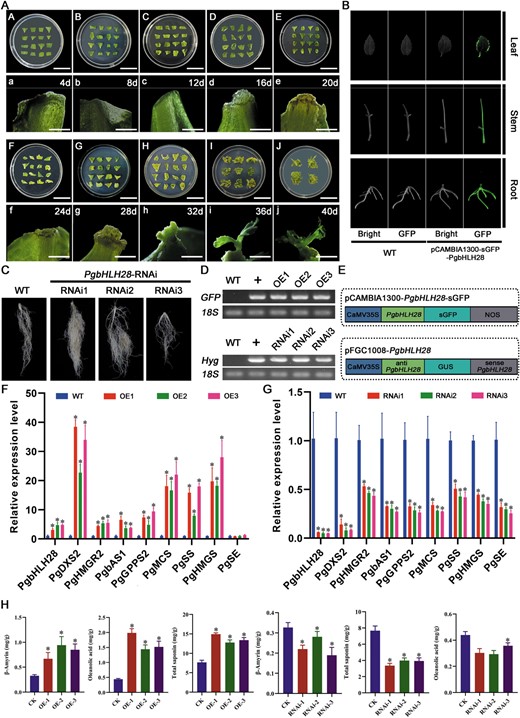
PgbHLH28 modulates saponin accumulation by regulating genes involved in the saponin biosynthesis pathway. (A) Adventitious bud induction in P. grandiflorus. (B) GFP fluorescence detection in PgbHLH28 overexpression lines. Scale bar: 3 cm (A–J), 5 mm (a–j) (C) Hairy root development via RNA interference of PgbHLH28 in P. grandiflorus. (D) RT-PCR confirmation of transgenic plants. (E) Schematic diagram of the construction of overexpression and RNA interference vectors. (F) Relative expression levels of genes involved in saponin biosynthesis pathway in PgbHLH28 overexpression lines. PgbHLH28-OE1, PgbHLH28-OE2, and PgbHLH28-OE3 are three independent PgbHLH28 overexpression lines. (G) Relative expression levels of genes involved in saponin biosynthesis pathway in PgbHLH28 silencing lines. PgbHLH28-RNAi1, PgbHLH28-RNAi2, and PgbHLH28-RNAi3 are three independent PgbHLH28 silencing lines. (H) Total saponin, oleanolic acid, and β-amyrin content in overexpression and RNA silencing lines.
Measurements of saponin content and expression of structural genes involved in saponin biosynthesis in the PgbHLH28 transgenic hairy roots and transgenic plants demonstrated that the results obtained from both methods were consistent (Fig. 5F–H; Supplemental Material Fig. S1C–D), suggesting that both methods could be used to validate P. grandiflorus gene functions. Furthermore, the hairy root transformation method offered transformation time and operability advantages. Therefore, the hairy root transformation method was selected as the primary approach to validate gene function in further experiments. These experiments established PgbHLH28 as a positive regulator of saponin biosynthesis in P. grandiflorus.
3.6. PgbHLH28 activates the transcription of downstream genes (PgHMGR2 and PgDXS2)
Transgenic experiments identified PgbHLH28 as a key regulator of MeJA-induced saponin accumulation in P. grandiflorus. Regulatory network analysis revealed structural genes (PgGGPS2, PgHMGR2, PgHMGR1, and PgDXS2) as downstream targets regulated by PgbHLH28 (Fig. 6A).
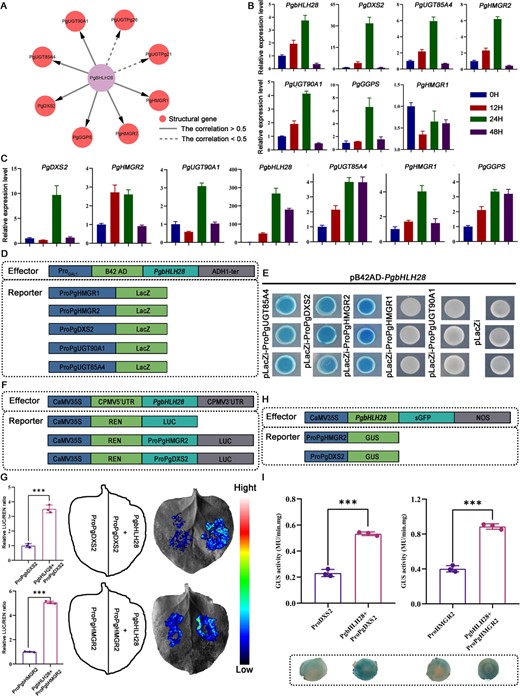
The PgbHLH28 gene promotes the expression of the terpenoid backbone biosynthetic genes PgDXS2 and PgHMGR2. (A) PgbHLH28 is a key regulatory factor in the synthesis of terpenoid compounds. The pink circles denote structural genes. Solid lines represent a correlation >0.5 between PgbHLH28 and the structural genes, while dashed lines represent a correlation <0.5. (B) RT-qPCR of PgbHLH28 TF and six highly correlated structural genes under MeJA treatment. (C) Transient overexpression of PgbHLH28 TF in P. grandiflorus roots increases the expression levels of six structural genes. (D) Schematic diagram of Y1H construction structure. (E) PgbHLH28 binds to the promoters of PgUGT85A4, PgDXS2, and PgHMGR2 genes. (F) Schematic diagram of dual-luciferase vector structure. (G) Dual-luciferase assays show PgbHLH28 activating the transcriptional activity of PgDXS2 and PgHMGR2 genes. (H) Schematic diagram of GUS activity vector structure. (I) GUS reporter assays demonstrate PgbHLH28 activating the transcription activity of PgDXS2 and PgHMGR2 genes.
In order to determine if the PgbHLH28 gene regulates the structural gene, the pCAMBIA1300-PgbHLH28-sGFP overexpression vector was constructed and transiently overexpressed in the roots. The relative expression levels of PgbHLH28 and six structural genes in the regulatory network were significantly upregulated, and MeJA could also induce the expression of these genes, as validated by qRT-PCR (Fig. 6B–C). Secondly, Y1H assays confirmed PgbHLH28 binding to the cis-elements in the promoter regions of PgDXS2, PgHMGR2, and PgUGT85A4 (Fig. 6 D–E). While PgHMGR2 and PgDXS2 are key rate-limiting enzymes involved in saponin biosynthesis, further investigations focused on the regulation between PgbHLH28 and two structural genes (PgHMGR2 and PgDXS2). Furthermore, dual-luciferase reporter assays in tobacco demonstrated that PgbHLH28 significantly enhanced the transcriptional activity of PgDXS2 and PgHMGR2 compared to the control (Fig. 6F–G). The GUS staining experiment also demonstrated that PgbHLH28 enhanced the transcriptional activity of PgDXS2 and PgHMGR2 genes (Fig. 6H–I). Taken together, these findings demonstrated that the PgbHLH28 TF regulated the expression of the saponin biosynthesis pathway functional genes, PgDXS2 and PgHMGR2, via directly binding to their cis-regulatory elements in the promoter regions, thereby modulating saponin accumulation.
3.7. PgDXS2 and PgHMGR2 regulate saponin accumulation in P. grandiflorus
PgDXS2 and PgHMGR2 serve as critical rate-limiting enzymes in the MEP and MVA pathways for saponin biosynthesis [23]. PgbHLH28 gene overexpression was found to promote saponin accumulation. The dual-luciferase reporter assays, GUS staining, and Y1H experiments proved that the PgbHLH28 gene could bind to the promoters of PgDXS2 and PgHMGR2 genes to regulate their expression. To validate the functions of key rate-limiting enzymes PgDXS2 and PgHMGR2, overexpression and RNA interference vectors for two rate-limiting enzymes were constructed as described in above methods in this article. The vectors were then transformed into the P. grandiflorus via A. tumefaciens K599, yielding positive transgenic hairy roots (Figs. 7A–C and8A–C). Transgenic hairy roots were assayed for total saponin content, intermediate product β-amyrin, and oleanolic acid content. The results demonstrated that these components were significantly higher in the transgenic hairy roots than in WT. In contrast, in silenced hairy roots, the content of these three components was significantly lower than that in WT (Figs. 8F and9F). Gene expression data revealed that saponin biosynthesis pathway genes were upregulated in overexpressed hairy roots and suppressed in silenced hairy roots (Figs. 7D–E and8D–E). These results indicated that both PgDXS2 and PgHMGR2 could enhance the saponin accumulation in P. grandiflorus.
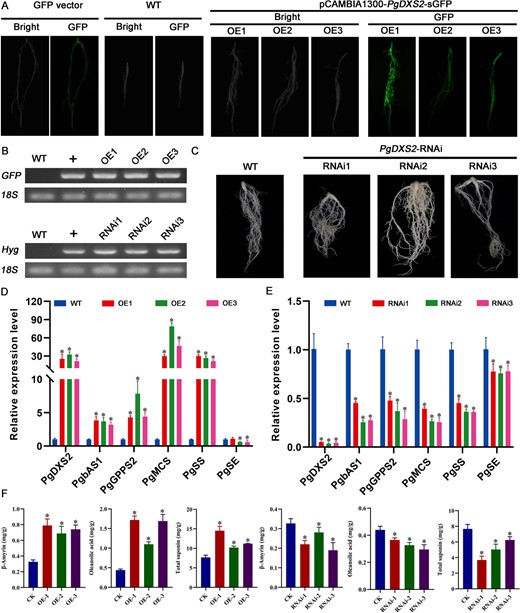
PgDXS2 modulates saponin accumulation in P. grandiflorus. (A) Hairy root generation through overexpression of PgDXS2 in P. grandiflorus. (B) RT-PCR validation of transgenic hairy root. (C) Hairy root generation via RNA interference of PgDXS2 in P. grandiflorus. (D) Relative expression of genes involved in saponin biosynthesis in PgDXS2 overexpression lines. PgDXS2-OE1, PgDXS2-OE2, and PgDXS2-OE3 are three independent PgDXS2 overexpression lines. (E) Relative expression of genes involved in saponin biosynthesis in PgDXS2 silencing lines. PgDXS2-RNAi1, PgDXS2-RNAi2, and PgDXS2-RNAi3 are three independent PgDXS2 silencing lines. (F) Total saponin, oleanolic acid, and β-amyrin content in overexpression and RNA silencing lines.
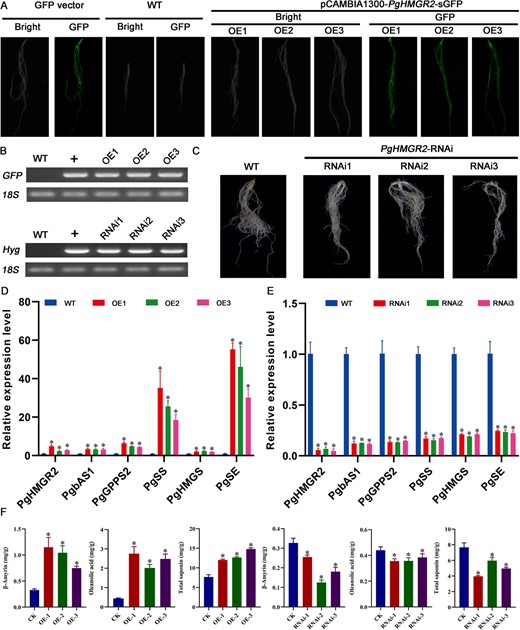
PgHMGR2-modulated saponin accumulation in P. grandiflorus. (A) Hairy root generation through overexpression of PgHMGR2 in P. grandiflorus. (B) RT-PCR validation of transgenic hairy root. (C) Hairy root generation via RNA interference of PgHMGR2 in P. grandiflorus. (D) Relative expression of genes involved in saponin biosynthesis in PgHMGR2 overexpression lines. PgHMGR2-OE1, PgHMGR2-OE2, and PgHMGR2-OE3 are three independent PgbHLH28 overexpression lines. (E) Relative expression of genes involved in saponin biosynthesis pathway in PgHMGR2 silencing lines. PgHMGR2-RNAi1, PgHMGR2-RNAi2, and PgHMGR2-RNAi3 are three independent PgHMGR2 silencing lines. (F) Total saponin, oleanolic acid, and β-amyrin content in overexpression and RNA silencing lines.
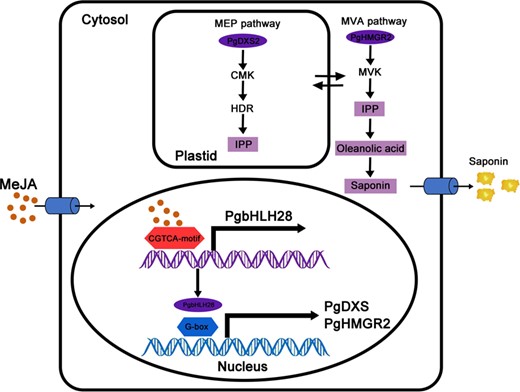
Working model of PgbHLH28 in the regulation of saponin biosynthesis in P. grandiflorus. PgbHLH28 specifically binds with the promoters of the key structural genes involved in saponin biosynthesis (including PgDXS2 and PgHMGR2) and activates their transcription, resulting in enhanced saponin accumulation in P. grandiflorus. Furthermore, intermediate products produced by both the MVA and MEP pathways can be utilized in both pathways via transmembrane transport.
4. Discussion
Specific concentrations of exogenous MeJA enhanced secondary metabolite content in medicinal plants. In Leucas aspera and Panax quinquefolium, exogenous MeJA increased oleic acid, triterpenoid, and ginsenoside content [24, 25]. However, different concentrations of MeJA exhibit varying effects on the content of the secondary metabolites [26]. Four concentration gradients were examined to investigate the influence of MeJA on saponin accumulation and identify the optimal inducing concentrations. The four different concentrations increased saponin content (Fig. 1B). When the concentration of exogenous MeJA was 100 μmol/l, the induction effect was optimal, and the saponin content was highest (Fig. 1B), similar to previous studies in P. ginseng [27, 28]. Therefore, this concentration can be applied in practical production to enhance saponin content in P. grandiflorus. Soluble sugars and starch, acting as substrates in glycolysis, directly impact the biosynthesis and accumulation of saponins via MVA and MEP pathways [1]. After MeJA treatment, the soluble sugar and starch content decreased compared to the control (Fig. 1C), while saponin content showed an increasing trend, suggesting that MeJA accelerated the conversion of soluble sugar and starch to saponins. Due to the differences in MeJA-induced accumulation patterns of various metabolites [26, 29], the content of various saponin monomers and intermediate biosynthesis products under 100 μmol/l treatment was measured. A significant increase was observed in β-amyrin and oleanolic acid content (Fig. 1C). The triterpenoid saponins in P. grandiflorus belong to the oleanane type, with oleanolic acid as the main sapogenin [30]. Therefore, total saponin content was directly promoted due to increased oleanolic acid content (Fig. 1C). Various saponin monomers are formed via hydroxylation, oxidation, and glycosylation of β-amyrin and oleanolic acid by the actions of CYP450 and GTs [3]. Measurements of the content of the three saponin monomers identified that platycodin D and platycoside E exhibited an increasing trend while platycodin D3 was suppressed (Fig. 1C). MeJA may reduce platycodin D3 content by inhibiting the expression of genes that modify of platycodin D3, including PgCYP450 and PgUGT genes (Supplementary Table S4). MeJA not only affected secondary metabolite content in plants but also had inhibitory effects on root growth [31, 32]. The MeJA-treated roots of P. grandiflorus were browned, and the degree of browning deepened with increasing MeJA concentration, demonstrating root growth inhibition (Fig. 1A).
Recent advances in high-throughput sequencing have made big data analysis a key approach for exploring genes involved in triterpenoid saponin biosynthesis. Preliminary transcriptomic analyses have been conducted on triterpenoid saponin biosynthetic pathways across different plant species, including Glycyrrhiza uralensis [33], P. ginseng [34], and Clinopodium gracile [35]. These studies have enhanced the gene library resources for triterpenoid saponin biosynthesis. KEGG enrichment and WGCNA analysis of MeJA-treated P. grandiflorus root transcriptomic data (Figs. 2A–C,3A–C) identified the alpha-linolenic acid metabolism pathway, which was involved in the MeJA biosynthesis pathway [36], the glycolysis/gluconeogenesis, and starch and sucrose metabolism, which provide the key substrates pyruvic acid, glyceraldehyde 3-phosphate, and acetyl-coenzyme A for the MVA and MEP pathways [1]. Significant enrichment was also observed in the triterpene saponin backbone biosynthesis and the ABC transporters pathways, which were involved in saponin transportation and biosynthesis [37]. These data suggested that MeJA induced the expression of genes related to the glycolysis/gluconeogenesis, starch and sucrose metabolism, and triterpene saponin backbone synthesis pathway to promote the biosynthesis of triterpene saponins that finally activate the expression of genes related to ABC transporters pathway involved in transporting saponins from the cytoplasm to the extracellular compartment to increase saponin content in the roots.
The bHLH TFs play significant roles in the biosynthesis of terpenoid metabolites triggered by MeJA signaling, including monoterpenes, sesquiterpenes, diterpenes, and triterpenes [38, 39]. RNA-seq analysis revealed that the PgbHLH28 gene exhibited upregulation in response to MeJA induction and held a central position in the regulatory network (Fig. 6A). In Medicago truncatula, TSAR1 (triterpene saponin biosynthesis activating regulator 1) and TSAR2 activation of bAS, CYP93E2, and CYP72A61v2 genes increased hemolytic saponin content [13]. TSARL1 and TSARL2 also enhanced the accumulation of saponin content by activating HMGR1 and MKB1 genes [40]. Furthermore, bHLH TFs that regulate triterpenoid biosynthesis have been identified in Cucumis sativus L., P. ginseng, and B. platyphylla [17]. These data suggest that bHLH TFs participate in regulating saponin accumulation. To confirm PgbHLH28 function, overexpression and RNA interference experiments were performed. The results demonstrated that PgbHLH28 overexpression increased saponin accumulation and the expression of structural genes involved in saponin biosynthesis, while RNA silencing had the reverse effect (Fig. 5F–H). In addition to promoting saponin accumulation, some bHLH TFs also inhibit the accumulation of related metabolites. SmbHLH60 suppressed the transcription of SmTAT1 and SmDFR, thereby inhibiting the biosynthesis of the phenolic acids and anthocyanins [41]. In Salvia miltiorrhiza, SmbHLH92-RNAi transgenic hairy roots showed increased salvianolic acid and tanshinone content, indicating that SmbHLH92 acts as a negative regulator [42]. We also identified a negatively regulated transcription factor, PgbHLH36, in the regulatory network that may inhibit saponin synthesis by negatively regulating the saponin biosynthesis structural gene PgHMGR1 (Supplementary Table S7). These results emphasize the complex nature of the bHLH gene family in regulating metabolic processes and the necessity of identifying the underlying transcriptional regulatory mechanisms of saponin biosynthesis.
Terpenoids represent the largest group of secondary metabolites in plants [43]. In higher plants, the basic five-carbon units of all terpenoids, IPP, and dimethylallyl pyrophosphate (DMAPP), are synthesized via two independent pathways in different cellular compartments. Monoterpenes, diterpenes, and tetraterpenes are products from the MEP pathway, which is located in the cytoplasm. Sesquiterpenes, sterols, and triterpenes are derivatives from the mevalonate (MVA) pathway located in the plastids [44]. Despite their separation in different organelles, evidence shows intermediates from these pathways can cross the plastid membrane, indicating mutual utilization [45]. Isotopic labeling of intermediates in Antirrhinum majus L. and Solidago canadensis L., sesterterpenes (primarily synthesized by the MVA pathway), rely on intermediates produced by the MEP pathway, which is primarily responsible for generating monoterpenes. This demonstrates that the intermediates between the two pathways can be exchanged and utilized through the plastid membrane [45–47]. Transcriptome analysis of MeJA-treated roots revealed that genes involved in the MEP and MVA pathways were induced and upregulated to varying degrees (Fig. 4A–B). Furthermore, the analysis revealed that PgDXS2 and PgHMGR2, encoding enzymes in the MEP and MVA pathways, respectively, were the core structural genes in the regulatory network, suggesting that both pathways may be involved in saponin biosynthesis (Fig. 6A). Overexpression of PgDXS2 and PgHMGR2 resulted in elevated content of oleanolic acid, β-amyrin, and total saponins, accompanied by upregulation of saponin biosynthesis pathway genes (Figs. 7 and 8). In contrast, inhibition of these genes suppressed intermediate metabolite levels, as well as the expression levels of pathway genes. These findings suggested that the intermediates of the two pathways may be mutually utilized through the plastid membrane, synthesizing downstream terpenoids in P. grandiflorus.
5. Conclusion
This study aimed to elucidate the underlying molecular mechanisms of the role of MeJA in promoting saponin biosynthesis in P. grandiflorus. RNA-seq data analysis demonstrated that PgbHLH28 acts as a positive regulator of saponin biosynthesis. Y1H, dual-luciferase, and transgenic experiments showed that PgbHLH28 promoted saponin accumulation by activating the expression of PgHMGR2 and PgDXS2. Moreover, overexpression and silencing of PgDXS2 and PgHMGR2 demonstrated that both MVA and MEP pathways contribute to saponin biosynthesis, indicating the pathways’ intermediates are mutually utilized through the plasma membrane (Fig. 9). These research results have increased the understanding of the saponin biosynthesis mechanism induced by MeJA. These findings, therefore, support future research on saponin biosynthesis and molecular breeding strategies.
2. Materials and methods
2.1. Plant material and measurements
Platycodon grandiflorus plants used in this study were cultivated in Harbin, Heilongjiang Province, China. The sterile seedlings cultivated in the laboratory for 90 days were treated separately with MeJA concentrations: 0, 10, 50, 100, or 200 μM. The samples were collected at 0, 12, 24, and 48 hours, immediately frozen in liquid nitrogen, and stored at −80°C for further transcriptome and metabolite analyses. Total saponin content was measured using the vanillin–perchloric acid colorimetry assay [48]. Starch and soluble sugars were determined using the anthrone–sulfuric acid colorimetric assay [49, 50]. To ensure validity, three biological replicates per sample per experiment were performed. Each replicate contained 10 plants of uniform size.
2.2. Oleanolic acid, β-amyrin, and sapogenin monomers content analysis in P. grandiflorus
Oleanolic acid, β-amyrin and three saponin monomers (platycoside E, platycodin D3, and platycodin D) standards were purchased from Chengdu Desite Biotechnology Co., Ltd. (Chengdu, China). Metabolites were extracted from 0.5 g of P. grandiflorus root treated with MeJA concentrations of 0, 10, 50, 100, or 200 μM by adding 1 ml of methanol, sonicating for 30 min, centrifuging at 12 000 rpm for 10 min at 4°C, and filtering through a 0.22-μm syringe membrane for High-Performance Liquid Chromatography (HPLC) analysis. The HPLC detection was performed on the Agilent Series1260 system (Agilent Technologies Inc., USA), with Techmate C18 column (250 × 4.6 mm, 5 μm). The mobile phase for oleanolic acid and β-amyrin was methanol (A)-water (B) (90:10 v/v), with detection wavelengths of 203 and 215 nm, respectively. The mobile phase for saponin monomers was acetonitrile (A)-water (B) (27:73 v/v), with a detection wavelength of 215 nm. The sample injection volume was 10 μl, the flow rate was 1 ml/min, column temperature was 30°C [51]. Each sample was performed with three biological replicates.
2.3. RNA sequencing (RNA-seq) and data analysis
Total RNA from P. grandiflorus roots treated with 100 μM MeJA for 0, 12, 24, or 48 h was extracted using the RNeasy Plant Mini Kit (Qiagen, Venlo, Netherlands). The concentration and quality of the extracted RNA were assessed using the GXII Touch HT Nucleic Acid Analyzer (PerkinElmer, USA). After passing the quality control, the RNA library was constructed for each sample using the KAPA™ Single-Stranded RNA Library Preparation Kit (Illumina, USA). The libraries were sequenced on the Illumina HiSeq-2000 platform (Illumina, USA). The paired-end reads were mapped to the P. grandiflorus genome (https://ngdc.cncb.ac.cn/gwh) using HISAT2 [52]. Then, featureCounts was used to calculate transcripts per million (TPM) [53]. To ensure experimental accuracy, three biological replicates were used for RNA-seq and subsequent bioinformatic analyses. Each biological replicate included 10 individual plants of uniform size. The clean reads of 12 samples from this study were submitted to NCBI (accession number: PRJNA1032698).
2.4. Construction of saponin biosynthesis regulatory network under MeJA treatment
Differentially expressed genes (DEGs) (TPM > 1 and coefficient of variation (CV) > 0.1) were used to generate a regulatory network for saponin biosynthesis under MeJA treatment using the weighted correlation network analysis (WGCNA) package. The parameters were determined based on the previously described method [54]. The initial modules were merged using eigengenes to identify the key modules regulating saponin biosynthesis. This package was then used to calculate eigengene values for each module, to establish the relationship between metabolites and the modules associated with saponin biosynthesis. The Muffuz package [55] was used to partition the selected module sets into different clusters based on expression similarity patterns to obtain the core genes responsible for regulating saponin biosynthesis under MeJA treatment.
2.5. qRT-PCR
Gene expression analysis via quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed according to the established protocols, including RNA extraction, reverse transcription, PCR amplification, and data analysis [20]. Three biological replicates per sample were analyzed with the 18S RNA gene as the internal control [1]. The gene-specific primers for qRT-PCR were presented in Supplementary Table S1.
2.6. Yeast one-hybrid assay
Yeast one-hybrid assay (Y1H) assay was performed using the pLacZi One-Hybrid system. The full-length PgbHLH28 TF was inserted between Kpn I and Xho I sites in the pB42AD vector as the bait. The promoters of PgUGT85A4, PgDXS2, PgHMGR2, PgHMGR1, and PgUGT90A1 genes were ligated between Kpn I and Xho I sites in the pLacZi vector as the prey. The bait and prey vectors were then co-transformed into yeast strain EGY48. The pB42AD-PgbHLH28 and an empty vector pLacZi were used as controls. The SD/−Trp/-Ura medium was used for the growth of transformants. After 3 days, yeast-exhibiting positive clones were transferred to SD/−Trp/-Ura yeast media containing X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) and cultured in the dark at 30°C for 2–3 days. The color changes of the recombinant yeast on the agar plates were examined to identify an interaction between the PgbHLH28 and the downstream functional genes [56].
2.7. Dual-luciferase assay
The PgbHLH28 coding sequence was inserted between BamH I and Hind III sites in the pGreenII62-SK vector as the effector. PgDXS2 and PgHMGR2 promoter sequences were inserted between BamH I and Hind III sites in the pGreen 0800-LUC vector as the double-reporter. The constructed reporter and effector plasmids were transformed into Agrobacterium tumefaciens strain GV3101 (psoup-p19). Cell suspensions with effector and reporter vectors were then transiently injected into 4-week-old tobacco leaves [57, 58]. The pGreen 0800-ProPgDXS2-LUC and pGreen 0800-ProPgHMGR2-LUC vectors were used as the control. After 2–3 days of cultivation, the results of the dual-luciferase experiments were photographed using an automated chemiluminescence image analysis system (Tanon 5200, Shanghai, China), and the LUC/REN activity was measured using a Luciferase Reporter Gene Assay Kit (Yeasen Biotechnology, Shanghai, China) [59]. Three biological replicates were performed for the dual-luciferase assay. Each biological replicate included five different tobacco leaves.
2.8. PgbHLH28 overexpression in P. grandiflorus
The full-length PgbHLH28 gene, lacking a termination codon, was inserted between BamH I and Kpn I sites in the pCAMBIA1300-sGFP overexpression vector. The correctly sequenced pCAMBIA1300-PgbHLH28-sGFP vector was then used in the A. tumefaciens GV3101 transformation. Platycodon grandiflorus leaf explants were precultured on differentiation medium (MS medium containing 1 mg/l 6-BA, 0.1 mg/l NAA, 30 g/l sucrose, and 5 g/l agar, at pH 5.8) for 7 days and then transferred into A. tumefaciens GV3101 cell suspension containing the pCAMBIA1300-PgbHLH28-sGFP construct (OD = 0.6). After 20 min of infection at 28°C, the excess bacterial solution was removed using filter paper and co-cultured for 4 days under dark conditions. The explants were then transferred to a shoot-inducing medium (MS medium containing 1 mg/l 6-BA, 0.1 mg/l NAA, 30 g/l sucrose, 5 g/l agar, 30 mg/l hygromycin B, and 400 mg/l cefotaxime sodium). Upon shoot formation on the explant cultures, the shoots were transferred to selective rooting medium (1/2 MS medium containing 0.5 mg/l NAA, 30 g/l sucrose, 5 g/l agar, 30 mg/l hygromycin B, and 400 mg/l cefotaxime sodium) to induce root formation. Each separate shoot represented an independent line. After ~30 days, the rooted plants exhibiting hygromycin resistance were transferred to pots containing a 1:1 volume ratio of peat and vermiculite for growth under a 16/8 h light/dark photoperiod. Positive transgenic plants were identified according to the following three steps. Firstly, GFP fluorescence was detected in hairy root using an automated chemiluminescence image system (Tanon 5200, Shanghai, China) to confirm their transgenic status. Secondly, the RNA was extracted from positive transgenic hairy roots and amplified using GFP and 18S RNA primers. Lastly, qRT-PCR was used to quantify transcript levels of PgbHLH28 and related pathway genes. Primers used in this study are presented in Supplementary Table S7. HPLC and vanillin-perchloric acid colorimetry assay were used to measure saponin content as described in this article.
2.9. Functional verification of PgbHLH28, PgDXS2, and PgHMGR2 genes using hairy root
The full-length PgbHLH28, PgDXS2 and PgHMGR2 genes, without termination codons, were inserted between BamH I and Kpn I sites in the pCAMBIA1300-sGFP overexpression vector. The 300-bp specific forward and reverse fragments of PgbHLH28, PgDXS2, and PgHMGR2 genes were inserted into the pFGC1008 vector between Asc I and BamH I, as well as Spe I and Swa I sites to construct RNA interference vectors, respectively. The correctly sequenced overexpression and RNA interference vectors were transformed into A. tumefaciens strain K599, then cultivated on LB solid medium (containing 50 mg/l kanamycin) for 36–48 h. Selected single colonies were cultured in 5 ml of LB liquid medium, followed by overnight incubation in 200 ml of LB medium containing 50 mg/l kanamycin and 200 μmol/l AS. When the OD600 of the culture reached ~0.8, it was centrifuged at 4000 rpm for 10 min. The pellet was suspended in an infiltration buffer (containing 200 μmol/l AS, 10 mmol/l MES, and 10 mmol/l MgCl2) and then allowed to activate by incubating for 2 h at room temperature in the dark. This infiltration buffer was then used for infiltration. Thirty-day-old seedlings without roots were submerged in infiltration buffer twice and subjected to vacuum infiltration at −0.05 MPa, each for 90 s. The infected plants were washed with distilled water before transplanting into a 1:1 (v/v) mixture of peat and vermiculite for hairy root induction [60, 61]. Positive plant identification was conducted 60 days later. Transgenic hairy roots were detected using GFP green fluorescence detection and PCR. Transcript levels of PgbHLH28, PgDXS2, PgHMGR2, and related pathway genes in positive hairy roots were quantified using qRT-PCR. HPLC was used to measure the saponin content. Primers used in this study were presented in Supplementary Table S7.
2.10. GUS activity staining
The full-length PgbHLH28 gene lacking termination codons was inserted between BamH I and Kpn I sites in the pCAMBIA1300-sGFP overexpression vector. The 2000-bp sequences of both PgDXS2 and PgHMGR2 promoters were inserted between BamH I and Hind III sites in the pBI121-GUS vector. The recombinant vector pCAMBIA1300-PgbHLH28-sGFP, proPgDXS2-GUS, and proPgHMGR2-GUS were transformed into GV3101. The pCAMBIA1300-PgbHLH28-sGFP and proPgDXS2-GUS, proPgHMGR2-GUS were respectively co-expressed in the 0.5-cm-thick root segments submerged in the cell suspension (containing 200 μmol/l AS, 10 mmol/l MES, and 10 mmol/l MgCl2) for 20 min. The excess bacterial solution was removed with filter paper, and the treated root segments were transferred onto plates for incubation in the dark for 3 days. For histochemical staining, the root segments were immersed in GUS staining buffer (2 mM ferricyanide, 0.1% Triton X-100, 10 mM EDTA, 2 mM ferricyanide, phosphate buffer, and 2 mM X-Gluc, at pH 7.2) at 37°C overnight under dark conditions. Finally, the color changes and GUS activity were examined to determine interactions between the PgbHLH28 and the downstream structural genes PgDXS2 and PgHMGR2.
2.11. Statistical analysis
All experiments were conducted with three biological replicates. Results are reported as mean ± standard deviation (SD). Statistical significance was indicated by asterisks, as determined by Student’s t-test or one-way analysis of variance (*, P < .05).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31971700) and the Joint Guiding Project of Natural Science Foundation of Heilongjiang Province (No. LH2020C014). In addition, we thank MJEditor (www.mjeditor.com) for providing English editing services during the preparation of this manuscript.
Author contributions
D.C., W.Z., J.Z., and T.Y. conceived and designed the experiments. W.Z., Y.F., and W.J. performed the experiments. W.Z., J.Z., J.D., and P.G. performed the data analysis. W.Z. wrote the manuscript. All authors read and approved the final manuscript.
Data availability
The data presented in this study are available in the article or supplementary material. The raw RNA-seq data generated in this study are available in the NCBI-SRA database (PRJNA1032698). The sequences of PgbHLH28, PgDXS2 and PgHMGR2 are available in the NGDC database (C_AA057966.1- C_AA057968.1).
Conflict of interest statement
The authors have no conflicts of interest to declare.
Supplementary Data
Supplementary data is available at Horticulture Research online.
References
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}