




Abstract
UBQLN2 mutations cause amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD), but the pathogenic mechanisms by which they cause disease remain unclear. Proteomic profiling identified ‘mitochondrial proteins’ as comprising the largest category of protein changes in the spinal cord (SC) of the P497S UBQLN2 mouse model of ALS/FTD. Immunoblots confirmed P497S animals have global changes in proteins predictive of a severe decline in mitochondrial health, including oxidative phosphorylation (OXPHOS), mitochondrial protein import and network dynamics. Functional studies confirmed mitochondria purified from the SC of P497S animals have age-dependent decline in nearly all steps of OXPHOS. Mitochondria cristae deformities were evident in spinal motor neurons of aged P497S animals. Knockout (KO) of UBQLN2 in HeLa cells resulted in changes in mitochondrial proteins and OXPHOS activity similar to those seen in the SC. KO of UBQLN2 also compromised targeting and processing of the mitochondrial import factor, TIMM44, resulting in accumulation in abnormal foci. The functional OXPHOS deficits and TIMM44-targeting defects were rescued by reexpression of WT UBQLN2 but not by ALS/FTD mutant UBQLN2 proteins. In vitro binding assays revealed ALS/FTD mutant UBQLN2 proteins bind weaker with TIMM44 than WT UBQLN2 protein, suggesting that the loss of UBQLN2 binding may underlie the import and/or delivery defect of TIMM44 to mitochondria. Our studies indicate a potential key pathogenic disturbance in mitochondrial health caused by UBQLN2 mutations.
Introduction
Mutations in UBQLN2 cause amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD) (1,2). The encoded ubiquilin-2 (UBQLN2) protein is one of a small family of proteins in humans that function in protein quality control (3–6). The proteins regulate proteostasis by clearing ubiquitinated proteins through the proteasome and autophagy–lysosomal pathways (7–16). Most ALS/FTD mutations in UBQLN2 map in or near a PXX repeat, a domain unique to UBQLN2 (2). Accumulating evidence suggests that the mutations compromise proteasomal degradation and impede autophagy (1,9,13,17–19). However, how these defects cause pathogenesis remains unclear.
A key disturbance implicated in the etiology of many neurodegenerative diseases, particularly ALS, is mitochondrial dysfunction. Gross abnormalities in mitochondrial structure are seen in motor neurons (MNs) of ALS patients (20,21). Furthermore, structural and functional deficits in mitochondria are commonly seen in different rodent models of ALS (reviewed in 22). One speculation is that the dysfunction may lead to MN demise because of the vital bioenergetic role mitochondria have in ATP production (22). Mitochondria house the electron transport chain, composed of five multisubunit complexes that generate ATP through oxidative phosphorylation (OXPHOS) (23). Damage or loss of a single subunit in any complex can drastically hinder ATP production (24,25).
UBQLN proteins have been implicated in quality control of mitochondrial proteins (26,27). Most mitochondrial proteins are synthesized outside of the mitochondria and then imported into the organelle (28,29). Cells utilize a complex system of signaling and factors to stringently regulate the type, amount and quality of proteins imported into mitochondria (30). UBQLN proteins were shown to triage mitochondrial proteins for import (26,27). However, the consequence of mutation or loss of UBQLN2 in affecting mitochondrial function has not been established.
We recently described transgenic (Tg) mice with neuron-specific expression of UBQLN2, showing that a P497S line, expressing ALS/FTD mutant P497S UBQLN2, recapitulates hallmark features of the human disease. In contrast, a WT356 line, expressing equivalent amounts of wild-type (WT) UBQLN2, has minimal signs of disease (31,32). Here, we describe evidence of major perturbations in the structure, function and biochemical properties of mitochondria in the P497S mutant line. We show that inactivation of UBQLN2 expression in cells recapitulates the mitochondrial deficits found in the animals and can be rescued by reexpression of WT but not ALS/FTD mutant UBQLN2 proteins. Mechanistically, we show that UBQLN2 is required for mitochondrial protein import and that ALS/FTD mutations in UBQLN2 are compromised in this activity.
Results
Alteration in mitochondrial protein levels in P497S mutant mice
We recently reported on global proteomic changes to the hippocampus and lumbar SC of 8-week-old isogenic P497S, WT356 and non-Tg mice (33). Gene ontology analysis revealed the largest category (i.e. number) of protein changes in P497S animals was ‘mitochondrial proteins’ (Supplementary Material, Fig. S1A). The Log2 ratios of the mitochondrial proteins whose levels were significantly (P < 0.05) changed in P497S versus either the WT356 or non-Tg animals are shown (Supplementary Material, Fig. S1B). All of these changes were of a reduction in the levels of proteins involved in diverse mitochondrial activities. They included multiple subunits in all five OXPHOS complexes, ribosome subunits as well as proteins involved in mitochondria complex assembly, dynamics and import. The magnitude of their reduction was greater when P497S mutant animals were compared with WT356 animals than with non-Tg animals. We reasoned that the increase could stem from positive and negative effects of expression of the WT and mutant UBQLN2 transgenes on mitochondrial function. Importantly, the changes manifested mainly in the SC and not in the hippocampus (33), which we speculate could be due to differential vulnerability of neurons in the two tissues to pathogenesis.
We also examined the ANSWER ALS proteomic database to see whether the mitochondrial protein changes found in our P497S mutant animals also occur in ALS cases and found a similar general trend of a decrease in the levels of many mitochondrial proteins in ALS cases (Supplementary Material, Fig. S2). However, the magnitude of many of the changes were quite different in P497S animals, suggesting the mechanism of interference of mitochondrial protein levels in P497S animals is different to the cases in the ALS repository, or because of intrinsic differences between the species, or for some other reason. Particularly noteworthy is the larger magnitude of decrease in the TOMM import complex in the mutant UBQLN2 animals, compared to the human cases (mostly sporadic cases). Nevertheless, this comparison shows that mitochondrial protein changes are major alterations seen in ALS.
Our subsequent studies focused on the identity, timing and functional consequence of these changes.
Validation of mitochondrial protein changes in the P497S animals
We first determined whether alteration in mRNA expression could underlie the protein changes seen in P497S animals. However, quantification of mRNA expression of select OXPHOS subunits in spinal MN by translational profiling revealed little difference between P497S and non-Tg animals (Supplementary Material, Fig. S1C), suggesting the change likely arises by a posttranslational mechanism.
We next conducted immunoblots of SC lysates of age-matched P497S, WT356 and non-Tg animals to validate and determine the timing of the changes. To ensure reproducibility of our findings, lysates from at least three independent animals for each genotype (for this and most subsequent determinations) were probed for proteins involved in different mitochondrial functions (Fig. 1A–F).
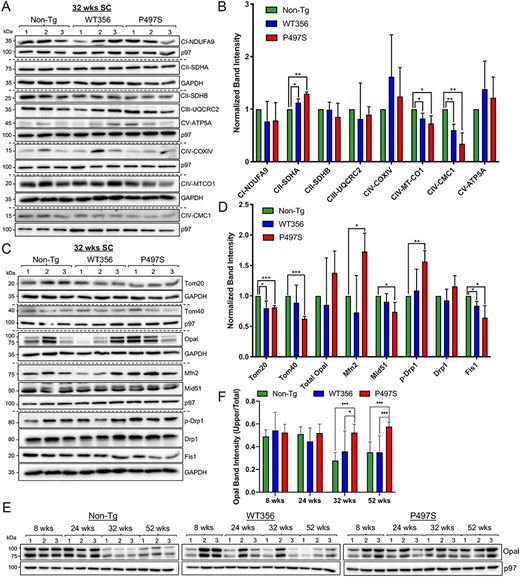
Mitochondrial protein changes in the P497S UBQLN2 mouse model of ALS/FTD. (A) Immunoblots of SC lysates from three independent mice for the three genotypes probed for mitochondrial electron transport chain proteins. (B) Quantification of the blots shown in A. *P < 0.05, **P < 0.01. (C) Immunoblots of SC lysates from 32-week-old animals probed for mitochondrial dynamics and import proteins. (D) Quantification of the blots shown in C. *P < 0.05, **P < 0.01, ***P < 0.001. (E) Immunoblots of SC lysates from mice of different ages and genotypes probed for Opa1. (F) Quantification of uncleaved Opa1 (100 kDa) relative to total Opa1 levels. *P < 0.05, ***P < 0.001.
Analysis of a select set of OXPHOS subunits revealed that levels of both MT-CO1 and CMC1 of Complex IV were significantly decreased in 32-week-old P497S animals compared with non-Tg animals. Further examination of the lysates for proteins that regulate mitochondrial fission and fusion revealed a consistent pattern of change in P497S animals, suggestive of a tilt in the normal dynamics favoring fusion (Fig. 1C). For example, levels of the fusion factor, Mfn2, were significantly increased in P497S compared with non-Tg animals (Fig. 1D). In contrast, levels of Fis1, a protein that mediates stress-induced mitochondrial fission (34), were decreased in P497S compared with non-Tg animals. Furthermore, while overall levels of the fission dynamin-related proteins Drp1 were unchanged, its phosphorylation at Serine637, which blocks GTPase required for its fission activity (35,36), was increased in the P497S mutant.
To determine when disturbances in mitochondrial fusion manifest in the mutant P497S line, SC lysates of animals at 8, 24, 32 and 52 weeks of age were probed to monitor the levels of Opa1 and its cleaved forms (Fig. 1E). Opa1 is required for fusion of the inner mitochondrial membrane and for formation of mitochondrial cristae (37,38). The protein exists as two major forms: an active 100 kDa full-length form and an inactive 80 kDa cleaved form (39,40). Summation of both Opa1 bands, after normalization for protein loading, revealed little differences across age or genotype. However, calculation of the proportion of active Opa1 to total Opa1 protein levels in the samples revealed statistically higher levels of the 100 kDa form in P497S animals at and beyond 32 weeks of age (Fig. 1F). Interestingly, the amount of Opa1 in P497S animals remained relatively constant across ages, whereas there were large fluctuations in the WT356 and non-Tg groups.
The third group of mitochondrial proteins we analyzed was composed of Tom20 and Tom40, components of the import channel (30,41,42). Tom20 is the major receptor for protein import, whereas Tom40 is the central component of the import channel. Levels of both Tom20 and Tom 40 were decreased in the SC of P497S mutant animals compared with non-Tg (Fig. 1C and D).
These immunoblot results demonstrate the global extent of the alterations in mitochondrial proteins in the SC of mutant P497S animals. We believe that because the P497S mutant transgene is only expressed in neurons, the changes in whole tissue lysates are likely to be an underestimate of their true magnitude.
Age-dependent decline of mitochondrial function in P497S animals
We next examined whether mitochondria isolated from animals of the three genotypes have any differences in functional activity. Accordingly, mitochondria were purified from the SC of 52-week-old animals and their purity was confirmed by observation of enrichment by electron microscopy (EM) and by concentration of mitochondrial markers compared to cytosolic proteins (Supplementary Material, Fig. S3A and B). Immunoblots of equal amounts of protein lysate of these purifications revealed significant reduction in levels of several OXPHOS subunits in the isolations from P497S animals, compared to those from either WT356 or non-Tg animals (Fig. 2A and B).
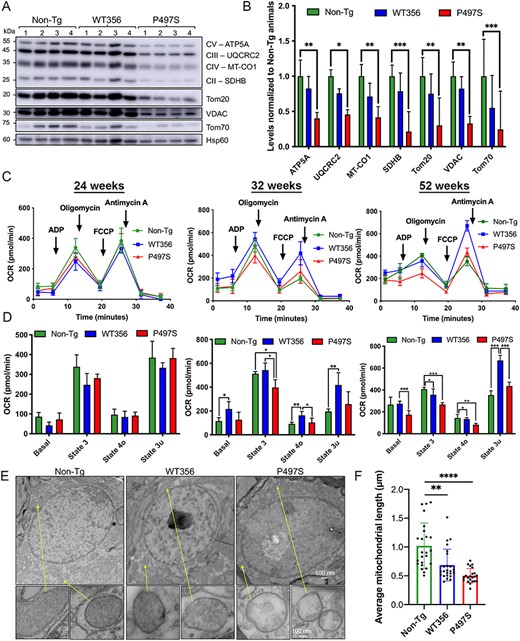
Structural and functional mitochondrial defects in the SC of P497S animals. (A) Immunoblots of equal protein lysates of mitochondria purified from the SC from four 52-week-old animals for the three genotypes probed for the proteins shown. (B) Quantification of the blots shown in A. *P < 0.05, **P < 0.01, ***P < 0.001. (C) Seahorse mitochondria respiration assays of freshly isolated mitochondria from the lumbar SC of mice from the three genotypes at 24, 32 and 52 weeks of age; oxygen consumption rate (OCR) curves are shown. n = 3–5. (D) Bar charts depict basal, State 3 (ADP-stimulated), State 4o (proton leak) and State 3u (maximal) respiration rates from traces shown in C. *P < 0.05, **P < 0.01, ***P < 0.001. (E) TEM images of the cell body regions of MN in the SC of 52-week-old mice for the three genotypes. Also shown are the magnified images of particular mitochondria in the MN (arrows). Scale bars for representative images, 500 and 100 nm for zoomed in mitochondria. (F) Average mitochondria length (μm) in SC motor neurons. **P < 0.01, ****P < 0.0001.
Mitochondria respiratory function was measured using a Seahorse XF24 respirometer (43–45). Freshly isolated mitochondria, with equivalent protein content, from the SC of 24-, 32- and 52-week-old animals were plated into XF24 wells with seven technical replicates for each genotype. The Complex I–linked substrates glutamate and malate were added along with ADP to induce phosphorylating respiration. Oligomycin, FCCP and antimycin A + rotenone were then added sequentially to allow estimation of coupling, maximum respiration and background oxygen consumption, respectively (46). Mitochondria from P497S animals exhibited an age-dependent decrease in mitochondrial respiration, compared with WT356 and non-Tg animals (Fig. 2C and D). Decreased phosphorylating State 3 respiration was detected as early as 32 weeks in the P497S animals. At 52 weeks of age, mitochondria from the P497S animals showed statistically significant decreases in State 4o (oligomycin-sensitive respiration) and basal respiration. An electron flow assay was then conducted to assess the activities of individual OXPHOS complexes by sequentially adding the Complex I inhibitor rotenone, the Complex II substrate succinate, the Complex III inhibitor antimycin A and the Complex IV electron donor TMPD/ascorbate to mitochondria initially incubated with the Complex I–linked substrates pyruvate and malate. Mitochondria from P497S animals showed reduced rates of Complex II– and Complex IV–dependent oxygen consumption (Supplementary Material, Fig. S3C).
Repetition of the functional assays using mitochondria purified from the hippocampus revealed a similar age-dependent manifestation of respiration deficits in P497S animals compared to WT356 and non-Tg animals (Supplementary Material, Fig. S4A and B). The lower yield of isolated mitochondria from the hippocampus limited the scope of these assays. Nevertheless, the results suggest that the mitochondrial respiration deficits extend throughout the central nervous system in P497S animals.
Structural abnormalities of mitochondria in spinal MN of P497S animals
Because MNs are lost in the SC in ALS and in the P497S mouse model, we examined SC sections of 52-week-old animals by EM for mitochondria structural deformities. We also focused on MNs because of their large nucleus (6–10 μm) and easy identification compared to other cell types where the mutant UBQLN2 transgene might not have been expressed. The examination revealed that mitochondria in MNs of P497S animals were noticeably shorter and had many large voids in cristae invaginations compared to non-Tg and WT356 animals (Fig. 2E and F).
UBQLN2 inactivation in cultured cells results in mitochondrial deficits similar to those seen in diseased P497S animals
We next used HeLa and NSC34 cell lines inactivated of UBQLN2 expression, the derivation of which was previously described (13), to determine whether the mitochondria aberrations in P497S animals arise from loss of UBQLN2 function. We used the HeLa UBQLN2 knockout 8 (KO8) line for the majority of our investigations because, unlike the other lines, it did not display compensatory changes in other UBQLN isoforms (13). The two UBQLN2 KO NSC34 lines (KO20 and KO69) were mainly used for confirmatory purposes because of their poor survival in many functional assays.
We first examined whether the levels of OXPHOS subunits and mitochondrial import proteins were changed in the KO8 line. To ensure reproducibility, lysates from four independent cultures were analyzed. Similar to the decrease in OXPHOS proteins found in P497S animals, the KO8 line had significant reduction in the levels of SDHB (Complex II), MT-CO1 and MT-CO2 (Complex IV), and ATP5A (Complex V) compared with the parental WT UBQLN2-expressing HeLa line. In addition, levels of both Tom20 and Tom40 import proteins were reduced in the KO8 line (Fig. 3A and B). A reduction in many mitochondrial proteins was similarly observed in the UBQLN2 KO NSC34 lines (Supplementary Material, Fig. S6A and B).
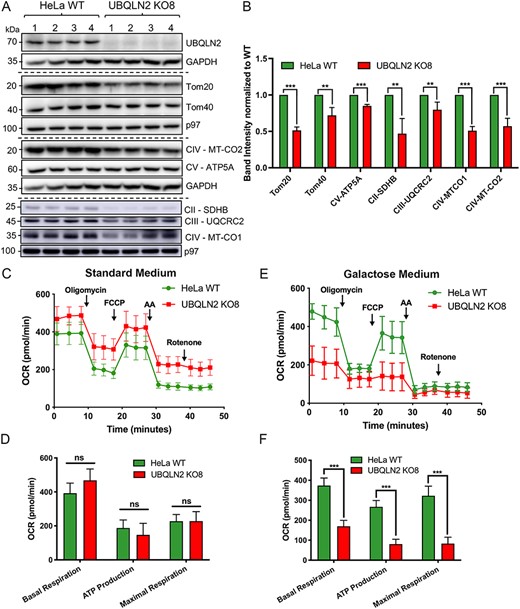
Inactivation of UBQLN2 expression in cells recapitulates the mitochondrial deficits seen in P497S animals. (A) Immunoblots of four independent lysates from parental HeLa WT and CRISPR/cas9 UBQLN2 KO8 lines probed for the indicated mitochondrial import and OXPHOS proteins. (B) Quantification of the blots shown in A. **P < 0.01, ***P < 0.001. (C) OCR curves depicting respiration rates of HeLa WT and UBQLN2 KO8 cell lines grown in standard DMEM media supplemented with glucose. (D) Bar chart of basal, ATP (oligomycin-sensitive) and maximal respiration. n = 4, ns, not significant. (E) OCR curves of HeLa WT and UBQLN2 KO8 lines grown in oxidative DMEM media supplemented with galactose. (F) Bar chart of basal, ATP (oligomycin-sensitive) and maximal respiration. n = 4, ***P < 0.001.
We next examined whether the mitochondrial protein changes in the KO8 line arise from differences in protein stability. To assess this, we conducted cycloheximide (CHX)-chase experiments to monitor the turnover of a select set of proteins (Supplementary Material, Fig. S5). Turnover of MCL-1, a protein that is rapidly degraded by the proteasome, was little changed in the KO8 line, suggesting that the proteasomal degradation pathway is not blocked in the line. Additionally, the rate of turnover of all the nuclear-encoded mitochondrial proteins we analyzed (UQCRC2, ATP5A and Tom20) did not differ substantially between the KO8 and WT HeLa lines. Interestingly, however, the rate of turnover of the mitochondrial-encoded MT-CO1 was faster in KO8 cells, suggesting UBQLN2 knockout may destabilize mitochondrial proteins that are synthesized within the organelle. Based on prior studies (47,48), we investigated the possibility that the instability is linked to a mitochondrial import defect (see below).
KO of UBQLN2 leads to functional defects in mitochondrial respiration
We next examined whether mitochondrial respiration is altered in KO8 cells. A comparison of the mitochondrial respiration states in WT HeLa and KO8 cultures grown in standard glucose-containing DMEM media revealed no significant differences (Fig. 3C and D). However, when cultured in galactose medium to force cells to rely on the OXPHOS system for ATP production (49–51), we saw a robust and statistically significant decrease in basal respiration, oligomycin-sensitive respiration, which is the fraction of respiration that corresponds to ATP production, and maximal respiration in the KO8 line (Fig. 3E and F). These functional deficits are consistent with depletion of key OXPHOS subunits found in the KO8 line. Repeating these assays in the NSC34 cell lines revealed major deficits in mitochondrial basal respiration and ATP production in both UBQLN2 KO lines even when cultured in standard glucose medium (Supplementary Material, Fig. S6C and D), suggesting that this motor neuron–like cell line may rely more on the OXPHOS system than HeLa cells.
We next examined whether the morphology and dynamics of mitochondria fusion and fission are changed in KO8 cells. The mitochondrial network in cells was visualized by Tom20 staining and categorized into three classes based on whether it was predominantly elongated (>80% elongated), had intermediate length (>50% short rod-like mitochondria) or was fragmented (>50% oval mitochondria). The analysis revealed that the vast majority of cells in the WT HeLa line had a predominantly elongated network (~73%), while the remaining contained mitochondria of intermediate length and few, if any, contained fragmented mitochondria (Fig. 4A and B). In contrast, the population of cells with elongated mitochondria in the KO8 line was reduced to 49% and the populations with intermediate and fragmented mitochondria were increased to 34 and 17%, respectively.
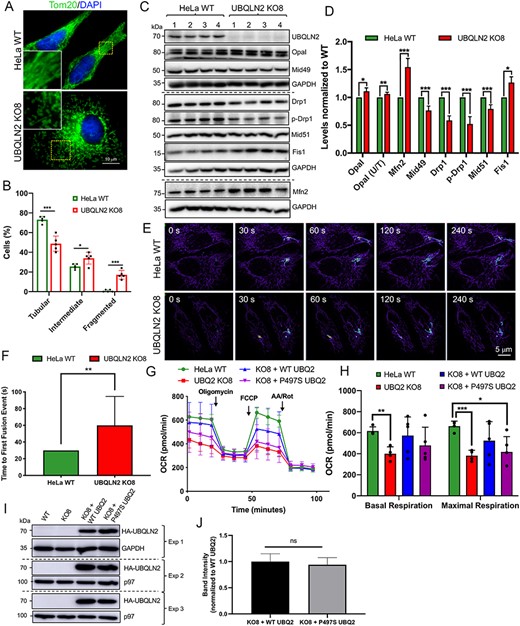
Alterations in mitochondria proteins, structure, function and dynamics in UBQLN2 KO cells and rescue of the functional deficits by reexpression of WT UBQLN2, but not by mutant P497S UBQLN2 proteins. (A) Parental HeLa WT and UBQLN2 KO8 lines stained with Tom20 to decorate mitochondria. Scale bar, 10 μm. (B) Quantification of the mitochondria network morphology of WT and KO8 cells into the percentage with tubular (>80% elongated mitochondria), intermediate (>50% short rod-like mitochondria) or fragmented (>50% fragmented mitochondria) mitochondria. n = 5, 50 cells per group. (C) Immunoblots of four independent lysates from WT and KO8 cultures probed for proteins shown. (D) Quantification of mitochondrial dynamic proteins shown in C. *P < 0.05, **P < 0.01, ***P < 0.001. (E) Time course images showing the extent of dissipation of GFP fluorescence in two focally activated regions in WT and KO8 cells expressing the mito-PAGFP reporter. (F) Quantification of the time of first fusion from the assay shown in E. n = 12, **P < 0.01. (G) OCR curves depicting parental HeLa WT and UBQLN2 KO8 cultures transfected with empty vector control, HA-tagged WT UBQLN2 or HA-tagged P497S UBQLN2 grown in oxidative DMEM media supplemented with galactose. (H) Quantification of basal and maximal respiration are shown for traces in G. n = 5, *P < 0.05, **P < 0.01, ***P < 0.001. (I) Immunoblots to show expression of the HA-tagged UBQLN2 constructs for the three separate experiments shown in G and H. (J) Quantification of HA-tagged WT and P497S mutant UBQLN2 proteins shown in I.
Modulation of the mitochondrial network in cells is governed by changes in the amount and/or posttranslational modification of factors that alter the dynamics of mitochondrial fusion and fission (37). Immunoblot comparison of the HeLa cell lines for proteins that regulate mitochondrial dynamics revealed an increase in the levels of factors that promote mitochondrial fusion in KO8 line (Fig. 4C and D). The change was similar to that seen in P497S animals but was unexpected considering the morphological findings that the mitochondria in the KO8 line were more fragmented. For example, levels of Mfn2 (38,52,53) and the proportion of the active 100 kDa Opa1 band were increased in the KO8 line. Similarly, while Fis1 and MiD49 function together with Drp1 to regulate mitochondrial fission (34,54), Fis1 was increased, while MiD49 was decreased in the KO8 line compared with the parental HeLa line. Furthermore, both, the levels of Drp1 and its phosphorylation (at Ser616), which stimulates mitochondrial fission (55), were reduced in the KO8 line.
We next used live cell imaging to compare the rate of mitochondrial fusion between the KO8 and WT HeLa lines (56). Accordingly, the rate of dissipation of fluorescence of two focally activated regions in WT and KO8 HeLa cells transfected with a photoactivated mito-PAGFP reporter were compared (56,57). Visualization of the mito-PAGFP fluorescence indicated that the dissipation of the fluorescent signal was substantially slower in KO8 cells compared to normal HeLa cells (Fig. 4E). Quantifications further revealed that the time taken for the first mitochondrial fusion event was considerably delayed in the KO8 cells (Fig. 4F). The slower rate of mitochondrial fusion, higher levels of factors that promote fusion but increased fragmentation of mitochondria in the KO8 cells suggest a futile attempt of the cells to restore mitochondria length.
Rescue of mitochondrial respiration deficits by reexpression of WT, but not ALS/FTD P497S mutant UBQLN2
To confirm UBQLN2 requirement for mitochondrial function, we tested whether reexpression of WT or ALS mutant UBQLN2 proteins can rescue the respiration deficits in KO8 cultures. Transfection of KO8 cultures with cDNA encoding HA-tagged WT-UBQLN2 partially restored basal and maximal mitochondrial respiration levels compared with parallel cultures transfected with cDNA encoding the P497S UBQLN2 mutant or with the empty vector (Fig. 4G and H). The inability of the mutant in the rescue of the respiration deficits was not lower expression of the construct, as immunoblots showed the protein was expressed to similar levels as the WT construct (Fig. 4I and J). The results provide strong evidence that the mitochondrial deficits in KO8 cells originate from loss of UBQLN2 expression and that the P497S mutation is a loss-of-function mutation.
Aberrant targeting of TIMM44 to mitochondria in UBQLN2 KO cells
Because mitochondrial respiration and the levels of several mitochondrial proteins were reduced in KO8 cells, we examined the possibility that UBQLN2 is required for mitochondrial protein import. Accordingly, we studied TIMM44, an essential nuclear-encoded mitochondrial protein (42,58). We selected TIMM44 for several reasons. First, proteomic analysis indicated its levels were reduced in the SC of P497S animals (Supplementary Material, Fig. S1B). Second, TIMM44 was recovered in a screen of UBQLN2 interactors (59). Third, mitochondrial targeting of the protein can easily be scored by virtue of a 5 kDa reduction in its size due to proteolytic cleavage of its N-terminal leader sequence upon mitochondrial import (58,60).
We first examined whether levels of TIMM44 were changed in P497S animals and in the HeLa KO8 cell line. Immunoblot analysis of SC lysates of mice at 32 weeks of age revealed TIMM44 levels were indeed significantly reduced in P497S animals compared to non-Tg and WT356 animals (Fig. 5A). Further quantification of TIMM44 levels in the SC of 14 independent 24-week-old non-Tg and P497S animals revealed an ~50% reduction in P497S animals (Supplementary Material, Fig. S7A and B). Likewise, immunoblots revealed TIMM44 levels were significantly reduced in KO8 cells (Fig. 5C and D).
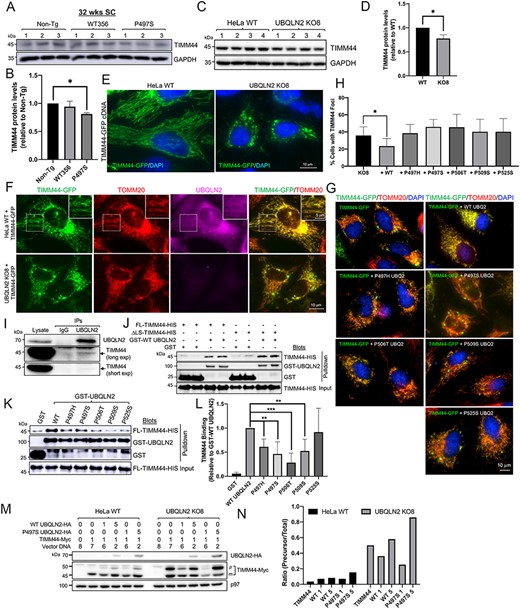
Alteration of TIMM44 protein levels in P497S animals and differences in mitochondrial targeting of the protein in cells containing and lacking UBQLN2. (A) Immunoblots of SC lysates from three independent 32-week-old animals for the three mouse genotypes probed for TIMM44 and GAPDH. (B) Quantification of TIMM44 levels in the blots shown in A after normalization of protein loading. *P < 0.05. (C) Immunoblot of four different HeLa WT and UBQLN2 KO8 cultures probed for TIMM44 and GAPDH. (D) Same as B, but for the blots shown in C. *P < 0.05. (E) Representative images of TIMM44-GFP fluorescence and of DAPI staining in HeLa WT and UBQLN2 KO8 cells. Scale bar, 10 μm. (F) Different color fluorescence images of cells stained for the proteins shown to monitor targeting of TIMM44-GFP to mitochondria in HeLa WT (1st row) and UBQLN2 KO8 cells (2nd row). Magnified images of boxed area in the 1st row are shown on the upper right-hand side. Scale bar, 10 and 5 μm. (G) Merged images of KO8 cells showing extent of colocalization of GFP fluorescence of the TIMM44-GFP reporter with TOMM20 staining in cells cotransfected with empty vector, or with WT UBQLN2, or with five different ALS/FTD mutant UBQLN2 cDNAS. Scale bar, 10 μm. (H) Quantification of cells with TIMM44 foci for experiment presented in G. n = 150–300 cells, *P < 0.05. (I) Immunoprecipitation of endogenous proteins from HeLa cells with either IgG control antibody or anti-UBQLN2 antibody and immunoblotted for either TIMM44 (short and long exposures) or UBQLN2. (J) Pulldown assays showing direct binding between both FL and the ΔLS TIMM44-HIS recombinant proteins with GST-UBQLN2 fusion protein. (K) Same as J, but comparing binding of FL TIMM44-HIS protein with WT and ALS/FTD mutant GST-fusion proteins. (L) Quantification of the amount of TIMM44 pulldown relative to the amount of GST-fusion protein recovered and normalized to the binding of WT GST-UBQLN2 protein. n = 4, **P < 0.01. ***P < 0.001. (M) Demonstration that UBQLN2 promotes maturation of TIMM44. HeLa and UBQLN2 KO8 cultures were transfected with cDNAs (μg) as shown (equivalent amount of total cDNA). Lysates were immunoblotted and probed with anti-Myc and anti-HA antibodies. (N) Quantification of precursor TIMM44 (50 kDa) over total TIMM44 levels in blot presented in M.
We next examined whether TIMM44 targeting to mitochondria differs between WT and KO8 cells. Targeting was analyzed by determining whether GFP fluorescence of a TIMM44-GFP reporter construct colocalized with Tom20 staining, a known mitochondrial marker. Microscope analysis revealed almost complete colocalization of GFP fluorescence with Tom20 staining in HeLa WT cells (Fig. 5F). Careful examination of the magnified images revealed hints of UBQLN2 staining with mitochondria (Fig. 5F, insert). In contrast, in the KO8 cells, GFP fluorescence in mitochondria decorated by Tom20 staining was weaker and instead was concentrated in multiple foci dispersed in the cytosol, which by confocal microscopy and line scan analysis were negative for Tom20 and ubiquitin staining (Fig. 5F and Supplementary Material, Fig. S8A and C). Approximately 40% of KO8 cells transfected with the TIMM44 reporter contained these abnormal GFP foci compared to 2% in normal HeLa cells. The lack of ubiquitin staining of the GFP foci was puzzling, since we predicted the non-targeted TIMM44 protein had misfolded and aggregated in the cytoplasm. To examine this paradox, we examined staining of endogenous TIMM44 and ubiquitin in HeLa WT and KO8 cells. The confocal staining revealed KO8 contained many more TIMM44 puncta, which were ubiquitin positive, based on line scan overlap of the peaks of their fluorescence. This observation was unlike in the HeLa WT cells where fewer TIMM44 puncta were observed, and the puncta were also negative for ubiquitin staining (Supplementary Material, Fig. S8B, D and E). Taken together, the results suggest that loss of UBQLN2 compromises proper targeting of TIMM44 to mitochondria, which results in the accumulation and possible aggregation of the protein in the cytosol.
Rescue of TIMM44-GFP targeting to mitochondria by reexpression of WT but not ALS/FTD mutant UBQLN2 proteins
We next examined whether proper targeting of TIMM44-GFP to mitochondria in KO8 cells can be restored by reexpression of either WT UBQLN2 or ALS/FTD mutant UBQLN2 proteins. Microscope examination revealed that significantly fewer KO8 cells cotransfected with cDNA encoding WT-UBQLN2 contained abnormal GFP fluorescent foci compared with cells transfected with the GFP-TIMM44 reporter alone (Fig. 5G and H and Supplementary Material, Fig. S9). Importantly, the number of cells containing abnormal foci was not altered by cotransfection with any of the five different ALS/FTD mutant UBQLN2 cDNA we tested, suggesting that the ALS/FTD mutations compromise UBQLN2 targeting of TIMM44 to mitochondria (Fig. 5G and H and Supplementary Material, Fig. S9). Interestingly, the GFP foci in the P497S-transfected cells were devoid of UBQLN2 staining.
UBQLN2 mutants bind weaker with TIMM44
We considered two main possibilities for why TIMM44 is not properly targeted to mitochondria in KO8 cells. First, UBQLN2 may be required to regulate TIMM44 turnover and, in its absence, the protein misaccumulates. Second, UBQLN2 may be required to chaperone TIMM44 to the mitochondrial membrane for insertion.
To address the first possibility, we compared the turnover of TIMM44 in normal and KO8 HeLa cells by cycloheximide-chase experiments. The results revealed little difference in the turnover of TIMM44 in the WT and KO8 HeLa lines, providing lack of support for the first possibility (Supplementary Material, Fig. S7C and D).
To evaluate the second possibility, we conducted immunoprecipitation and GST pulldown assays examining whether UBQLN2 and TIMM44 bind one another. Immunoprecipitation of UBQLN2 from HeLa cells resulted in coprecipitation of a small amount of TIMM44, indicating the two endogenous proteins bind together in a complex (Fig. 5I). To examine if ALS mutations in UBQLN2 alter binding with TIMM44, we conducted additional immunoprecipitations of HeLa cell cultures cotransfected with expression constructs either encoding HA-tagged WT UBQLN2 or carrying the P497S mutation with C-terminus GFP-tagged TIMM44. Although TIMM44-GFP was coimmunoprecipitated by both the WT and the P497S UBQLN2 proteins, slightly less TIMM44-GFP was coimmunoprecipitated by the P497S mutant, suggesting weakened interaction between the proteins (Supplementary Material, Fig. S7E).
We next examined whether UBQLN2 and TIMM44 bind directly with one another by conducting GST pulldown assays to analyze binding between recombinant purified GST-WT UBQLN2 and His-tagged TIMM44 proteins.
Interestingly, although full-length (FL) human TIMM44 was expressed as a C-terminus His-tag protein in Escherichia coli, the largest His-purified protein ran anomalously on gels, highly suggestive of cleavage of the TIMM44 leader sequence. To confirm this, we deleted the leader sequence (ΔLS-TIMM44-His) (61) and found the shorter expressed protein was similar in size to the FL-purified product, consistent with our prediction. We used both TIMM44 proteins for pulldown assays (Fig. 5J). The assays revealed equivalent and 25 times stronger binding of both TIMM44-His ligands with GST-WT UBQLN2 than with GST alone (Supplementary Material, Fig. S7F). We repeated the assays analyzing if the TIMM44 leader sequence alone binds UBQLN2 but found little specificity of its binding (Supplementary Material, Fig. S7G).
We next analyzed whether GST-UBQLN2 proteins containing one of five different ALS/FTD mutations (P497H, P497S, P506T, P509S and P525S) differ in their binding with TIMM44. We ensured that the quality of the GST-purified proteins was similar for the comparison (Supplementary Material, Fig. S7H). The pulldown assays revealed a general trend of weaker binding for all the mutants tested, with four of them (P497H, P497S, P506T and P509S) displaying significantly weaker binding than WT UBQLN2 (Fig. 5K and L), suggesting that loss of the binding may compromise TIMM44 targeting to mitochondria. Interestingly, the P525S mutant, which was the only mutant that did not show significantly weaker binding with TIMM44, is the least aggressive of the five ALS/FTD UBQLN2 mutants we tested, based on the age of clinical onset of ALS in carriers of the mutations (1,2).
Facilitation of TIMM44 maturation in KO8 cells by WT but not ALS mutant UBQLN2 proteins
Because the TIMM44 leader sequence is cleaved subsequent to its import into mitochondria, we analyzed the amount of the precursor that remains in HeLa cells lacking or expressing WT and mutant UBQLN2 proteins. Accordingly, normal HeLa or UBQLN2 KO8 cell cultures were cotransfected with a constant amount of Myc-tagged TIMM44 cDNA with increasing amounts of either HA-tagged WT or P497S mutant UBQLN2 cDNAs (1 or 5 μg). Lysates from the cultures, 24 h posttransfection, were then immunoblotted for TIMM44-Myc and UBQLN2-HA (Fig. 5M). In WT HeLa cells, most of the TIMM44 protein detected was the mature form (44 kDa), with trace evidence of the precursor (49 kDa form). The ratio of these forms did not change significantly upon overexpression of either WT or P497S mutant UBQLN2 cDNAs in WT HeLa cells, although expression of higher amounts of the mutant slightly increased precursor accumulation. In contrast, the proportion of immature TIMM44 protein in non-UBQLN2–transfected KO8 cells was higher compared with WT HeLa cells, strongly suggesting UBQLN2 is required for import of TIMM44 into mitochondria. Overexpression of WT UBQLN2 in KO8 cells increased TIMM44 import, based on the reduction in the amount of TIMM44 precursor in the transfected cells. Importantly, overexpression of P497S mutant UBQLN2, particularly higher amounts of its cDNA, led to a dramatic rise in accumulation of the TIMM44 precursor (Fig. 5N). Curiously, expression of lower amounts of the mutant decreased both the mature and immature TIMM44 forms, for unknown reasons. Nevertheless, when combined with the GFP-targeting results, these results strongly suggest UBQLN2 is required for the efficient targeting and import of TIMM44 into mitochondria.
To rule out the alternative possibility that TIMM44 is sequestered by mutant P497S UBQLN2 protein in aggregates preventing its delivery to mitochondria, we stained brain and SC sections of P497S animals to determine whether UBQLN2 and TIMM44 colocalize with one another. Confocal microscopy revealed absence of colocalization of TIMM44 with the majority of the UBQLN2 inclusions in either the brain or SC of the mutant animals, consistent with lack of sequestration by the mutant UBQLN2 protein (Supplementary Material, Fig. S10A and B). Interestingly, however, we observed good colocalization of TIMM44 with UBQLN2 in inclusions within MN of the SC in P497S animals (Supplementary Material, Fig. S10B). To confirm this finding, we stained an SC section from 32-week-old P506T UBQLN2 mutant animal, from mice that we had previously analyzed (31), and found TIMM44 staining was also colocalized with UBQLN2 in MNs in the line (Supplementary Material, Fig. S10B). Although we do not know the identity of the puncta, we speculate that they could either be defective autophagosomes or aggregates containing the misfolded TIMM44 and UBQLN2 proteins. If they are aggregates, it is curious why TIMM44 is absent from the inclusions outside the MNs. To ensure the mistargeting of mitochondrial proteins is not unique to TIMM44, we conducted additional staining examining localization of TIMM50 and found similar colocalization of the protein in inclusions in MNs (Supplementary Material, Fig. S11), suggesting a common pattern of mistargeting of mitochondrial proteins in UBQLN2 mutant animals.
Discussion
Mitochondrial dysfunction has been implicated in the pathogenesis of many neurodegenerative diseases, including ALS, Parkinson’s and Alzheimer’s diseases (62–64). Here, we have provided compelling evidence through studies of the P497S mouse model of ALS/FTD and rescue experiments of UBQLN2 KO cells that the ALS/FTD-associated UBQLN2 P497S mutation is involved in mitochondrial dysfunction, likely through a loss-of-function mechanism. Multiple lines of evidence, obtained through biochemical, structural, immunological, pathologic and functional examination all suggest mitochondrial abnormalities are intimately tied to pathogenesis in the P497S mutant animals.
We found clear evidence that mitochondrial health is severely compromised in SC tissue of P497S animals. First, proteomic and biochemical analysis revealed that P497S animals have reduced levels of many key OXPHOS subunits required for energy production compared to non-Tg animals. Second, mitochondria isolated from P497S animals displayed age-dependent deficits in ATP production and respiration. Third, mitochondria in spinal MNs of P497S had gross distortions and large voids of mitochondria cristae. Fourth, SC lysates of P497S animals had major changes in the abundance and properties of factors that regulate mitochondrial dynamics, with a tilt toward those that favor fusion. Fifth, contrary to the increase in fusion machinery, P497S animals had reduced mitochondria length. Sixth, P497S animals had diminished expression of mitochondrial import factors. Collectively, these findings all point to mitochondrial dysfunction as being intimately linked to disease pathogenesis in P497S animals. We cannot currently determine if the mitochondrial dysfunction is the primary driver of disease, although the major alteration of mitochondrial properties found in UBQLN2 knockout cells provides strong evidence that this may be true.
Inactivation of UBQLN2 expression in HeLa and NSC34 cells led to mitochondrial alterations very similar to those found in late-stage P497S mice. KO of UBQLN2 in HeLa cells reduced levels of OXPHOS complex subunits, decreased respiration used for ATP generation, decreased mitochondrial length, reduced the rate of mitochondria fusion and increased the ratio of proteins promoting mitochondrial fusion. The similarity between the mitochondrial defects seen in UBQLN2 KO cells to those observed in aged P497S mice suggests that the defects seen in the animals arise from loss of UBQLN2 function.
The alterations in mitochondrial properties found in the WT356 line provide additional support linking mitochondrial health to UBQLN2 expression. In the WT356 line, levels of mitochondrial proteins and respiration were slightly increased or enhanced compared to non-Tg animals, which are reciprocal to the changes seen in P497S animals. These results suggest that increased WT UBQLN2 expression may provide some benefit to mitochondria during aging. However, because high level of UBQLN2 overexpression in mice is toxic (65), there is probably a limit to which overexpression is beneficial.
Disturbances in mitochondrial protein quality control have been observed following inactivation of various UBQLN isoforms. Itakura et al. (26) showed that simultaneous KO of UBQLN 1, 2 and 4 in HEK293 cells results in aberrant accumulation of mitochondrial proteins in the cytosol (26). Similar misaccumulation was seen upon ablation of UBQLN1 in cells (27). However, changes in mitochondrial function were not identified. Here, we have shown that inactivation of UBQLN2 expression, by itself, leads not only to changes in levels of proteins critical for mitochondrial health but also to functional impairment of mitochondrial respiration. The functional impairments were mainly found in cells cultured in galactose medium, which forces energy reliance on the OXPHOS system. It is not surprising that this respiration deficit was not always observed in cells cultured in glucose-containing medium owing to OXPHOS inhibition by the Crabtree effect (66). However, in animals, the mitochondrial deficiency could have major consequences because neurons in vivo are heavily reliant on energy production by the OXPHOS system (67).
An important functional difference found between WT UBQLN2 and the P497S mutant protein was the capability of the former, but not the latter, to rescue the respiration defect seen in KO8 cells. The lack of rescue by the mutant may underlie the mitochondrial defect that develops in P497S animals. The question is how does it arise, considering that the animals also express endogenous WT mouse UBQLN2. A possible mechanism is that, over time, the P497S mutant protein progressively inactivates the WT UBQLN2 protein by coaggregating with it. A similar dominant-negative mechanism of inactivation was proposed to underlie the autophagy defect in P497S animals (31).
UBQLN2 KO mice develop mild motor impairments but not neuron disease (33), suggesting ALS/FTD mutations in UBQLN2 are unlikely to cause ALS through a simple loss-of-function mechanism and likely involve additional gain-of-function mechanisms (13). The mitochondrial defects found in the P497S animals were more pronounced in the SC than in the hippocampus. The reason for this may stem from differential vulnerability of mitochondria to the UBQLN2 mutation between neurons of the two tissue types and/or because of differences in the efficiency of mitochondria purification between the tissues. The first possibility fits with the predominant loss of MN cells seen in the SC in ALS. Both the shape and physiology of MN are thought to contribute to their selective demise. MNs are not only very large and have extremely long processes, but they also have unusually high energy demands. These features make them heavily reliant on healthy mitochondria for their survival. Thus, the increase of mitochondrial defects seen in the SC may simply reflect differences in mitochondria fatigue or oxidative damage intrinsic to the tissue. Another possibility is that different cell types exhibit differential susceptibility to the UBQLN2 mutation due to intrinsic differences in gene expression, including different UBQLN2 isoforms. We also cannot rule out the possibility that mitochondria in the hippocampus of P497S animals are also functionally impaired but were too damaged to be purified for the assays. Nevertheless, considering the mitochondria used in our assays were purified from tissues containing a mixture of cell types, not just neurons to which transgenic expression of the P497S mutant was restricted, it is likely that the deficits observed in mitochondria from our preparations are an underestimate of their true dysfunction in vivo.
Mitochondrial function is known to be closely tied to changes in mitochondrial morphology and dynamics (37,68). Further proof that mitochondria in the SC of P497S animals were damaged was evident by the large voids and the highly distorted cristae in mitochondria within spinal MNs of the animals. Such morphological aberrations were not evident in the WT356 and non-Tg animals. The sparsity and distortions of the cristae are consistent with the functional reductions in respiration activity and loss of OXPHOS protein subunits measured in purified mitochondria and tissue lysates. The underlying mechanisms driving these changes are unclear but may be tied to the alterations in the mitochondrial fusion and fission machinery seen in the animals. In contrast to the smaller size of mitochondria seen in the MNs, P497S SC lysates contained an elevation in proteins that promote mitochondrial fusion and a reduction in those that promote fission. A similar phenomenon was seen in the UBQLN2 KO cells, where mitochondria were more fragmented yet had an elevation of factors that promote fusion. Insight into this paradox was obtained by live cell imaging of changes in mitochondrial dynamics, which revealed that UBQLN2 KO cells have significantly slower rates of mitochondrial fusion. Interestingly, similar studies of mitochondrial dynamics in cells expressing mutant proteins involved in different neurodegenerative diseases, such as Huntington’s, Parkinson’s, Alzheimer’s and ALS, have linked a more fragmented mitochondrial network to reduced mitochondrial fusion (69–73). Based on these findings, we speculate that the elevation of fusion factors in the P497S animals and UBQLN2 KO cells may reflect a futile attempt made by cells to restore proper mitochondrial length.
The mitochondrial import defect found of TIMM44 in UBQLN2 KO cells and rescue of the defect by overexpression of WT UBQLN2 but not by the P497S mutant provide important clues into the potential mechanism by which loss of UBQLN2 expression and ALS mutations in UBQLN2 compromise mitochondrial health. TIMM44 is an essential nuclear-encoded protein that functions in mitochondrial protein import (42,58). The mature TIMM44 protein resides in the inner mitochondrial membrane, where it regulates protein import by bridging interaction of the transit polypeptide with the molecular motor involved in protein translocation (74–77). Studies have shown that TIMM44 depletion, or antibody neutralization of its activity, impedes protein import (74,77,78). Based on these observations, we speculate that reduction of TIMM44 levels in UBQLN2 KO cells and in mutant P497S animals compromises mitochondrial protein import, including its own. Consistent with this idea, KO8 cells transfected with TIMM44-Myc cDNA accumulated more TIMM44 precursor than normal HeLa cells. Moreover, more GFP-tagged TIMM44 protein accumulated in foci in the cytosol of UBQLN2 KO8 cells compared to normal HeLa cells expressing UBQLN2. A puzzling property of the GFP foci was absence of ubiquitin staining, which would be expected for proteins that are misfolded and aggregated. In contrast, endogenous TIMM44 protein accumulated in ubiquitin-positive puncta in KO8 cells, providing support for the idea that lack of its import into mitochondria results in its aggregation in the cytosol. It will be interesting in the future to determine the reason for this paradox. A key question that arises from these observations is how loss of UBQLN2 compromises TIMM44 mitochondrial import. Although we do not know the answer to this question, we speculate it may involve loss of UBQLN2 chaperone and/or regulation of TIMM44 degradation. Turnover studies indicated little difference in TIMM44 turnover in KO8 and normal HeLa cells, providing lack of support for the latter idea. However, because the half-life of TIMM44 is very long, small changes in protein turnover could have been missed. Instead, we favor the chaperone idea because of the strong binding we observed between recombinant WT UBQLN2 and TIMM44 proteins. Interestingly, all five ALS/FTD mutant UBQLN2 proteins we tested bound weaker with TIMM44 than WT UBQLN2, suggesting a potential molecular explanation for why TIMM44 targeting to mitochondria is compromised. We speculate that UBQLN2 binding may be required to either chaperone or prevent TIMM44 from misfolding prior to its insertion into mitochondria. Another possibility is that import is blocked because of accumulation of misfolded proteins at the pore, which are normally cleared by UBQLN2.
It is possible that the decline in mitochondrial function in mutant P497S animals stems directly from the defect in mitochondrial protein import. Mitochondria rely on import of over 1000 nuclear-encoded proteins for their health and function. Therefore, the protein import defect is predicted to have dire consequences for mitochondria function. Indeed, defects in mitochondrial protein import were recently shown to cause mitochondrial stress, which triggers the mitochondria-compromised protein import response (mito-CPR), a protective signaling pathway to alleviate the stress (79). It will be interesting in the future to determine if this signaling pathway is at all altered, or compromised by UBQLN2 mutations. It is also conceivable that the defect in TIMM44 import arises for some other reason, for example a general decline in mitochondria health. The decline in mitochondria health may arise from accumulation of damaged mitochondria due to failure to be cleared by mitophagy, a possibility we entertain because UBQLN2 mutations impede autophagy (13). Interestingly, the block in autophagy was linked to autophagosome acidification defect, which in turn has been linked to mitochondrial activity, suggesting the intriguing possibility that the two defects may be connected (80).
Finally, our findings, showing mitochondrial dysfunction is linked to loss of UBQLN2 expression in cells and to expression of the P497S ALS/FTD mutant UBQLN2 protein in mice, highlight the importance of factors that regulate proteostasis, like UBQLN2, play in maintaining healthy organelle function and how the loss of this function could ultimately lead to devastating diseases, like ALS and FTD.
Materials and Methods
Antibodies and reagents
The following primary antibodies were used: anti-OXPHOS antibody cocktail (Abcam, ab110413; contains antibodies against ATP5A, UQCRC2, COX1, SDHB and NDUFB8), NDUFA9 (Abcam, ab14713), SDHA (ProteinTech, 14865-1-AP), COX2 (ProteinTech, 55070-1-AP), COX4 (ProteinTech, 11242-1-AP), CMC1 (ProteinTech, 11448-1-AP), TOMM20 (LSBio, LS-C417027; ProteinTech, 11802-1-AP), TOMM40 (SantaCruz, sc-365466 and sc-365467), TOMM70 (Santa Cruz, sc-390545),Opa1 (LSBio, LS-C143234), Mfn1 (Abcam, ab75602), Mfn2 (ProteinTech, 12186-1-AP), Drp1 (Cell Signaling, 8570S), p-Drp1 (Cell Signaling, 4867S and 4494S), Fis1 (ProteinTech, 10956-1-AP), MiD49 (ProteinTech, 16413-1-AP), MiD51 (ProteinTech, 20164-1-AP), TIMM44 (ProteinTech, 13859-1-AP), TIMM50 (Abcam, ab204486), MCL1 (SantaCruz, sc-12756), UBQLN2 (NovusBiologicals, NBP2-25164), GFP (eBioscience, 14-6674-82 and UM281, made in-house), His (Invitrogen, MA1-135), HA (Sigma-Aldrich, H3663 and UM80, made in-house), myc (UMY81, made in-house), GAPDH (ProteinTech, 60004-1-Ig), β-actin (SantaCruz, sc-1616), p97/VCP (UMY475, made in-house) and ChAT (#AB144P, IF, Millipore-Sigma, Burlington, MA). The following secondary antibodies were used: goat antirabbit IgG secondary antibody, HRP (ThermoScientific, 31460) and goat antimouse IgG (H + L) secondary antibody, HRP (ThermoScientific, 31460), donkey antirabbit IgG Alexa Fluor 488 (Abcam, ab150073), donkey antimouse IgG Alexa Fluor 594 (Abcam, ab150108) and donkey antigoat IgG Alexa Fluor 647 (Abcam, ab150131).
Tissue culture and transfections
HeLa (ATCC CCL-2) and NSC34 (Cedarlane CLU140) cultures were grown and maintained in DMEM media supplemented with Pen-Strep, L-glutamine and 10% FBS. For cells cultured in oxidative media, cells were grown in DMEM media without glucose (ThermoScientific™, A1443001) and supplemented with 5 mm galactose, 1 mm sodium pyruvate and 10% FBS (24). The generation of the HeLa and NSC34 UBQLN2 KO lines used in the study was described previously (13). They were cultured similar to their parental lines. Transient transfection experiments were conducted with Lipofectamine 3000 (ThermoFisher) or through standard CaPO4 transfection (81).
UBQLN2 transgenic mouse lines
Three different congenic C57BL/6 mouse lines, a non-Tg line, a Tg P497S line with Thy1.2 neuron-specific expression of FL human UBQLN2 carrying the ALS/FTD P497S mutation and a WT356 line with Thy1.2 expression of equivalent amounts of WT UBQLN2 were used for all the studies. Detailed characterization of the behavior and pathology in the three lines has been described (31,32). The lines were backcrossed with C57BL/6 mice for at least 10 generations prior to use for the current experiments.
RNAseq
Translational RNAseq profiling of genes in MN cells in P497S and non-Tg mice were performed using BacTRAP technology of GFP-immunoprecipitated complexes extracted from SC tissue homogenates of 8-week-old double Tg mice also carrying the Chat-BacTRAP-Rpl110a transgene was conducted as described previously (82,83). The Log2 fold ratio change in mRNA expression (averaged for three different animals) of candidate mitochondrial proteins found between P497S versus non-Tg is shown.
Mitochondrial isolations
Tissues were isolated from UBQLN2 transgenic mice and homogenized at 200 rpm for ~5 strokes and then at 400 rpm for ~3 strokes in mitochondrial isolation buffer (220 mm mannitol, 70 mm sucrose, 2 mm HEPES, 1 mm EGTA). Crude nuclear pellet was removed through centrifugation at 1330g for 10 min at 4°C. Supernatant was centrifuged at 21 200g for 10 min at 4°C for isolation of a crude mitochondrial pellet. An Optiprep™ layered gradient was constructed with densities of 1.079 and 1.175 g/ml solutions. The crude mitochondrial pellet was resuspended in a 5:1 solution of Optiprep (100%) to isolation buffer and layered under the 1.175 g/ml solution layer. The gradient was centrifuged at 30 700g for 1 h at 4°C. The mitochondria-containing band at the interface of the 1.079 and 1.175 g/ml layers was extracted and resuspended in mitochondrial assay solution (MAS; 70 mm sucrose, 220 mm mannitol, 10 mm KH2PO4, 5 mm MgCl2, 2 mm HEPES, 1 mm EGTA, 0.2% BSA). Suspensions were repelleted through centrifugation at 12 500g for 5 min at 4°C. The pellet was washed with 1 ml of MAS and repelleted by centrifugation at 12 500g for 5 min (three times). Final pellet was then resuspended in 200 μl of MAS.
Transmission electron microscopy
Ultrathin sections of ~70 nm of perfused SC and hippocampal tissues were mounted onto grids, and images were captured with an FEI Tecnai T12 high-resolution transmission electron microscope (UMB Core Facility, Baltimore MD). Images were acquired with an AMT-XR611 digital camera and AMTV600 software (Advanced Microscopy Techniques).
Seahorse respiration assays + complex activity
Seahorse XF24 V7 plates (Agilent Technologies) were coated with polyethylenimine for 1 h and washed with 95% ethanol three times and then air-dried. The plates were used for performing Seahorse assays with isolated mitochondria. For cells, polyethylenimine coating was unnecessary. For isolated mitochondria, 10 μg of mitochondrial protein suspension, quantified by Bicinchoninic acid assay (BCA) (Thermo Scientific™, Waltham, MA), was plated in XF24 wells in a volume of 50 μl. Plating was randomized to avoid plating effects and each sample was plated as 5–7 replicates. Plates were spun at 2300g for 20 min and, after centrifugation, 450 μl of Seahorse assay media, supplemented with 10 mm pyruvate and 2 mm malate, was carefully added to each well as not to disturb the homogenous mitochondrial layer. During centrifugation, the XF24 Seahorse cartridge was switched to XF calibrant media after overnight equilibration in distilled water. The Seahorse cassette was loaded with ADP, oligomycin, FCCP and antimycin A/rotenone with optimal concentrations for ADP and FCCP predetermined by titration.
For HeLa cells, 60 000 cells/100 μl were found to be the optimal seeding density by titration, while for NSC-34 cells, 80 000 cells/100 μl achieved an optimal OCR signal. Cells were plated into the XF24 wells and, after observing that the cells were attached, an additional 150 μl of media was added to each well 4–5 h later. A separate small aliquot of cells was plated into six dishes and were used for normalization after the Seahorse assay to compensate for differences in growth rates. Cells were grown overnight in the Seahorse plate, and Seahorse XF24 cartridges were equilibrated in water overnight. On the subsequent day, cells were washed with Seahorse assay media and equilibrated in media for 1 h prior to running the assay. Cell density, morphology and health were monitored under a light microscope. At the same time, Seahorse cartridges were switched to XF calibrant solution and cassettes were loaded with oligomycin, FCCP and antimycin/rotenone. Optimal concentrations of drugs were titrated prior to experiments. Optimal final concentrations for oligomycin and antimycin A/rotenone were found to be 1 μm for both HeLa and NSC34 cells, while FCCP concentration for HeLa cells and NSC-34 cells was 2 and 1 μm, respectively. Depending on group allocations, each cell line was analyzed as 5–7 replicates.
Immunofluorescence
Immunofluorescence was performed as previously described (31). HeLa WT and UBQLN2 KO cells were stained with primary antibodies against proteins shown and detected with complementary antimouse conjugated Alexa Fluor 488, 594 and/or 647 secondary antibodies. SC and brain tissues were dissected and stored at −80°C prior to embedding in O.T.C. compound (Sakura Finetek USA, Inc., Torrance, CA) and cut with a cryostat (Leica Biosystems, Buffalo Grove, IL). SC and brain sections were stained with primary antibodies against UBQLN2, TIMM44 and Tom20 and detected with complementary antirabbit Alexa Fluor 488 and antimouse Alexa Fluor 594 secondary antibodies. Cells and tissues were visualized with 40× Apo objective lens using a Nikon Eclipse Ti A1R confocal microscope. Images were processed with iVision software (BioVision Technologies, PA). Line scans to evaluate colocalization within regions of interest were performed using ImageJ software. Lines of interest were drawn to include at least two representative puncta. The plot profile function was used to analyze the pixel intensity across a defined line of interest across each color channel. Intensity profiles were overlaid to analyze the degree of immunofluorescence signal colocalization between proteins.
SDS-PAGE and immunoblotting
Protein lysates from cells or tissues were prepared by homogenization in protein lysis buffer (50 mm Tris pH 6.8, 150 mm NaCl, 20 mm EDTA, 1 mm EGTA, 0.5% SDS, 0.5% NP40, sarkosyl 0.5%, 1 mm Prefabloc [AEBSF, Roche, Indianapolis, IN], 10 mm orthovanadate, 2.5 mm NaF) (31). Protein concentration of lysates was measured by Bicinchoninic acid assay (BCA) (Thermo Scientific™, Waltham, MA). Samples with equivalent protein levels were mixed with SDS sample buffer and separated by SDS-PAGE. Proteins were transferred to 0.45 μm PVDF membranes (Millipore, Billeria, MA) for 20 min using Invitrogen Power Blotter semidry transfer system (Invitrogen, ThermoScientific™, Waltham, MA). Membranes were probed with primary antibodies and appropriate complementary secondary horseradish peroxidase-conjugated antibodies. The chemiluminescence signal produced after incubation with the Supersignal West Pico reagent (Thermo Fisher Scientific, Rockford, IL) was captured using a Fluoro-Chem M Imager (Protein Simple, Santa Clara, CA). Densitometric quantification of protein levels was performed using Alpha View software (Protein Simple, San Jose, CA).
Cycloheximide chase
Cycloheximide (CHX)-chase experiments were performed by adding CHX to a final concentration of 100 μm to cell cultures grown in standard DMEM media, followed by incubation for different lengths of time at 37°C in 5% CO2. The cultures were then removed at appropriate intervals and washed three times with 1× PBS buffer and the cells were lysed using our standard lysis buffer. Lysates were sonicated, the protein concentrations were determined by the BCA and equal amounts of the proteins were separated by SDS-PAGE and immunoblotted for different proteins.
Bacterial expression and purification of HIS and GST-tagged recombinant proteins
The same procedures that were described previously for bacterial expression and purification of recombinant HIS and GST-fusion proteins were used (9,13). The GST-UBQLN2 fusion proteins used in this study all had GST-fused in-frame at the N-terminus of the complete human UBQLN2 open reading frame (ORF). A description of the constructs encoding WT UBQLN2 and each of the five different familial ALS/FTD mutations (P497H, P497S, P506T, P509S and P525S) identified by Deng et al. (1) were described previously (9). The ligands for the GST-pulldown assays were all C-terminally HIS-tagged proteins expressed using a pET21a expression vector. The entire FL ORF of human TIMM44 (FL-TIMM44-HIS), as well as the protein deleted of the mitochondrial leader sequence (ΔLS-TIMM44-HIS), and the leader sequence fused to GFP (LS(TIMM44)-GFP-HIS) were expressed in Rosetta(DE3) E. coli and purified using Ni-NTA agarose according to the procedure described by the manufacturer (Qiagen, Germantown, MD). All of the purified GST and HIS-tagged recombinant proteins were dialyzed against a 20 mm Tris pH 8.0, 150 mm NaCl and 10% glycerol solution and frozen at −80°C until use.
GST pulldown assays were performed by incubating 10 μg of the GST-UBQLN2 fusion proteins, or 4.5 μg of GST alone, with 12 μg of particular TIMM44-HIS tagged or GFP-HIS tagged proteins in 1 ml binding buffer (20 mm Tris pH 8.0, 150 mm NaCl, 10% glycerol, 0.1% NP-40) at room temperature for 2 h. The concentration of NP-40 was increased from 0.1 to 0.3% and 5% milk was added to the beads for the pulldowns involving mutant UBQLN2 proteins to prevent non-specific binding of TIMM44 in the assays. A 1/10 portion of the mixture was saved for analysis of the input ligand. Then 100 μl glutathione beads were added and this protein/bead mixture was rotated for 2 h at room temperature. The beads were washed three times in binding buffer and resuspended in 100 mm Tris, pH 6.8 and 1× protein loading buffer. Equal portions of the input were similarly probed for the added ligands. The samples were resolved on SDS-PAGE gels and analyzed by immunoblotting. All pulldowns were repeated with similar results at least three times.
ANSWER ALS proteomics analysis
ALS patient proteomics data were provided through request of access to the ANSWER ALS data portal (dataportal.answerals.org). Mitochondrial protein hits were identified within the patient proteomics data set and categorized based on their roles in mitochondria. Log2 fold change ratios of individual mitochondrial proteins between ALS patients and healthy controls were calculated and compared to log2 fold changes in those identical proteins between P497S mutant and non-Tg animals, identified in the UBQLN2 P497S proteomics data set.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.0 software (GraphPad Software Inc., San Diego, CA). Western blot analysis, mitochondria length, respiration and complex activity were statistically evaluated using one-way or two-way ANOVA with post hoc Tukey tests (95% confidence intervals). Experiments with only two groups, such as a comparison of mitochondrial length in WT parental cells versus UBQLN2 KO #8 cells, were evaluated with a student t-test. Statistical significance was determined as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Acknowledgements
We thank Dr. Monica Hall for conducting the RNAseq experiments. We thank Dr Alexandra M. Whiteley, University of Colorado Boulder, for insightful and helpful comments on the manuscript.
Conflict of Interest statement. The authors declare no competing interests.
Funding
The Robert Packard Center for ALS Research at Johns Hopkins and NIH NINDS grants (R01NS098243, R01NS100008 to M.J.M.); NIGMS grant (R01GM129584 to M.K.).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}