




Abstract
Mutations in the Kunitz-type serine protease inhibitor HAI-2, encoded by SPINT2, are responsible for the pathogenesis of syndromic congenital sodium diarrhea (SCSD), an intractable secretory diarrhea of infancy. Some of the mutations cause defects in the functionally required Kunitz domain 1 and/or subcellular targeting signals. Almost all SCSD patients, however, harbor SPINT2 missense mutations that affect the functionally less important Kunitz domain 2. How theses single amino acid substitutions inactivate HAI-2 was, here, investigated by the doxycycline-inducible expression of three of these mutants in HAI-2-knockout Caco-2 human colorectal adenocarcinoma cells. Examining protein expressed from these HAI-2 mutants reveals that roughly 50% of the protein is synthesized as disulfide-linked oligomers that lose protease inhibitory activity due to the distortion of the Kunitz domains by disarrayed disulfide bonding. Although the remaining protein is synthesized as monomers, its glycosylation status suggests that the HAI-2 monomer remains in the immature, lightly glycosylated form, and is not converted to the heavily glycosylated mature form. Heavily glycosylated HAI-2 possesses full anti-protease activity and appropriate subcellular targeting signals, including the one embedded in the complex-type N-glycan. As predicted, these HAI-2 mutants cannot suppress the excessive prostasin proteolysis caused by HAI-2 deletion. The oligomerization and glycosylation defects have also been observed in a colorectal adenocarcinoma line that harbors one of these SPINT2 missense mutations. Our study reveals that the abnormal protein folding and N-glycosylation can cause widespread HAI-2 inactivation in SCSD patents.
Introduction
Congenital sodium diarrhea (CSD) is a clinically and genetically heterogenous secretory diarrhea of infancy [1–3]. The disease is caused by impaired sodium absorption leading to an increase in electrolyte and water flux toward the intestinal lumen. Mutations in three different genes, SLC9A3, GUCY2C, and SPINT2, have been identified as causing these genetic disorders [3]. SLC9A3 encodes a Na+/H+ exchanger 3 (NHE3), which is responsible for the majority of intestinal brush-border sodium absorption and is important for acid-base homeostasis. The SLC9A3 mutations identified in CSD patients either abolish protein expression or decrease the basal Na+/H+ exchange activity of NHE3, which is consistent with the clinical presentation of this syndrome which includes high fecal loss of Na+ and metabolic acidosis [4]. GUCY2C encodes receptor guanylyl cyclase C (GC-C), which can catalyze the conversion of guanosine triphosphate to cyclic guanosine 3′,5′-monophosphate (cGMP) [5, 6]. cGMP in turns inhibits NHE3 function [7]. The sodium diarrhea caused by activating and hyper-stimulating mutations in GUCY2C is, therefore, attributed to the inhibition of NHE3 [8]. In contrast to the mutations in SLC9A3 and GUCY2C, the mechanisms underlying the pathogenesis of CSD caused by SPINT2 mutations appear to be much more complicated. The defective brush-border Na+/H+ exchange observed in patients with SPINT2 mutations indicates the involvement of ion channels and transporters [9]. However, the intestinal morphological alterations seen in these patients, including mild to moderate villus atrophy, which is not observed in patients with mutations in SLC9A3 and GUCY2C, strongly suggest that the impact of SPINT2 mutation likely extend beyond altered ion channels and transporter activity. Furthermore, patients with SPINT2 mutations exhibit extraintestinal defects, including choanal atresia, anal atresia, hypertelorism and corneal erosions [9]. The defects caused by SPINT2 mutation are, therefore, not limited to the gastrointestinal (GI) tract and likely impact proteins other than ion channels and transporters. Based on these similarities and differences, patients with SPINT2 mutations are characterized as syndromic congenital sodium diarrhea (SCSD), as distinguishing from classic or non-syndromic CSD, caused by mutations in SLC9A3 and GUCY2C.
SPINT2 encodes an integral membrane, Kunitz-type serine protease inhibitor, named hepatocyte growth factor activator inhibitor (HAI)-2 [10]. The pathogenesis of SCSD presumably starts with the uncontrolled activity of HAI-2 target proteases in the enterocytes, assuming that the mutations found in such patients cause the loss or a reduction in HAI-2 expression or function. Although the serine proteases matriptase and prostasin have been recognized as the primary physiological targets of HAI-2 in the human [11], there is significant disagreement in the literature as to which protease, matriptase or prostasin, is dysregulated and likely responsible for the pathogenesis of SCSD [12–16]. HAI-2 suppresses active proteases through the formation of stable enzyme-inhibitor complexes [11, 17], the analysis of which has revealed that prostasin and not matriptase is the key HAI-2 target protease in human intestinal tissues and Caco-2 colorectal adenocarcinoma cells [13, 14]. Consistent with this finding, targeted deletion of HAI-2 in Caco-2 cells results in the uncontrolled proteolytic activity of prostasin but not matriptase [14]. In contrast to this finding, others have reported that the enzymatic activity of matriptase and not prostasin is responsible for the intestinal defects caused by targeted deletion of HAI-2 in mouse intestines [15, 16]. Genuine physiological differences in matriptase biology between human and mouse have been also observed in the skin, which largely to the opposite seem to relate to differences in the matriptase expression pattern during the course of epidermal differentiation between the two species [18]. This physiological difference between humans and rodents appears to also occur in the GI tract, which may explain the apparent difference at whether it is matriptase or prostasin dysregulation, that is involved in the pathogenesis of SCSD, particularly with respect to the sodium-driven diarrhea. A functional linkage between matriptase and HAI-2 has also been reported in transfection-based overexpression studies using human cells, such as HEK293 [12]. Due to the differences in subcellular localization and distribution, HAI-2 which is typically has a primarily intracellular disposition, HAI-2 does not have full access to matriptase on the cell surface. As a result, the functional linkage between HAI-2 and matriptase varies among different cells, ranging from essentially no linkage in resting mammary epithelial cells to a limited and ineffective interaction in neoplastic B-cells [17, 19]. Although the forced expression of HAI-2 can result in the recapitulation of a biochemical relationship between HAI-2 and matriptase, enforced overexpression studies are notorious for producing artifactual interactions between proteins, and it seems likely that overexpression of HAI-2 might lead to it artificially gaining access to matriptase. The conclusion drawn from these studies might, therefore, be of limited relevance to understanding the functional relationship between endogenous matriptase and HAI-2 at cellular levels and in vivo.
The widespread expression of HAI-2 in epithelial cells contrasted with the characteristic phenotype that results from SPINT2 mutation in SCSD suggests that the impact of those mutations on the molecular and cellular mechanisms responsible for SCSD pathogenesis are restricted somehow to the cells of the GI-tract and some cell lineages during embryonic development [10, 20]. The enterocyte-selective functional linkage between HAI-2 and prostasin in conjunction with the atypically high levels of prostasin zymogen activation in enterocytes probably renders these cells significantly more susceptible to HAI-2 inactivation than other epithelial cells, such as keratinocytes [14]. The hypothesis that uncontrolled prostasin proteolytic activity in the enterocytes of SCSD patients is responsible for the phenotype observed is, however, predicated on the assumption that SCSD-associated SPINT2 mutations result in the inactivation of HAI-2. SCSD patients harbor several different SPINT2 mutations. The impact that each of the mutations has on HAI-2’s ability to inhibit prostasin is, however, not as simple as in the case of the targeted deletion of HAI-2. In addition to the Kunitz domain 1, which provides the structural basis for HAI-2 binding with and inhibiting the serine protease domain of prostasin [21–23], HAI-2 also relies on its sub-cellular targeting motifs for translocation into sufficiently close proximity with prostasin for effective inhibition [13, 24]. These subcellular targeting signals include one embedded in the intracellular domain and one involving the complex-type N-glycan modification of HAI-2 [20, 24, 25]. In the current study, we set out to examine the impact of SPINT2 mutations on the structural integrity of the Kunitz domain 1 and the targeting signals with particular attention to a sub-group of SPINT2 mutations comprised of point mutations of the functionally less important Kunitz domain 2. This group of SPINT2 mutations is particularly important, being found in almost all SCSD patients. Whether and how the altered Kunitz domain 2 inactivates HAI-2 is highly relevant to the question of whether uncontrolled prostasin proteolytic activity is involved in SCSD pathogenesis.
Results
Some SPINT2 mutations found in almost all SCSD patients are not in the functional elements required for HAI-2 action as prostasin inhibitor
HAI-2 protein as synthesized is comprised of a signal peptide, the extracellular domain with two protease-inhibitory Kunitz domains, the transmembrane domain, and the cytoplasmic domain (Fig. 1, WT). Kunitz domain 1 and not Kunitz domain 2 have been shown to be the part of the molecule responsible for the inhibitory activity against prostasin [21–23]. Subcellular targeting signals, embedded both in the cytoplasmic domain and encompassing the complex-type N-glycan attached to Asn-57 (N57), direct HAI-2 to large intracellular vesicles [20, 24, 25]. In human enterocytes, HAI-2 performs its physiological function via the control of prostasin proteolysis [13, 14]. There are at least 13 different SPINT2 mutations have been identified in SCSD patients (Fig. 1) [9, 26–31]. The potential impact of these SPINT2 mutations on HAI-2 function were assessed with regard to their ability to alter the structure of Kunitz domain 1 and/or HAI-2 subcellular targeting.
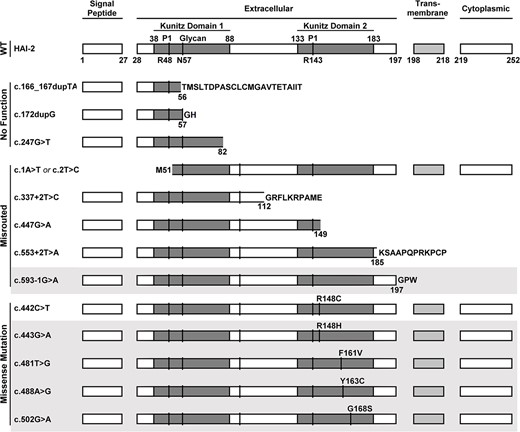
SPINT2 mutations in Kunitz domain 2 are found in almost all SCSD patients. The effect of SCSD-associated SPINT2 mutants are compared with the structure of the wild-type protein (WT) with the protein domain structures deduced using the NCBI ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) and are schematically presented. These SPINT2 mutants are classified into three groups: No function, misrouted, and missense mutation that causes single amino acid substitutions in Kunitz domain 2. The affected residues caused by the missense mutations are indicated, for example, as R148C. In addition to being present in a compound-heterozygous manner, some of these mutations also present in a homozygous manner, which are indicated in gray shadow. The N-glycosylation site Asn-57 is indicated by glycan and N57. The P1 sites of both Kunitz domain are also indicated, R48 and R143, respectively.
Three of the mutations, c.166_167dupTA, c.172dupG, and c.247G>T, would be expected to result in synthesis of a truncated form of HAI-2 lacking a functional Kunitz domain 1 (Fig. 1, No Function). Furthermore, the protein expression levels of these truncated HAI-2 may be reduced by the effect of nonsense-mediated mRNA decay triggered by the mutations creating premature stop codons.
Five of them, including c.1A>T or c.2T>C, c.337+2T>C, c.447G>A, c.553+2T>A, and c.593-1G>A, would result in the production of HAI-2 variants lacking a complete set of the required subcellular targeting signals and structural elements, such as the signal peptide, transmembrane domain, and/or cytoplasmic domain. As a result, they would be expected to be misrouted and thus have no access to prostasin. For example, the gene product of SPINT2 variant, c.1A>T or c.2T>C, does not have signal peptide and so would not be synthesized and undergo subcellular targeting via the endoplasmic reticulum (ER) and the secretory pathway. The remaining four SPINT2 mutants in this group would be synthesized via the ER as a secreted rather than an integral membrane protein, due to the lack of a transmembrane domain (Fig. 1, Misrouted). Although these five HAI-2 mutants contain an unaltered Kunitz domain 1, they would be denied access to prostasin in enterocytes. In contrast to these eight SPINT2 mutants, which either have no Kunitz domain 1 or are misrouted, five missense SPINT2 mutants, found in SCSD patients, c.442C>T (R148C), c.443G>A (R148H), c.481T>G (F161V), c.488A>G (Y163C), and c.502G>A (G168S), would be expected to result in the synthesis of HAI-2 protein only containing point mutations in Kunitz domain 2 (Fig. 1, Missense Mutation). This is unexpected as they otherwise would seem to contain all the necessary domains required for normal HAI-2 function as a prostasin inhibitor.
The prototypic Kuntz domain is that found in bovine pancreatic trypsin inhibitor (BPTI, also known as aprotinin) and contains approximate 51 residues [32]. Usually, there are three pairs of disulfide bonds with the arrangement C1-C6, C2-C4, and C3-C5 which stabilize the folding of one antiparallel β-sheet consisting of two β-strands, one α-helix, and a solvent-exposed protease-binding loop (reactive site) which determines the specificity for different proteases [33]. Three of the four amino acid residues mutated in Kunitz domain 2: R148, F161, and Y163, are found within one of the two β-strands with the fourth: G168, located within the linker region (Fig. 2, as indicated). Although these mutations are not in the protease-binding loop, these mutated residues are highly conserved among different Kunitz domains found in different human proteins (Fig. 3, as indicated by a red triangle), indicating the importance of these residues in the function and structure of the Kunitz domain. For example, in human TFPI2 Kunitz domain 1, the Arg at P5’, the Phe at P18’, and the Tyr at P20’ have been shown to be essential to stabilize the reactive site structure through the formation of an internal hydrophobic core [34]. Mutations at the corresponding residues in HAI-2 (R148, F161, Y163) could, therefore, similarly affect the structure and function of the HAI-2 Kunitz domain 2 as protease inhibitor. Nevertheless, these mutations in Kunitz domain 2 should conceptually have no impact on the structure of Kunitz domain 1 that is responsible for the inhibition of prostasin in human enterocytes. More importantly, the majority of SCSD patients harbor one of the five missense SPINT2 mutants in a homozygous or compound-heterozygous manner (Fig. 1). These SCSD-associated missense SPINT2 mutants might be expected, therefore, to be translated into HAI-2 variants similar to their wild-type counterpart with a functional Kunitz domain 1 and a complete set of subcellular targeting signals in almost all SCSD patients. If one presumes that SCSD is caused by uncontrolled prostasin proteolysis due to the inability and/or inaccessibility of these widespread SCSD-associated HAI-2 mutants to inhibit prostasin, there must be some unconventional mechanisms, by which the simple substitution of one amino acid residue in Kunitz domain 2 results in the loss of a functional Kunitz domain 1 and/or the targeting signals.
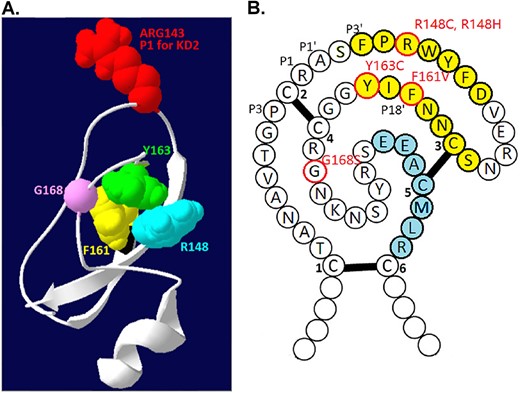
The residues involved in the SCSD-associated SPINT2 mutations are not in the reactive site of the Kunitz domain 2. (A) The 3D structure of HAI-2 Kunitz domain 2 was built using the AlphaFold protein structure database and Swiss-PdbViewer 4.10. The P1 site ARG143 (shown in red) and the four residues that are found to be mutated in SCSD patients, including Arg-148 (R148 in blue), Phe-161 (F161 in yellow), Tyr-163 (Y163 in green) and Gly-168 (G168 in light purple), are indicated. (B) The schematic 2D structure of HAI-2 Kunitz domain 2 is presented using single letter amino acid notation. The two β strands are indicated in yellow, one α helix in blue, and the three pairs of disulfide bonds as C1-C6, C2-C4, and C3-C5. The amino acid residues mutated in SCSD patients are indicated in red circle and the mutations are also indicated as R148C, R148H, F161C, Y163C, and G168S in red).

The residues involved in the SDSD-associated SPINT2 mutations are conserved among the Kunitz domains present in different human proteins. The amino acid sequences of 27 Kunitz domains from 18 human proteins are aligned and compared with bovine pancreatic trypsin inhibitor (BPTI). The six cysteine residues involved in the three disulfide bonds (black or gray shadow), the two β strands (yellow), one α helix (blue) are indicated. The P1 site (red shadow), putative N-glycosylation site (blue letter), the residues mutated in SCSD patients (red arrow point and red letter) are also marked. The alignment was performed using MUSCLE. The 2D structures were obtained from UniProtKB (marked by *) or predicted by PROMALS3D.
Such a mechanism could be present in LoVo colorectal adenocarcinoma cells, in which one of the five SCSD-associated SPINT2 missense mutations, c.442C>T (R148C) has been found (https://cancer.sanger.ac.uk/cell_lines/sample/overview?id=907790) and which results in a proportion of the HAI-2 being expressed in nonfunctional oligomers in these cells [25]. HAI-2 is synthesized into one of two different N-glycosylated forms: lightly glycosylated HAI-2 likely with oligomannose-type N-glycan and heavily glycosylated HAI-2 with complex-type N-glycan, both of which modifications are on Asn-57 (N57) [17, 20, 25]. In LoVo cells, a proportion of the lightly glycosylated HAI-2 has been detected in disulfide-linked oligomers, which have not been observed in more than 20 other cell lines analyzed [20]. These disulfide-linked HAI-2 oligomers exhibit no anti-trypsin activity due to the distorted conformation of the Kunitz domains caused by disarrayed disulfide linkages [25].
A significant decrease in heavily glycosylated HAI-2 in LoVo colorectal adenocarcinoma cells, which harbor a naturally occurred c.442C>T SPINT2 mutation
Lightly glycosylated HAI-2 represents the product of N-glycosylation in the endoplasmic reticulum (ER), which serves as a precursor for N-glycan branching in the Golgi apparatus to generate heavily glycosylated HAI-2 [20]. The N-glycan branching found in heavily glycosylated HAI-2 not only results in enhanced anti-trypsin activity, but also provides the subcellular targeting signal to direct HAI-2 to the large vesicles, both of which are essential for the control of prostasin proteolysis [13, 14, 20, 24, 25]. Given that the lightly N-glycosylated HAI-2 is the precursor form, its oligomerization results in a reduction in the levels of heavily glycosylated HAI-2, which is the mature end product and the functional form of HAI-2 for the control prostasin proteolysis. The impact of the c.442C>T SPINT2 mutation (R148C) on the expression of heavily glycosylated HAI-2 was, therefore, next investigated by comparing the ratio of lightly versus heavily glycosylated HAI-2 among four different human colorectal cancer lines, including LoVo cells (Fig. 4). Two different HAI-2 mAbs have previously been developed that exhibit differential binding affinity to the two HAI-2 forms: the mAb XY9, which recognizes lightly glycosylated HAI-2 but not heavily glycosylated HAI-2 and the mAb DC16, which recognizes both HAI-2 forms but apparently with a greater binding affinity for heavily glycosylated HAI-2 than lightly glycosylated HAI-2 [17, 20]. Immunoblot analyses reveal a parallel trend of expression of the two HAI-2 species detected by the mAb XY9 (Fig. 4A) and the HAI-2 species detected by the mAb DC16 (Fig. 4B) with the highest expression in HT29, followed by Caco-2 and then LS174T. A similar and more obvious co-expression trend can be found in our previous study that examined more than twenty different cell lines [20]. LoVo cells appear to be the only exception with a disproportionally low level of HAI-2 signal detected by the mAb DC16 (Fig. 4B, lane 7). The majority of the lightly glycosylated HAI-2 detected by the mAb XY9 were in its monomer form doublet with sizes of between 25–30 kDa (Fig. 4A, as indicated). In addition to the monomer, lightly glycosylated HAI-2 was also detected as signals of apparently greater sizes, particularly in LoVo cells (Fig. 4A, lane 3). These lightly glycosylated HAI-2 species with larger apparent mass include disulfide-linked HAI-2 oligomers and stable enzyme-inhibitor complexes with activated proteases [25]. The enzyme-inhibitor complexes can be dissociated by boiling treatment due to the noncovalent nature of the interaction between HAI-2 and proteases. In contrast, boiling treatment cannot dissociate the disulfide-linked oligomers, which can only be dissociated by exposure to reducing agents. The biochemical characterizations of the two types of HAI-2 complexes can be found in our previous study [25] and in Fig. 6.
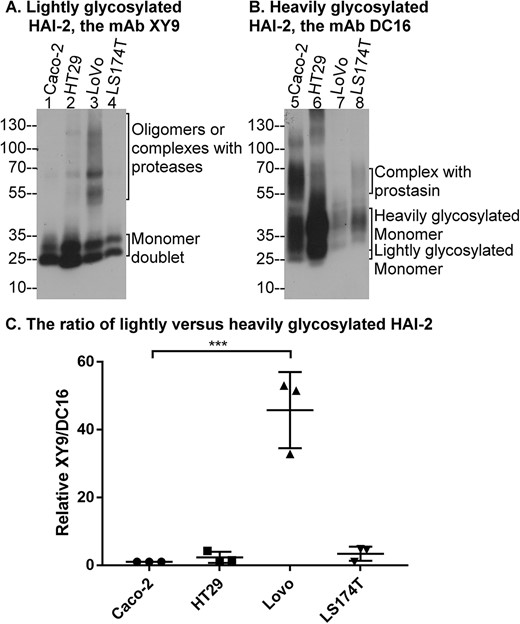
The level of heavily glycosylated HAI-2 is disproportionally low in LoVo colorectal adenocarcinoma cells that harbor SPINT2 mutation. Lysates containing equal amounts of protein from four colorectal cancer lines, as indicated, were analyzed by immunoblot for the expression of lightly glycosylated HAI-2 using the mAb XY9 (A) and heavily glycosylated HAI-2 using the mAb DC16 (B). The various HAI-2 species are indicated. The ratios of lightly versus heavily glycosylated HAI-2 for each cell line were estimated from the band density using three independent experiments using ImageJ (C). ***P < 0.001.
The monomer form of heavily glycosylated HAI-2 detected by the mAb DC16 is seen as a smear pattern with sizes greater than lightly glycosylated counterpart. The diffuse appearance is consistent with its extensive N-glycan branching (Fig. 4B, as indicated). The mAb DC16 also recognizes the monomeric form of the lightly glycosylated HAI-2, as a minor species (Fig. 4B, as indicated). Stable enzyme-inhibitor complexes of heavily glycosylated HAI-2 with proteases are also observed in some of these colorectal cancer cells, particularly Caco-2 cells, in which prostasin-HAI-2 complex was detected at high levels (Fig. 4B, lane 5, as indicated). The identification and characterization of the prostasin-HAI-2 complex in Caco-2 cells can be found in our previous studies [13]. By comparing the expression levels of lightly glycosylated HAI-2 with the corresponding heavily glycosylated counterparts in the same cells, these colorectal cancer cells express the two HAI-2 forms at similar ratio, with the exception of LoVo cells (Fig. 4, lanes 3 and 7).
The ratio of lightly glycosylated HAI-2 (XY9) versus heavily glycosylated HAI-2 (DC16) can be estimated in a relatively quantitative manner by densitometry of the immunoblot signals, followed by dividing the total band density of lightly glycosylated HAI-2 by the total band density of heavily glycosylated HAI-2 for the same cell line. It is worth noting that although the mAb DC16 also detected the lightly glycosylated HAI-2, the level of this species was very low compared to the level of heavily glycosylated HAI-2. Indeed, the signal detected by the mAb DC16 is close to the expression levels of heavily glycosylated HAI-2 among the cells. The [XY9:DC16] signal ratio in Caco-2 cells was used as the primary reference and for comparison purposes adjusted to 1. With three independent experiments, the [XY9:DC16] ratio was estimated to be 2.3 for HT29, 46 for LoVo, and 3.4 for LS174T cells (Fig. 4C). The nearly 50-fold increase in the [XY9:DC16] ratio observed in LoVo cells largely results from the very low expression of heavily glycosylated HAI-2 in those cells. The HAI-2 expression characteristics observed in LoVo cells, therefore, suggests that the c.442C>T SPINT2 mutation may result in compromised expression of heavily glycosylated HAI-2.
SCSD-associated mutations in the Kunitz domain 2 increase disulfide bond-mediated oligomerization of lightly glycosylated HAI-2
As the functional form of HAI-2 for antiprotease activity, the significant decrease in heavily glycosylated HAI-2 expression should suppress, to some extent, the physiological function of HAI-2 as the cognate protease inhibitor of prostasin in human enterocytes. Given the fact that LoVo cells do not express prostasin, the impact of the c.442C>T SPINT2 mutation on prostasin proteolysis cannot be directly analyzed in LoVo cells. Furthermore, the c.442C>T SPINT2 mutation results in the addition of a new cysteine residue (R148C) in the HAI-2 variant. It is unclear how the addition of this cysteine residue contributes to the disarrayed disulfide linkages in the HAI-2 oligomers. We, therefore, set out to examine the impact of SPINT2 mutations located in Kunitz domain 2 on protein folding, N-glycosylation, and subsequently on the control of prostasin proteolysis using a Caco-2 variant in which the endogenous HAI-2 gene had been disrupted by CRISPR as a model system [14]. The HAI-2 knockout Caco-2 cells were first engineered to express either wild-type HAI-2 or one of three SCSD-associated SPINT2 Kunitz domain-2 mutants in a doxycycline-inducible manner. The SCSD-associated SPINT2 mutants included c.442C>T SPINT2 (R148C HAI-2), c.443G>A SPINT2 (R148H HAI-2) and c.448A>G SPINT2 (Y163C HAI-2). The c.443G>A SPINT2 mutant produces HAI-2 protein with a point mutation resulting in the replacement of Arg-148 with His, which does not introduce a new cysteine residue. The c.448A>G SPINT2 mutant produces HAI-2 with a Y163C substitution, which represents one of the SPINT2 mutations most commonly found in patients with SCSD.
Assessment of the impact of these mutations on HAI-2 oligomerization (Figs 5 and 6), N-glycan branching (Fig. 7), and function as prostasin inhibitor (Figs 8 and 9) were next investigated by comparing their effects to cells transfected with wide-type HAI-2. The expression and oligomerization of lightly glycosylated HAI-2 were first characterized by immunoblot analysis using the XY9 mAb (Fig. 5). As described above and in our previous publication [20], the HAI-2 mAb specifically recognizes lightly glycosylated HAI-2, which contains oligomannose-type N-glycan. The N-glycan branching, which generates the heavily glycosylated form of HAI-2, apparently causes the masking of the epitope recognized by the XY9 mAb. As a result, this mAb does not recognize heavily glycosylated HAI-2. Although only present at very low levels, lightly glycosylated HAI-2 was detectable in the HAI-2 KO Caco-2 cells transfected with the wildtype and mutant HAI-2 constructs, even in the absence of doxycycline treatment (Fig. 5 lanes 1). This is likely due to either trace amounts of doxycycline frequently present in FBS, or the inherent leakiness of the system (Fig. 5. lanes 1). Treatment with doxycycline resulted in a rapid dose-dependent increase in the levels of lightly glycosylated HAI-2 present in the cells (Fig. 5, lanes 2–5). Modest induced expression of HAI-2 was seen at 0.02 ug/ml doxycycline (Fig. 5, lanes 2), rising to a level comparable with that in parental Caco-2 cells by 0.1 ug/ml doxycycline (Fig. 5, lanes 3), and to a supra-physiological level at 1, and 10 ug/ml doxycycline (Fig. 5 lanes 4 and 5). This wide dose-dependent expression of lightly glycosylated HAI-2 suggests that the mutant proteins can be synthesized in a similar fashion to its wild-type counterpart, at least so far as in the endoplasmic reticulum prior to transit to the Golgi apparatus for N-glycan branching.
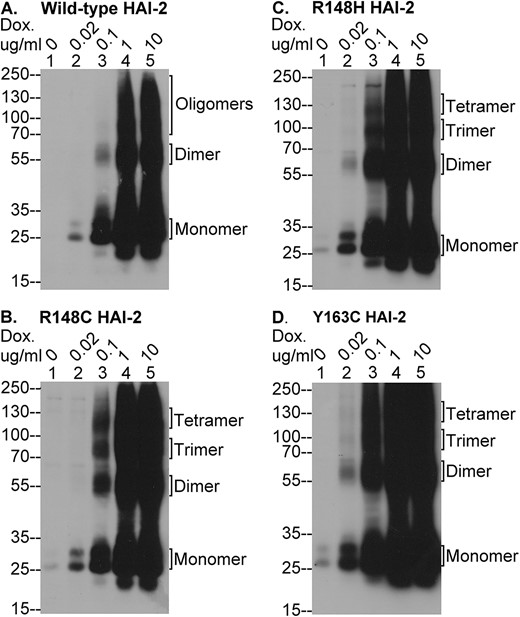
The SCSD-associated SPINT2 mutants are expressed as lightly glycosylated HAI-2 in monomer and multiple complex forms. Expression of wild type and mutant HAI-2 proteins was induced by treatment with different concentrations of doxycycline (Dox.), as indicated, in HAI-2 knockout Caco-2 variants engineered to express wild-type HAI-2 (A) or one of the three SCSD-associated mutants, R148C HAI-2 (B), R148H HAI-2 (C), and Y163C HAI-2 (D). Equal amounts of lysate protein was analyzed by immunoblot under non-reducing and non-boiled conditions for lightly glycosylated HAI-2 species using the HAI-2 mAb XY9. HAI-2 monomer and its oligomer forms (see fig. 6) were indicated.
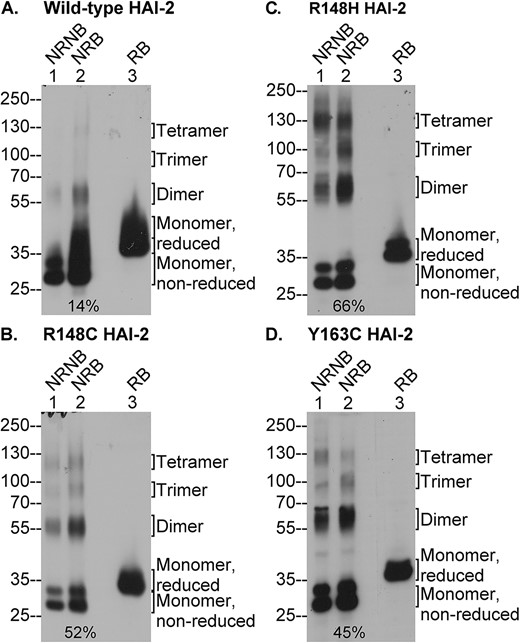
SCSD-associated SPINT2 mutations greatly increase the disulfide bond-mediated oligomerization of lightly glycosylated HAI-2. Expression of wild-type HAI-2 and the three SCSD-associated HAI-2 mutants (B, C, and D) was induced by 0.1 μg/ml doxycycline treatment of the modified Caco-2 cells. Equal amounts of lysate proteins were analyzed by immunoblot under the non-reducing and non-boiled (lanes 1, NRNB), non-reducing and boiled (lanes 2, NRB), or the reducing and boiled (lanes 3, RB) conditions for lightly glycosylated HAI-2 species using the HAI-2 mAb XY9. The various species of lightly glycosylated HAI-2 are indicated. The ratio of HAI-2 oligomers relative to total HAI-2 under the non-reducing and boiled conditions were estimated and indicated (lanes 2).
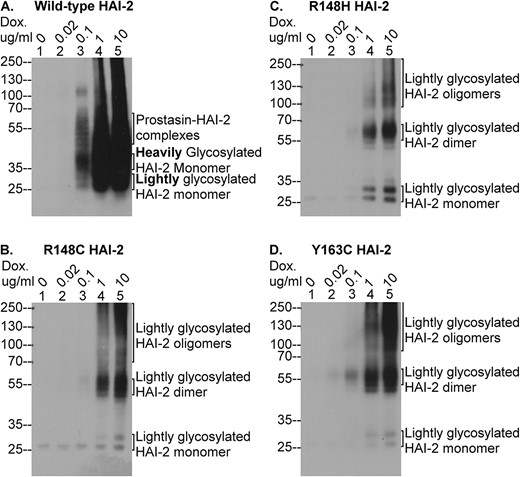
The SCSD-associated SPINT2 mutants are not expressed as heavily glycosylated HAI-2. HAI-2 protein expression was induced using different concentrations of doxycycline (Dox.), as indicated, in HAI-2 knockout Caco-2 variants engineered to express wild-type HAI-2 (A) and the three SCSD-associated mutants, R148C HAI-2 (B), R148H HAI-2 (C), and Y163C HAI-2 (D). Equal amounts of cellular proteins were analyzed by immunoblot under non-reducing and non-boiled conditions for HAI-2 species using the HAI-2 mAb DC16. The various HAI-2 species were indicated.
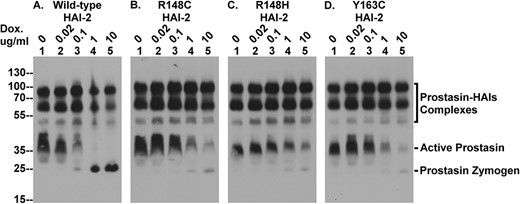
The SCSD-associated HAI-2 mutants cannot effectively suppress the prolonged lifespan of active prostasin caused by HAI-2 loss in Caco-2 cells. The four Caco-2 variants were treated with increasing concentrations of doxycycline (Dox.) to induce expression of wild-type HAI-2 (A) and the three SDSC-associated HAI-2 mutants (B, C, and D). Equal amounts of cellular proteins were analyzed by immunoblot for prostasin using the prostasin mAb YL11. The various prostasin species, including the complexes with the HAIs, active prostasin, and prostasin zymogen are indicated.
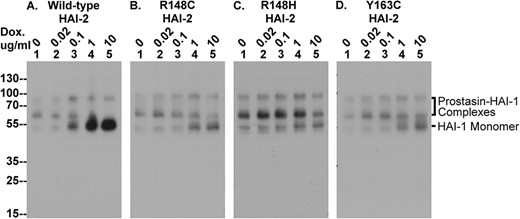
The SCSD-associated HAI-2 mutants cannot effectively reverse the depletion of HAI-1 monomer caused by targeted HAI-2 deletion in Caco-2 cells. The four Caco-2 variants were treated with increasing concentrations of doxycycline (Dox.) to induce expression of wild-type HAI-2 (A) and the three SDSC-associated HAI-2 mutants (B, C, and D). Equal amounts of cellular proteins were analyzed by immunoblot for HAI-1 using the mAb M19. HAI-1 complexes with prostasin and HAI-1 monomer are indicated.
Examination of the protein band profile, however, reveals evidence of interesting and important effects of these SCSD-associated mutations on HAI-2 synthesis and protein folding. Firstly, in non-modified cells such as the parental Caco-2 cells, the vast majority of the lightly glycosylated HAI-2 is consistently detected as a monomer doublet band with a size around the 25-kDa marker, based on the study with more than 20 different cell lines [20]. In the knockout cells transfected with wild type or mutant HAI-2, a significant proportion of lightly glycosylated HAI-2 is detected in multiple complexes, particularly when supra-physiological levels of HAI-2 is expressed (Fig. 5, lanes 4 and 5). Secondly, although wild-type HAI-2 is also detected in this system in these complex forms, the ratio of the complexes relative to the total HAI-2 appeared to be much lower for the wild-type than the mutants (Fig. 5, lanes 4 and 5). The difference in the ratio was even more obvious when the level of HAI-2 was expressed at a level similar to the endogenous levels found in the parental Caco-2 cells by treatment of the transfected cells with 0.1 μg/ml doxycycline (Fig. 5, lanes 3). Although the ratio was decrease in the mutant transfected cells much lower level treated with 0.02 μg/ml doxycycline (Fig. 5, lanes 2), this lower ratio in part results from a dilution effect on the detection of signals distributing to the multiple complexes (Fig. 5, lanes 2). Finally, the sizes of these HAI-2 complexes, particularly those seen in the mutant expressing cells, are consistent with the predicted sizes of HAI-2 oligomers: the 55-kDa band for the dimer; the 85-kDa for the trimer; and the 125-kDa band for the tetramer. Another biochemical characteristic of HAI-2 oligomerization is that these oligomers are held together by disulfide bonds.
In order to verify these HAI-2 complexes are the product of disulfide bond-mediated oligomerization and to better estimate the proportion of HAI-2 oligomers relative to total HAI-2 expressed, lysates prepared from the transfected cells being stimulated with 0.1 μg/ml doxycycline were analyzed by immunoblot under two additional conditions: 1) non-reducing and boiled conditions, under which the stable, but non-covalent enzyme-inhibitor HAI-2 complexes will be dissociated and 2) reducing and boiled conditions under which the disulfide-linked oligomers will be dissociated into monomers. The boiling treatment did not appear to cause dissociation and disappearance of the HAI-2 complexes (Fig. 6, comparing lanes 2 with lanes 1). An interesting observation that has consistently been made is that the overall amount of signal detected, particularly for the wild-type HAI-2, increased in the boiled samples (Fig. 6A, comparing lanes 2 with lanes 1). This increase in the total signal detected suggests that some proportion of the lightly glycosylated HAI-2 may be present in even larger complexes or aggregates, which cannot be completely dissociated by 1% SDS at room temperature and so do not migrate into the SDS-gel. Alternatively, the boiling treatment may cause the exposure of more epitope recognized by the mAb. Nevertheless, the band profiles under non-reducing and boiled conditions allowed us to estimate the ratio of the oligomers relative to total in the absence of potential interference from stable HAI-2 complexes with proteases. Using densitometry and ImageJ analysis, the ratio of HAI-2 oligomers to the total was estimated to be 14% for the wide-type, 52% for the R148C mutant, 66% for the R148H mutant, and 45% for the Y163C mutant (Fig. 6, lanes 2, as indicated). The involvement of disulfide linkages in the formation of these HAI-2 complexes was corroborated by the dissociation and disappearance of the oligomers under reducing conditions and a corresponding increase in the levels of the monomer (Fig. 6, lanes 4). The slower migration of HAI-2 monomer on SDS-PAGE under reducing conditions compared to non-reducing conditions is due to the less compact conformation after breaking the disulfide bonds (Fig. 6, comparing lanes 3 with lanes 2).
SCSD-associated mutations in the Kunitz domain 2 cause loss of heavily glycosylated HAI-2 expression
The impacts of the SPINT2 mutations on HAI-2 N-glycan branching was next investigated by immunoblot analysis of the expression of heavily glycosylated HAI-2 using the DC16 mAb (Fig. 7). Although the DC16 also recognizes lightly glycosylated HAI-2, the diffuse appearance and the different size of these species make for easy and reliable discrimination between the heavily glycosylated HAI-2 monomer (> 35-kDa smear) and the lightly glycosylated HAI-2 monomer (> 25-kDa solid doublet). Upon treating the cells with 0.1 μg/ml doxycycline, the heavily glycosylated form of wild-type HAI-2 began to be expressed as both monomer and complexes. The monomer was detected as smeared bands with sizes slightly greater than 35-kDa and HAI-2 complexes with prostasin were also detected with a smeared appearance (Fig. 7A, lane 3A, as indicated). Lightly glycosylated wild-type HAI-2 monomer was also detected by the mAb DC16 as the characteristic doublet with sizes greater than 25-kDa and a weaker signal (Fig. 7A, lane 3, as indicated). There was a HAI-2 species with a size close to100-kDa, which is likely HAI-2 in complex with protease(s), such as matriptase [11]. With higher concentrations of doxycycline, expression of much higher levels of heavily glycosylated HAI-2 were induced, and the detection was overwhelmed with an intense smear ranging from 25-kDa to the top of the gel (Fig. 7A, lanes 4 and 5). In contrast to the wild-type, the protein band profile on analysis of lysates from the cells transfected with the three HAI-2 mutants reveals no evidence of heavily glycosylated HAI-2 expression, although signal was detected by the mAb DC16, particularly at the higher doxycycline concentrations (1 and 10 μg/ml) (Fig. 7B–D). This signal is the result of the cross-reactivity of the DC16 mAb with lightly glycosylated HAI-2, and is not directly from the heavily glycosylated HAI-2. We can make this assertion because: firstly, there was no signal corresponding to the heavily glycosylated HAI-2 monomer detected. The size of this species is slightly greater than the 35-kDa marker and appears as a smear. Secondly, only negligible levels of HAI-2 signal were detected using the mAb DC16 in the cells treated with 0.1 μg/ml doxycycline (Fig. 7B–D, lanes 3). Thirdly, fairly high levels of HAI-2 signal were observed in the cells induced with 1 and 10 μg/ml doxycycline (Fig. 7B–D, lanes 4 and 5), which is likely the combined result from the high level of lightly glycosylated HAI-2 expression (Fig. 5, lanes 4 and 5) and the weak cross-reactivity of the mAb DC16 to lightly glycosylated HAI-2. Finally, the size of the HAI-2 bands detected by the mAb DC16 under these conditions match those of various lightly glycosylated HAI-2 species, as detected by the mAb XY9, including 1) the 25-kDa doublets corresponding to the lightly glycosylated HAI-2 monomer, 2) the 55-kDa bands corresponding to the lightly glycosylated HAI-2 dimer, and 3) the 100-kDa and 125-kDa bands corresponding to the trimer and tetramer of lightly glycosylated HAI-2, respectively. Taken together, these data suggest that no detectable heavily glycosylated HAI-2 was expressed by the three SCSD-associated missense SPINT2 mutants.
The SCSD-associated HAI-2 mutants cannot effectively revert the high level of free active prostasin or the depletion of HAI-1 monomer that result from the targeted deletion of HAI-2 in Caco-2 cells
Although lightly glycosylated HAI-2 monomer is synthesized, the loss of heavily glycosylated HAI-2 suggests that the enterocytes in SCSD patients might experience uncontrolled prostasin proteolysis similar to what is seen in HAI-2 knockout Caco-2 cells [14]. In human intestinal tissue and Caco-2 human colorectal adenocarcinoma cells, prostasin is under the tight control of HAI-1 and HAI-2 [13]. It is worth noting that HAI-1 functions as the default prostasin inhibitor in many types of epithelial cell and cell lines, but that HAI-2 serves as the primary prostasin inhibitor in human enterocytes, in a cell-type selective manner [13, 14]. Although prostasin undergoes constant high-level zymogen activation in Caco-2 cells, the resultant free active prostasin is only short-lived due to the rapid inhibition by HAI-1 and HAI-2. Targeted HAI-2 deletion in Caco-2 cells leaves the fraction of the newly activated prostasin that is inhibited by HAI-2 in the parental cells either to be inhibited by binding with HAI-1 or to remain as the free, active enzyme. This leads to the depletion of the available HAI-1 monomer pool and, as a result, some of the active prostasin remains uncomplexed [14].
The presence of enzymatically active prostasin along with prostasin-HAI-1 complexes was easily detected by immune blot using the prostasin mAb YL11 in lysates prepared from the HAI-2 KO Caco-2 cells transfected with the wild-type HAI-2 construct, even prior to exposing the cells to doxycycline (Fig. 8A, lanes 1, as indicated). The detection of enzymatically active prostasin by immunoblot analysis was verified by the characteristic and the biochemical behavior of the corresponding prostasin bands detected by immunoblot analysis. The characterization and verification, as previously described [13], include 1) the rapid formation of the enzyme-inhibitor complex in solution using immunoaffinity-purified active prostasin and immunoaffinity-purified HAI-1 and 2) the detection of the amidolytic activity of the immunoaffinity-purified active prostasin using a synthetic fluorogenic substrate selective for prostasin. When wild-type HAI-2 expression was induced by treatment with increasing doxycycline concentrations, (Figs 5 and 7), the levels of free active prostasin decreasing in parallel with increasing HAI-2 expression (Fig. 8A, lanes 1–3, Active Prostasin) and then completely disappeared when very high level HAI-2 expression was induced (Fig. 8A lanes 4 and 5, Active Prostasin). The disappearance of the active prostasin was accompanied by the appearance of prostasin zymogen (Fig. 8A, lanes 3, 4, and 5, Prostasin Zymogen), suggesting that the supra-physiological levels of wild-type HAI-2 not only inhibit active prostasin but also prevent the zymogen activation of prostasin. This result and conclusion mirrors the observations made in our previous study [14].
In contrast to the effective inhibition of the free active prostasin present in HAI-2 knockout cells by the re-expression of wild-type HAI-2, expression of the three SCSD-associated HAI-2 mutants had little or no impact on the levels of free active prostasin when expressed at physiologically relevant levels (0.1 μg/ml doxycycline) (Fig. 8B–D, lanes 1–3, Active Prostasin). Furthermore, when supra-physiological levels of the HAI-2 mutants were expressed by treatment with 1 and 10 μg/ml doxycycline, the suppression of free active prostasin levels and the appearance of prostasin zymogen was far less noticeable than that produced by wild-type HAI-2 (Fig. 8, lanes 4 and 5, comparing B, C, and D with A for Active Prostasin and Prostasin Zymogen). As described above, these SCSD-associated HAI-2 mutants are expressed only in the lightly glycosylated form and not as heavily glycosylated HAI-2 (Figs 5 and 7). Although the lightly glycosylated HAI-2 oligomers have no anti-protease function, lightly glycosylated HAI-2 monomer does still possess anti-trypsin activity [25]. In spite of the very high levels of the lightly glycosylated HAI-2 monomer, they do not, however, appear to be able to effectively inhibit the free active prostasin (Fig. 8B–D, lanes 4 and 5). While it is true that the anti-trypsin activity of lightly glycosylated HAI-2 is weaker than that of the heavily glycosylated form [25], this does not satisfactorily explain why the levels of free, active prostasin persist. Rather, it is because lightly glycosylated HAI-2 lacks the appropriate sub-cellular trafficking signal to bring the inhibitor close proximity with the active prostasin. The decrease in the level of active prostasin observed in the mutant HAI-2 transfectants treated with 1 and 10 μg/ml doxycycline is likely the result of the extremely high levels of expression of lightly glycosylated HAI-2 monomer achieved overwhelming the trafficking system leading to “spillover” of the inhibitor into proximity with the active prostasin.
As described above and previously [14], in addition to the appearance of enzymatically active prostasin, targeted deletion of HAI-2 also caused the significant decrease in HAI-1 monomer. The reduction in the level of HAI-1 monomer observed in the HAI-2 knockout cells, was reverted by the induced re-expression of HAI-2, but once again, the HAI-2 mutant constructs were largely ineffective (Fig. 9). HAI-1 is synthesized as a functional monomer form with an apparent size close to the 55-kDa marker protein. In addition to the monomer, HAI-1 was also detected in complex with prostasin as a band of 100-kDa along with its degradation product with a size of 65-kDa. The HAI-1 in this enzyme-inhibitor complex is no longer able to inhibit the remaining active prostasin due to the occupation of the Kunitz domain 1 by active prostasin. Prior to the induction of wild-type HAI-2 expression, HAI-1 monomer is barely detectable or at very low levels (Fig. 9, lanes 1). As the level of wild-type HAI-2 is increased, the levels of HAI-1 monomer also increase (Fig. 9A). The increase in HAI-1 monomer was not nearly so obvious when the expression of the three SCSD-associated HAI-2 mutants was induced by treatment with 0.1 μg/ml doxycycline (Fig. 9B–D, comparing lanes 3 with lanes 1). Even when supra-physiological levels of the three SCSD-associated HAI-2 mutants were induced, the increase in HAI-1 monomer levels was modest (Fig. 9B–D, comparing lanes 4 and 5 with lanes 1). The inability of the SCSD-associated HAI-2 mutants to rescue the levels of HAI-1 monomer in HAI-2 KO Caco-2 cells is consistent with the notion that these HAI-2 mutants lack the function of the heavily glycosylated form of HAI-2 and the targeting of lightly glycosylated HAI-2 to a different subcellular location than that of prostasin.
It is worth noting that prostasin is not the only HAI-1 target protease in Caco-2 cells [13]. A significant proportion of the available HAI-1 is destined to control matriptase activity in the parental Caco-2 cells. Due to its rapid shedding, matriptase-HAI-1 complex can be found both in the cell lysate and the conditioned medium. Targeted HAI-2 deletion causes an increase in matriptase zymogen activation and shedding of matriptase-HAI-1 complexes [14]. The increased matriptase zymogen activation also contributes to the depletion of HAI-1 monomer. When wild-type HAI-2 expression was induced, although HAI-1 monomer levels increased also in response, that increase was not proportional to the decreased levels of HAI-1 complexes, as shown in fig. 9A. This occurs because in addition to the inhibition of prostasin by the HAI-2 it also reduces the zymogen activation of both prostasin and matriptase which combines to cause a rapid increase in the HAI-1 monomer pool.
Discussion
The presence of these five SPINT2 missense mutations in almost all SCSD patients examined suggests that the pathogenesis of SCSD must begin with mechanisms by which the altered secondary structure of Kunitz domain 2 results in a compromise of the functionally relevant Kunitz domain 1 and/or the altered subcellular targeting signals [9, 26–31]. Thereby, the physiological function of HAI-2 as a prostasin inhibitor is lost or significantly compromised. Based on the current study with three of the five SPINT2 missense mutations, the simple substitution of one amino acid residue unexpectedly causes abnormal protein folding and prevents N-glycan branching of these HAI-2 mutants. Consequently, these SCSD mutants cannot be synthesized into the heavily glycosylated form of HAI-2, which is the functional entity. The abnormal protein folding produced by these mutations was manifested by approximately one half of the mutant HAI-2 protein being synthesized as disulfide-linked oligomers, which possess no anti-protease activity, due to the distorted conformation of the Kunitz domains. The lack of N-glycan branching results in the remainder of the HAI-2 mutant protein being synthesized as the immature, lightly glycosylated form, which not only has weaker anti-protease activity, but which also contain incomplete subcellular targeting signals. As a result, the lightly glycosylated HAI-2 cannot function as an effective prostasin inhibitor. In spite of being normally translated, these SPINT2 mutants are not synthesized into mature heavily glycosylated HAI-2 and so their gene products are not able to control prostasin proteolysis in human enterocytes.
Oligomerization, one of the primary mechanisms underlying HAI-2 inactivation resulting from single amino acid substitutions, involves the formation of intermolecular disulfide bonds. The cysteine residues involved in this oligomerization are normally involved in the formation of the intramolecular disulfide bonds required to form the functional Kunitz domains [33]. In addition to the loss of functional Kunitz domains, some additional interesting features have been noticed for disulfide bond shuffling. The HAI-2 oligomers may represent a suboptimal conformation rather than a misfolded state. Misfolded HAI-2 should be translocated to the cytoplasm and degraded by the proteasome. The HAI-2 oligomers may “fool” the quality control mechanisms and so are not disposed efficiently when supra-physiological levels of wild type HAI-2 synthesis is induced (Fig. 5). The hypothesis that HAI-2 oligomers represent a state with suboptimal conformation is supported by the observation that HAI-2 oligomers in cell lysates can spontaneously dissociate into the monomer form when the cell lysates are stored in −80°C (data not shown). This spontaneous dissociation could involve disulfide bond shuffling and suggests that the oligomers are in a less stable thermodynamic state. N-glycosylation appears to be able to prevent oligomerization to some extent. Both disulfide bond formation and N-glycosylation are co-translational events in the ER. The N-glycan modification which involves the removal and addition of glucose represents an important mechanism to facilitate protein folding into the correct conformation [35]. Previously, when the N-glycosylation site, Asn-57, was mutated, it was shown that the vast majority of the non-glycosylated HAI-2 was synthesized as oligomers [25]. Our current study suggests that the integrity of other HAI-2 structural elements, such as Kunitz domain 2, also appear to be important for the N-glycosylation of HAI-2. In contrast to these SCSD-associated mutations, the R143L HAI-2 variant, in which the P1 site Arg-143 of the reactive center in Kunitz domain 2 is mutated to Leu, can be synthesized as the heavily glycosylated form (data not shown). The role of the altered secondary structure of the Kunitz domain 2 in the oligomerization and prevention of the synthesis of heavily glycosylated HAI-2 is likely to be specific for these SCSD-associated SPINT2 mutations.
The oligomerization of HAI-2 mutants is mediated by aberrant disulfide bond formation, which involves cysteine residues. This effect does not, however, depend on the additional cysteine residue caused by the R148C mutation as the oligomerization is not greater, and indeed possibly slightly less extensive than the R148H mutation. Our data does indicate that the R148H mutation does appear to cause the generation of HAI-2 oligomers at a slightly higher ratio than the R148C mutation. The dominant forces that guide protein folding are hydrophobic effects, conventional hydrogen bonding, Coulombic interactions, and van der Waals interactions. Other interactions that also contribute to protein folding can be classified into two groups: 1) abundant weak interactions of the protein backbone, and 2) less frequent strong interactions involving protein side chains, including interactions involving aromatic rings (cation–π, X–H···π, π–π, anion–π, and sulfur-arene) [36]. Although the R148C mutation could cause unexpected disulfide bonds during the folding process, these disulfide bonds could be reversed especially with the help of protein disulfide-isomerase (PDI) in the ER. In the R148H mutant, the His residue contains an imidazole group, which is partially protonated at physiological pH. In contrast, the Arg residue in wild-type HAI-2 contains a 3-carbon aliphatic straight chain with a guanidinium group at its distal end. In spite of being a basic residue like Arg, the imidazole group of His presents a planar ring containing six π-electrons, and thus the side chain of His can be involved in strong interactions introduced by aromatic rings. This notion is further supported by a stronger effect of the R148H mutation than the R148C mutation on the fold stability change (ΔΔG) upon single amino acid mutations, based on the prediction using STRUM (https://zhanggroup.org/STRUM/), an online service for prediction of the fold stability change (ΔΔG) upon single amino acid mutations on protein structure [37]. In combination with the predicted 3D structure of HAI-2 using AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/), the predicted ΔΔG are 0.27, 0.15 for R148H and R148C respectively. The predicted ΔΔG indicates that the R148H mutation has a stronger effect on HAI-2 protein structure than R148C. This predicted stronger impact on the fold stability change somewhat correlates with the higher oligomerization caused by the R148H vs R148C mutation.
In addition to the oligomerization which causes approximately one half of the HAI-2 synthesized to be inactivated, the lack of N-glycan branching causes the rest of the mutant HAI-2 to be unable to perform HAI-2’s physiological function as prostasin inhibitor. This is in spite of the fact that this portion of the HAI-2 can be detected as monomers with oligomannose-type N-glycan. It appears that, for some reason, the altered secondary structure in Kunitz domain 2 prevents the conversion and maturation of the lightly glycosylated HAI-2 into the mature counterpart with complex-type N-glycan. This defect may be the result of some problem with the transit from the ER to the Golgi apparatus and/or in the process of N-glycan branching in the Golgi apparatus. N-glycan branching is catalyzed by glycosidases, N-acetylglucosaminyltransferases, and a fucosyltrasferase [38–40]. The aberrant N-glycan branching frequently found in cancers primarily results from the dysregulated expression of these enzymes [40]. Our previous study with the N94Q HAI-2 mutant, however, suggests that the structure of the client protein can also affect the final products of these branching enzymes [25]. The substitution of Asn-94, which is positioned between the two Kunitz domains, unexpectedly changed the size of the complex-type N-glycan modified on the HAI-2 variant [25]. The complete loss of heavily glycosylated HAI-2 caused by the SCSD-associated mutations suggests that the altered secondary structure in the Kunitz domain 2 may either prevent the transit of newly synthesized lightly glycosylated HAI-2 monomer from the ER to the Golgi apparatus and/or the interaction of the HAI-2 with these N-glycan branching enzymes. The expression of unusually low levels of heavily glycosylated HAI-2 in LoVo cells (Fig. 4) may also shed light on how the defect with N-glycan branching could occur. LoVo cells are heterozygous with respect to SPINT2 with one allele being wild-type and the other allele carrying a point mutation at Arg-148. If one assumes that both alleles are transcribed equally, the presence of much less than half of the HAI-2 synthesized being of the heavily glycosylated form strongly suggests that the mutated HAI-2 protein negatively affects N-glycan branching of the wild-type HAI-2 protein. The dominant-negative effect hypothesized for the low expression of heavily glycosylated HAI-2 found in LoVo cells may, however, not be relevant to the situations in some heterozygous parents of the SCSD patients with one normal SPINT2 allele. These parents were not reported to be affected by clinical or intermediate signs of this disorder, indicating that the physiological function of HAI-2 may not be compromised. This potential discrepancy between LoVo cells and the heterozygous parents could result from the obvious difference between the non-cancerous enterocytes in vivo and the cultured colorectal adenocarcinoma cells cancer, such as the expression of prostasin. Alternatively, in these heterozygous parents the dominant-negative effect could affect the synthesis of heavily glycosylated HAI-2 somehow to less severe degree than that found in LoVo cells. At certain lower levels of heavily glycosylated HAI-2 expressed in conjunction with the existing high levels of HAI-1 protein, the prostasin proteolytic activity could still well-maintained or the imbalance in prostasin proteolytic activity sufficiently mild to not significantly disrupt Na+ homeostasis.
The mechanistic details regarding how loss of HAI-2 function, and/or how unrestrained prostasin proteolysis causes the failure in sodium absorption in the GI-tract of SCSD patients remains elusive. The reduced suppression of prostasin activity results in an increase in the functional half-life of the free, active enzyme which could result in increased proteolytic activation and/or processing of prostasin substrates, which could enhance the physiological function of prostasin. If this is the case, the prostasin must play a suppressive role in sodium absorption in human enterocytes. Alternatively, the prolonged prostasin proteolytic activity could cause the failure of sodium absorption via the undesired cleavage of physiologically irrelevant substrates, including some ion channels/transporters involved in sodium homeostasis. Although miss-regulated prostasin activity may impact sodium absorption via a specific or non-specific mechanism, studies with the fr/fr mouse model with a V170D prostasin missense mutation and the frCR/frCR rat, which has a G54-P57 prostasin deletion, suggest that prostasin mutations primarily impact the local control of water homeostasis, based on a dehydration phenotype in the skin of affected mice and an increase in stool hydration (diarrhea) in affected rats [41]. The skin barrier function of prostasin has been further confirmed by the study of prostasin knockout mice [42]. These animal studies suggest that prostasin may have roles in the regulation of ion channels/transporters involved in water homeostasis. For example, an increase in the sodium current across the plasma membrane was seen when prostasin and the epithelial sodium channel (ENaC) were co-expressed in xenopus oocytes [43]. Furthermore, prostasin-mediated ENaC activity can be inhibited by a variety of protease inhibitors [44–47]. As such, an increase rather than a decrease in sodium absorption would be the more likely result of prolonged prostasin proteolytic activity caused by the SPINT2 mutations.
In summary (Fig. 10), the pathogenesis of SCSD begins with the loss of HAI-2 physiological function as the primary prostasin inhibitor in a cell-type selective manner primarily in enterocytes. HAI-2 function relies on the subcellular targeting signals to gain access to prostasin and the Kunitz domain 1 for the ability to inhibit prostasin proteolytic activity. HAI-2 inactivation can result from the loss of or malfunction on these functionally important structural elements, which can be predicted for some SCSD-associated SPINT2 mutations. HAI-2 inactivation can also be caused by abnormal protein folding and N-glycosylation which is observed for some other SCSD-associated SPINT2 mutations that involve point mutations of the functionally less important Kunitz domain 2. The unexpected loss of HAI-2 function caused by these SCSD-associated SPINT2 mutations is essential for the pathogenesis of this type of SPINT2 mutation in almost all SCSD patients. HAI-2 inactivation probably causes uncontrolled proteolysis by the HAI-2 target proteases, primarily matriptase and prostasin. The cell-type selective functional relationship between HAI-2 and these two membrane-associated serine proteases means that the uncontrolled proteolysis primarily involves prostasin in enterocytes. Although dysregulation of ion channels/transporters is believed to be the culprit for the sodium diarrhea, the molecular mechanism, by which the excess prostasin proteolysis causes the dysregulation of ion channels/transporters, is not clear. The prolonged availability of enzymatically active prostasin caused by HAI-2 disfunction may lead to the hyperactivation of downstream substrates in a specific fashion, however, it is also possible that the dysregulation of sodium homeostasis is caused by the cleavage of proteins not typically involved in the regulation of these processes. The clinical presentation of SCSD patients with structural abnormalities such as choanal atresia, and anal atresia suggests that the uncontrolled proteolysis caused by HAI-2 inactivation probably also occurs in some cells during development. The underlying molecular and cellular mechanisms, including which proteases, matriptase or prostasin and the downstream effectors remain to be identified.
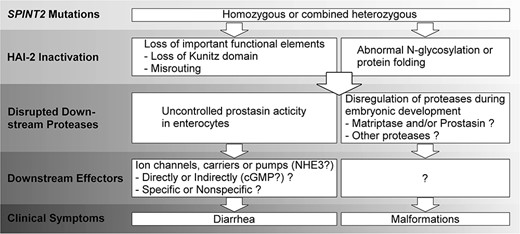
Schematic illustration of SCSD pathogenesis. The cellular and molecular mechanisms underlying the pathogenesis of SCSD caused by SPINT2 mutations begin with the inactivation of HAI-2, which causes uncontrolled proteolysis, followed by disrupted proteolysis and dysregulated downstream effectors, leading to sodium diarrhea and developmental defects. A description of the known and unknown molecular and cellular processes can be found in the discussion.
Materials and methods
Cell culture
The Caco-2 cell line was a gift from Dr Toni Antalis of the University of Maryland Baltimore. The preparation of a HAI-2 knockout Caco-2 variant by CRISPR has been described in our previous study [14]. The HT29, LS174T, and LoVo cell lines were obtained from the Tissue Culture and Biobanking Shared Resource (TCBSR) of the Georgetown Lombardi Comprehensive Cancer Center. All cell lines were grown in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere with 5% CO2 at 37°C.
Exogenous protein expression using the Tet-On system
A wide-type HAI-2 plasmid (pCMV-HAI2) was purchased from OriGene Technologies, Inc (CAT#: RC202044). The R148H, R148C and Y163C mutant HAI-2 plasmids were generated in the pCMV-HAI2 plasmid using the QuikChange XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instruction. The sequence of the primers used are listed below.
R148H: 5′-cacgtcaaagtaccagtgtgggaaggatgcacg-3′
5′-cgtgcatccttcccacactggtactttgacgtg-3′
R148C: 5′-caaagtaccagcatgggaaggatgcacggcaa-3′
5′-ttgccgtgcatccttcccatgctggtactttg-3′
Y163C: 5′-cggcagcctccacagatgaagttattgcaggagttc-3′
5′-gaactcctgcaataacttcatctgtggaggctgccg-3′
The wild-type and mutant HAI-2 cDNAs were subcloned into pLVX-TRE3G (Clontech/Takara) as previously described [48]. The pLVX-TRE3G-X plasmids or pLVX-Tet3G (TetON, Clontech/Takara) were co-transfected with pHRCMV8.2DR and pCMV-VSVG (both obtained from Dr Todd Waldman, Georgetown University) into HEK 293T cells to produce lentiviruses as previously described [14]. To generate exogenous HAI-2 (WT or mutant) expression with the Tet-On system, HAI-2 knockout Caco-2 cells was infected at an approximate MOI of 5 and polybrene at 6 μg/ml using two rounds of selection as previously described [14]. In the first selection, the Tet3G (TetOn) virus was used with G418 (1000 μg/ml, Life Technologies) selection for at least two passages. In the second selection, LVX-TRE3G-HAI-2, -R148H, -R148C or -Y163C virus was used with puromycin (2 μg/ml, Life Technologies) selection for at least two passages. Doxycycline at various concentrations was added to the media for 48 h to induce expression of the gene of interest prior to harvesting of the cells.
Western blotting and antibodies
The cells were harvested by scrapping on ice, then lysed with radioimmunoprecipitation assay (RIPA) buffer, containing 1 mM 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB). After removal of insoluble debris by centrifugation, the protein concentration in the supernatant was determined by Bio-Rad Protein Assay Dye Reagent (Bio-Rad Laboratories, Hercules, CA, USA). The cell lysates were diluted in 5x SDS sample buffer containing no reducing agent. For studies requiring processing samples under reducing condition, 1 μl 2-mercaptoethanol was added to 25 μl sample (with SDS-sample buffer), followed by incubation at 100°C (boiling) for 5 min. Proteins were separated by 10% SDS-PAGE, and transferred to 0.45 μm nitrocellulose membranes. The membranes were incubated with indicated primary mAbs and then horseradish peroxidase (HRP)-conjugated secondary antibodies (SeraCare—formerly Kirkegaard & Perry Laboratories). The results were visualized on x-ray films using Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences). The X-ray films were then scanned, and band intensity was determined using Image J. Three biological repeats were performed for each experiment. For statistical comparison, ordinary one-way ANOVA were performed using GraphPad Prism 9.4.0. Data are presented as mean ± SD. The primary mAbs used were the mAb YL11 for prostasin, the mAb M19 for HAI-1, and the mAbs XY9 and DC16 for HAI-2. The generation, validation, and characterization of these mAbs can be found in our previous publications [11, 17, 20, 49].
Conflict of interest statement: There is no conflict of Interest among all authors.
Funding
This study was supported by National Cancer Institute (NCI) Grant R01 CA 123223 (to M.D.J. and C.Y.L.), Grants (MAB-109-042, MAB-110-045, and MAB-111-060) from the Ministry of National Defense Medical Affairs Bureau, Taiwan and Grant (CMNDMC11110) from Chi-Mei Medical Center, Tainan, Taiwan (to J.-K.W.). The funders had no role in study design, data collection, and analysis, the decision to publish, or the preparation of the manuscript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}