




The atonal homolog 7 (ATOH7) gene encodes a transcription factor involved in determining the fate of retinal progenitor cells and is particularly required for optic nerve and ganglion cell development. Using a combination of autozygosity mapping and next generation sequencing, we have identified homozygous mutations in this gene, p.E49V and p.P18RfsX69, in two consanguineous families diagnosed with multiple ocular developmental defects, including severe vitreoretinal dysplasia, optic nerve hypoplasia, persistent fetal vasculature, microphthalmia, congenital cataracts, microcornea, corneal opacity and nystagmus. Most of these clinical features overlap with defects in the Norrin/β-catenin signalling pathway that is characterized by dysgenesis of the retinal and hyaloid vasculature. Our findings document Mendelian mutations within ATOH7 and imply a role for this molecule in the development of structures at the front as well as the back of the eye. This work also provides further insights into the function of ATOH7, especially its importance in retinal vascular development and hyaloid regression.
INTRODUCTION
The development of the vertebrate retina has been extensively studied as a simple and accessible model for neurogenesis. This work has shown that the seven major retinal cell types arise from a single population of progenitor cells through a tightly controlled sequential program of migration and differentiation (1). Basic helix-loop-helix (bHLH) transcription factors homologous to Drosophila achaete-scute and atonal have been shown to play key roles in specifying the neuronal fate of these multipotent retinal progenitors (2). One of the best characterized members of this family is ATOH7 (atonal homolog 7 [MIM *609875]), also known as Math5 in the mouse and Ath5 in other vertebrates.
ATOH7 is a single exon gene that encodes a 152-amino acid protein with a bHLH domain spanning residues 41–96 (3). The basic motif (residues 41–52) is essential for specific deoxyribonucleic acid (DNA) binding and the HLH domain (residues 53–96) forms homo- or hetero-dimers with other family members (4,5). It is one of the first transcription factors to be expressed in retinal progenitor cells and this expression coincides with the onset of neurogenesis (6). Loss-of-function and over-expression experiments in animals have shown that this protein is vital for retinal ganglion cell (RGC) development (7–10), but is also associated with differentiation of progenitors into photoreceptors, horizontal and amacrine cells (11,12). Given the hierarchical nature of retinal growth, with RGCs the first cell type to be specified, we might expect that removal of this protein would grossly disrupt eye development. However, studies in animal models have shown this is not the case. For example, ATOH7-deficient mice have normal sized eyes but have a >95% reduction in RGC number, and lack an optic nerve and chiasm. They also retain the overall retinal laminar structure though they are slightly thinner as a result of ganglion cell loss, and have an increased number of cone photoreceptors through a switch in cell specification (7,8). Similarly, zebrafish with a mutation in Ath5 have a comparable loss of RGCs and an increase in amacrine, bipolar and Muller cells which are consistent with cell fate differences in fish (9).
More recently, there has been increased interest in ATOH7 following several genome-wide association studies which have implicated variants in this gene as significantly associated with optic disc area (13–15) and open angle glaucoma (16,17). However, to date no human disease phenotype has been linked to Mendelian mutations within this gene. In this report, we have used a combination of autozygosity mapping and next generation sequencing to identify patients with homozygous mutations in ATOH7, and widen the reported role of this protein to include the development of the hyaloid and retinal vasculature as well as the anterior segment of the eye.
RESULTS
Clinical details and autozygosity mapping
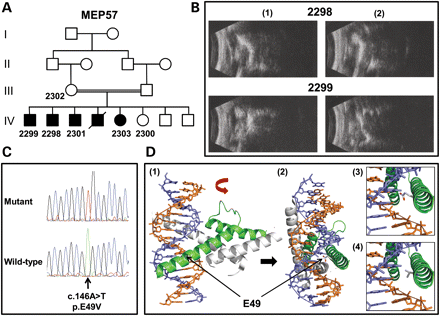
Analysis of MEP57. (A) Pedigree structure for family MEP57. The individuals from whom DNA is available are numbered. (B) B-scan ultrasound examination of the right (1) and left (2) eyes from patients 2298 (aged 33 years) and 2299 (aged 24 years). Note a reduced axial length (microphthalmia) together with disorganization of the vitreous and chronic retinal detachments. (C) Sequence chromatograms highlighting the ATOH7 mutation in an affected patient from MEP57 and the corresponding wild-type sequence from a normal individual. (D) Molecular modelling of the bHLH domain of human ATOH7 wild-type and p.E49V. Ribbon representation of the bHLH domain in ATOH7 (in green) dimerized with the transcription factor E2-alpha (in grey) and bound to double-stranded DNA (purple and orange helix) as a lateral (1) and, following a 90° rotation, frontal (2) view. Note residue E49 in the wild-type sequence is in close connection with the DNA helix. The proximity of the E49 (3) and V49 (4) residues to the DNA helix shows that the V49 is more distant and so is less likely to interact with the DNA.
Next generation sequencing
Given that the number of genes located in the mapped locus is nearly 160, we used a NGS approach to identify the pathogenic mutation in family MEP57. A customized liquid-phase target enrichment reagent was created to capture all the coding exons and flanking splice sites within the critical region. In total, 1490 of 1492 exons were covered, amounting to 278 kb of DNA sequence. Only 201 bp of exonic sequence could not be targeted due to homology with repetitive sequences. Genomic DNA from patient 2298 (IV:2 in Fig. 1A) was enriched using this reagent, and the resulting library was sequenced on an Illumina GAIIx sequencer. The processed sequencing files were aligned to the human reference sequence and polymerase chain reaction (PCR) duplicates were removed. Before their removal, the mean depth of coverage for targeted regions was 383 reads, but after duplicate removal the mean depth amounted to 110 reads with base phred quality scores of at least 17. In summary, 98% of the targeted bases were covered by at least five reads with this quality threshold. A total of 203 variants were detected within the locus but after exclusion of all those annotated as single nucleotide polymorphisms (SNPs) in dbSNP131 only 13 changes remained. Five were intronic (not splice site) variants, three were synonymous and five were non-synonymous variants. Of the five non-synonymous variants (Table 1), two were considered to be non-pathogenic or artefactual, as they were also identified in seven unrelated DNAs that were run in a parallel experiment, leaving three potentially pathogenic variants that were unique to MEP57; a homozygous G to A nucleotide change in FRMPD2 (ferm and pdz domains-containing protein 2 [MIM *613323]) that substitutes an arginine for a glycine residue (c.1051G>A; p.G351R), a homozygous A to T nucleotide change in ATOH7 that substitutes a valine for a glutamic acid residue (c.146A>T; p.E49V; Fig. 1C) and a homozygous G to T nucleotide change in PBLD (phenazine biosynthesis-like protein domain-containing protein [MIM *612189]) that substitutes an isoleucine for a serine residue (c.185G>T; p.S62I).
Summary of the non-synonymous variants identified following targeted capture and next generation sequencing for MEP57 (2298; IV.2)
| CHROM: POS | REF | MEP57 | Gene | cDNA, protein | Exon | Frequency in controls | Evolutionarily conserveda |
|---|---|---|---|---|---|---|---|
| chr10:49440275 | G | A | FRMPD2 | c.1051G>A, p.G351R | 10 | 3/340 chromosomes | NA |
| chr10:51768674 | CAA | Cb | AGAP6 | c.790_791del, p.K264RfsX10 | NA | Not determined | NA |
| chr10:62547870 | T | T/Gc | CDC2 | c.371T>G, p.V124G | NA | Not determined | NA |
| chr10:69991289 | A | T | ATOH7 | c.146A>T, p.E49V | 1A | 0/340 chromosomes | Yes |
| chr10:70056121 | G | T | PBLD | c.185G>T, p.S62I | 4 | 0/340 chromosomes | No |
| CHROM: POS | REF | MEP57 | Gene | cDNA, protein | Exon | Frequency in controls | Evolutionarily conserveda |
|---|---|---|---|---|---|---|---|
| chr10:49440275 | G | A | FRMPD2 | c.1051G>A, p.G351R | 10 | 3/340 chromosomes | NA |
| chr10:51768674 | CAA | Cb | AGAP6 | c.790_791del, p.K264RfsX10 | NA | Not determined | NA |
| chr10:62547870 | T | T/Gc | CDC2 | c.371T>G, p.V124G | NA | Not determined | NA |
| chr10:69991289 | A | T | ATOH7 | c.146A>T, p.E49V | 1A | 0/340 chromosomes | Yes |
| chr10:70056121 | G | T | PBLD | c.185G>T, p.S62I | 4 | 0/340 chromosomes | No |
The variants remaining after filtering against dbSNP131 are summarized. The chromosome and the position of the variant in base pairs based on the UCSC Genome Browser hg19 (CHROM: POS), reference sequence at this position (REF), MEP57 sequence at this position (MEP57), gene ID, cDNA and protein sequence for the variation, the exon harbouring the variant (the corresponding primer pairs for PCR amplification and direct DNA sequencing are depicted in Supplementary Data), the frequency of the variant in ethnically matched control DNAs and whether the normal amino acid residue at the position of the variation is evolutionarily conserved are shown.
NA, not applicable; b and c likely represent false positives as b was detected in 2/7 and c detected in 7/7 unrelated DNAs that were processed with the same enrichment reagent. aEvolutionary conservation is illustrated in Supplementary Data.
Summary of the non-synonymous variants identified following targeted capture and next generation sequencing for MEP57 (2298; IV.2)
| CHROM: POS | REF | MEP57 | Gene | cDNA, protein | Exon | Frequency in controls | Evolutionarily conserveda |
|---|---|---|---|---|---|---|---|
| chr10:49440275 | G | A | FRMPD2 | c.1051G>A, p.G351R | 10 | 3/340 chromosomes | NA |
| chr10:51768674 | CAA | Cb | AGAP6 | c.790_791del, p.K264RfsX10 | NA | Not determined | NA |
| chr10:62547870 | T | T/Gc | CDC2 | c.371T>G, p.V124G | NA | Not determined | NA |
| chr10:69991289 | A | T | ATOH7 | c.146A>T, p.E49V | 1A | 0/340 chromosomes | Yes |
| chr10:70056121 | G | T | PBLD | c.185G>T, p.S62I | 4 | 0/340 chromosomes | No |
| CHROM: POS | REF | MEP57 | Gene | cDNA, protein | Exon | Frequency in controls | Evolutionarily conserveda |
|---|---|---|---|---|---|---|---|
| chr10:49440275 | G | A | FRMPD2 | c.1051G>A, p.G351R | 10 | 3/340 chromosomes | NA |
| chr10:51768674 | CAA | Cb | AGAP6 | c.790_791del, p.K264RfsX10 | NA | Not determined | NA |
| chr10:62547870 | T | T/Gc | CDC2 | c.371T>G, p.V124G | NA | Not determined | NA |
| chr10:69991289 | A | T | ATOH7 | c.146A>T, p.E49V | 1A | 0/340 chromosomes | Yes |
| chr10:70056121 | G | T | PBLD | c.185G>T, p.S62I | 4 | 0/340 chromosomes | No |
The variants remaining after filtering against dbSNP131 are summarized. The chromosome and the position of the variant in base pairs based on the UCSC Genome Browser hg19 (CHROM: POS), reference sequence at this position (REF), MEP57 sequence at this position (MEP57), gene ID, cDNA and protein sequence for the variation, the exon harbouring the variant (the corresponding primer pairs for PCR amplification and direct DNA sequencing are depicted in Supplementary Data), the frequency of the variant in ethnically matched control DNAs and whether the normal amino acid residue at the position of the variation is evolutionarily conserved are shown.
NA, not applicable; b and c likely represent false positives as b was detected in 2/7 and c detected in 7/7 unrelated DNAs that were processed with the same enrichment reagent. aEvolutionary conservation is illustrated in Supplementary Data.
Segregation of each potentially pathogenic missense variant with the disease phenotype was confirmed by direct sequencing in all available family members. We then sequenced the variants in a cohort of 170 ethnically matched control individuals (340 chromosomes). The FRMPD2 variant was present in three control DNAs (and has now been entered into dbSNP132) and was therefore excluded as a pathogenic mutation. The ATOH7 and PBLD variants were not present in the control panel and were therefore assessed for functional impact with the PolyPhen-2 algorithm. Using a HumVar model, scores of 0.758 and 0.985 predicted that both variants were possibly damaging. However, alignment of orthologous protein sequences reveals that the E49 residue in ATOH7 is fully conserved, while the S62 residue in PBLD is conserved through mammals and birds but not in zebrafish (Danio rerio) and worm (Caenorhabditis elegans) (Supplementary Data). The E49 residue lies within the basic motif of the bHLH domain that is responsible for binding the transcription factor to DNA at the consensus hexanucleotide sequence known as the E box (19). In silico modelling shows that the missense mutation is likely to prevent DNA binding and abolish transcription factor activity (Fig. 1D).
Screening for further mutations by direct sequencing
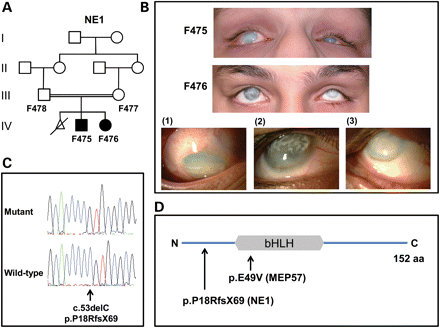
Analysis of NE1. (A) Pedigree structure for family NE1. The individuals from whom DNA is available are numbered. (B) Current photos of the anterior segment of affected family members F475 (aged 16 years) and F476 (aged 14 years). Detailed views of the right eye of patients F475 (1) and F476 (2) as well as the left eye of patient F476 (3) are shown. Note reduced globe size (microphthalmia), diffuse corneal haze with additional superficial, predominantly interpalpebral, discrete opacity secondary to calcium deposition (band keratopathy). The observed ocular deviation (strabismus) is a result of the poor vision. There are no signs associated with significant anterior segment inflammation. (C) Sequence chromatograms highlighting the ATOH7 mutation in an affected patient from NE1 and the corresponding wild-type sequence from a normal individual. (D) Diagrammatic representation of ATOH7 (NP_660161). The position of the bHLH domain and the mutations relative to the full-length 152 amino acid protein are shown.
Clinical examination of these patients revealed corneal opacities, microphthalmia, nystagmus, bilateral retinal detachments and vitreous degenerations, as in MEP57, but also cataracts, persistent fetal vasculature, hypoplastic optic nerves and calcifications present on the hyaloid membranes, retina and in the vitreous. The clinical phenotype was considered similar to that observed in Norrie disease (MIM #310600), but pedigree structure ruled out an X-linked condition. Both children had no microcephaly or mental retardation and no hearing problems, although the younger child made some sterotypic movements of the head. Fluorescein angiography examination of both parents showed no abnormalities. The patients were therefore given a diagnosis of recessive FEVR.
DISCUSSION
Here we report the identification of recessive human mutations within ATOH7 in two families with global eye developmental defects comprising severe vitreoretinal dysplasia, microphthalmia, corneal opacity, microcornea and nystagmus. Additional features reported in one of the families include congenital cataracts, optic nerve hypoplasia and persistent fetal vasculature. However, it is likely that both families have the same disorder and that the additional findings in NE1 were not observed in MEP57 due to the limited clinical data that was available. Our findings also support a recent study which identified a ∼6.5 kb homozygous deletion of an evolutionarily conserved remote enhancer located 20 kb upstream of ATOH7 in an inbred community of Kurdish descent from Iran in which 1% of the population had non-syndromic congenital retinal non-attachment as well as optic nerve aplasia, nystagmus and progressive corneal opacification (21). Although the authors used transgenic animal models with reporter constructs to show that the enhancer was required for ATOH7 expression, evidence that deletion of the enhancer region caused the disease phenotype remained circumstantial. However, the clinical features described in the Iranian patients show substantial overlap with that of the patients described in this study (Table 2). This study therefore provides further support for the assertion by those authors that the enhancer deletion is pathogenic in those patients. Furthermore, a locus for autosomal recessive persistent fetal vasculature has previously been mapped in a single family to a chromosomal region overlapping ATOH7 (22). Like the patients reported in this study, affected individuals in that family had retrolental masses, peripheral anterior synechiae, corneal opacities and cataracts. Examination of the youngest affected subject in that family, a 5-year-old boy, highlighted persistent fetal vasculature and retinal folds but no detachment suggesting that the other features are a consequence of disease progression. It therefore seems highly likely that this family will also harbour mutations in ATOH7. These studies highlight the varied clinical diagnoses assigned to patients who may be suffering from the same genetic disorder and emphasizes the need to interrogate thoroughly the phenotypic data presented in each case rather than focus on the label given at initial diagnosis.
Comparison of the clinical features of patients with homozygous ATOH7 mutations
| MEP57 (c.146A>T, p.E49V) | NE1 (c.53delC, p.P18RfsX69) | Ghiasvand et al.21 (∼6.5 kb deletion of ATOH7 enhancer) | |||||
|---|---|---|---|---|---|---|---|
| 2298 | 2299 | 2301 | 2303 | F475 | F476 | ||
| Age | 33 | 24 | ? | ? | 10 | 8 | 3–49 |
| Cornea | |||||||
| Corneal diameter | Small^ | Small^ | Small^ | Small^ | Small^ | Small^ | Small^ |
| Opacity | c>p | c>p | c>p | c>p | c>p | c>p | c>p |
| Secondary band keratopathy | − | − | − | − | + | + | ? |
| Globe | |||||||
| Microphthalmia OD/OS (mm) | 10/12 | 8/7 | ++^ | ++^ | 15/10 | +^ | ? |
| Iris | |||||||
| Irido-corneal adhesion | p | p | p | p | − | − | + |
| Iris coloboma | − | − | − | − | − | − | − |
| Irido-lenticular adhesions | − | − | − | − | − | − | − |
| Iris hypoplasia | − | − | − | − | − | − | − |
| Lens | |||||||
| Kerato-lenticular touch | − | − | − | − | − | − | − |
| Cataract | ? | ? | ? | ? | + | + | − |
| Vitreous | |||||||
| Vitreousa | Multifocal dot, retrolental mass | Multifocal dot, retrolental mass | ? | ? | Linear opacity, retrolental mass | Linear opacity, retrolental mass | Linear opacity, retrolental mass |
| Retina | |||||||
| Detacheda | ++ | ++ | ? | ? | ++ | ++ | ++ |
| Optic nerve | |||||||
| Sizea | ? | ? | ? | ? | Small^ | Small^ | Absent/small^ |
| Function | |||||||
| Nystagmus | + | + | + | + | + | + | + |
| Vision | NPL/NPL | NPL/NPL | PL/PL | PL/PL | PL/PL | PL/PL | NPL/NPL |
| MEP57 (c.146A>T, p.E49V) | NE1 (c.53delC, p.P18RfsX69) | Ghiasvand et al.21 (∼6.5 kb deletion of ATOH7 enhancer) | |||||
|---|---|---|---|---|---|---|---|
| 2298 | 2299 | 2301 | 2303 | F475 | F476 | ||
| Age | 33 | 24 | ? | ? | 10 | 8 | 3–49 |
| Cornea | |||||||
| Corneal diameter | Small^ | Small^ | Small^ | Small^ | Small^ | Small^ | Small^ |
| Opacity | c>p | c>p | c>p | c>p | c>p | c>p | c>p |
| Secondary band keratopathy | − | − | − | − | + | + | ? |
| Globe | |||||||
| Microphthalmia OD/OS (mm) | 10/12 | 8/7 | ++^ | ++^ | 15/10 | +^ | ? |
| Iris | |||||||
| Irido-corneal adhesion | p | p | p | p | − | − | + |
| Iris coloboma | − | − | − | − | − | − | − |
| Irido-lenticular adhesions | − | − | − | − | − | − | − |
| Iris hypoplasia | − | − | − | − | − | − | − |
| Lens | |||||||
| Kerato-lenticular touch | − | − | − | − | − | − | − |
| Cataract | ? | ? | ? | ? | + | + | − |
| Vitreous | |||||||
| Vitreousa | Multifocal dot, retrolental mass | Multifocal dot, retrolental mass | ? | ? | Linear opacity, retrolental mass | Linear opacity, retrolental mass | Linear opacity, retrolental mass |
| Retina | |||||||
| Detacheda | ++ | ++ | ? | ? | ++ | ++ | ++ |
| Optic nerve | |||||||
| Sizea | ? | ? | ? | ? | Small^ | Small^ | Absent/small^ |
| Function | |||||||
| Nystagmus | + | + | + | + | + | + | + |
| Vision | NPL/NPL | NPL/NPL | PL/PL | PL/PL | PL/PL | PL/PL | NPL/NPL |
All patients in families MEP57 and NE1 as well as the general features of the Iranian patients that were recently reported by Ghiasvand et al. (21) are depicted. The meanings of the symbols used in the table are as follows; ^, exact measurement unavailable; c, central; p, peripheral; −, absent; +, present; ++, present in a severe form; ?, unknown; PL, perception of light; NPL, no perception of light.
aThe descriptions for vitreous, retina and optic nerve were based on ultrasound or orbital MRI measurements.
Comparison of the clinical features of patients with homozygous ATOH7 mutations
| MEP57 (c.146A>T, p.E49V) | NE1 (c.53delC, p.P18RfsX69) | Ghiasvand et al.21 (∼6.5 kb deletion of ATOH7 enhancer) | |||||
|---|---|---|---|---|---|---|---|
| 2298 | 2299 | 2301 | 2303 | F475 | F476 | ||
| Age | 33 | 24 | ? | ? | 10 | 8 | 3–49 |
| Cornea | |||||||
| Corneal diameter | Small^ | Small^ | Small^ | Small^ | Small^ | Small^ | Small^ |
| Opacity | c>p | c>p | c>p | c>p | c>p | c>p | c>p |
| Secondary band keratopathy | − | − | − | − | + | + | ? |
| Globe | |||||||
| Microphthalmia OD/OS (mm) | 10/12 | 8/7 | ++^ | ++^ | 15/10 | +^ | ? |
| Iris | |||||||
| Irido-corneal adhesion | p | p | p | p | − | − | + |
| Iris coloboma | − | − | − | − | − | − | − |
| Irido-lenticular adhesions | − | − | − | − | − | − | − |
| Iris hypoplasia | − | − | − | − | − | − | − |
| Lens | |||||||
| Kerato-lenticular touch | − | − | − | − | − | − | − |
| Cataract | ? | ? | ? | ? | + | + | − |
| Vitreous | |||||||
| Vitreousa | Multifocal dot, retrolental mass | Multifocal dot, retrolental mass | ? | ? | Linear opacity, retrolental mass | Linear opacity, retrolental mass | Linear opacity, retrolental mass |
| Retina | |||||||
| Detacheda | ++ | ++ | ? | ? | ++ | ++ | ++ |
| Optic nerve | |||||||
| Sizea | ? | ? | ? | ? | Small^ | Small^ | Absent/small^ |
| Function | |||||||
| Nystagmus | + | + | + | + | + | + | + |
| Vision | NPL/NPL | NPL/NPL | PL/PL | PL/PL | PL/PL | PL/PL | NPL/NPL |
| MEP57 (c.146A>T, p.E49V) | NE1 (c.53delC, p.P18RfsX69) | Ghiasvand et al.21 (∼6.5 kb deletion of ATOH7 enhancer) | |||||
|---|---|---|---|---|---|---|---|
| 2298 | 2299 | 2301 | 2303 | F475 | F476 | ||
| Age | 33 | 24 | ? | ? | 10 | 8 | 3–49 |
| Cornea | |||||||
| Corneal diameter | Small^ | Small^ | Small^ | Small^ | Small^ | Small^ | Small^ |
| Opacity | c>p | c>p | c>p | c>p | c>p | c>p | c>p |
| Secondary band keratopathy | − | − | − | − | + | + | ? |
| Globe | |||||||
| Microphthalmia OD/OS (mm) | 10/12 | 8/7 | ++^ | ++^ | 15/10 | +^ | ? |
| Iris | |||||||
| Irido-corneal adhesion | p | p | p | p | − | − | + |
| Iris coloboma | − | − | − | − | − | − | − |
| Irido-lenticular adhesions | − | − | − | − | − | − | − |
| Iris hypoplasia | − | − | − | − | − | − | − |
| Lens | |||||||
| Kerato-lenticular touch | − | − | − | − | − | − | − |
| Cataract | ? | ? | ? | ? | + | + | − |
| Vitreous | |||||||
| Vitreousa | Multifocal dot, retrolental mass | Multifocal dot, retrolental mass | ? | ? | Linear opacity, retrolental mass | Linear opacity, retrolental mass | Linear opacity, retrolental mass |
| Retina | |||||||
| Detacheda | ++ | ++ | ? | ? | ++ | ++ | ++ |
| Optic nerve | |||||||
| Sizea | ? | ? | ? | ? | Small^ | Small^ | Absent/small^ |
| Function | |||||||
| Nystagmus | + | + | + | + | + | + | + |
| Vision | NPL/NPL | NPL/NPL | PL/PL | PL/PL | PL/PL | PL/PL | NPL/NPL |
All patients in families MEP57 and NE1 as well as the general features of the Iranian patients that were recently reported by Ghiasvand et al. (21) are depicted. The meanings of the symbols used in the table are as follows; ^, exact measurement unavailable; c, central; p, peripheral; −, absent; +, present; ++, present in a severe form; ?, unknown; PL, perception of light; NPL, no perception of light.
aThe descriptions for vitreous, retina and optic nerve were based on ultrasound or orbital MRI measurements.
Many of the ocular features for both the anterior and posterior segment described above in patients with ATOH7 mutations are also found in severe FEVR, osteoporosis pseudoglioma syndrome and Norrie disease (23–28). A recent study reported reduced optic nerve head size in a cohort of FEVR patients compared with controls (29). All of these disorders are known to be caused by mutations in genes which encode components of a signalling complex which activates the Norrin/β-catenin signalling pathway (30–32). This pathway is similar to the Wnt/β-catenin signalling pathway with the exception that the Wnt ligand is replaced by Norrin. Studies in human patients and mouse models have shown that the primary ocular defect in these disorders is insufficient development of the retinal vasculature (25,33–38), although dysgenesis of the ganglion cell layer was also noted in the Norrin knockout mouse (39). It is interesting to note that although Atoh7-deficient mice lack optic nerves and have a drastic reduction in ganglion cell abundance, they also have ectopic blood vessels within the posterior chamber that could represent persistent hyaloid vessels or reflect choroidal neovascularization, although a detailed examination of the vasculature in these mice was not performed (7). In addition to its role in the development of the retinal vasculature, Wnt signalling has also been shown to play a key role in the regression of the hyaloid vasculature. Wnt7b secreted by macrophages in the eye activates Wnt signalling in vascular endothelial cells to initiate cell death and hyaloid regression (40). This regression of the hyaloid vasculature precedes the development of the retinal vasculature, although the precise mechanism of this interaction is not known yet.
The main differences between the pathologies of the Atoh7-deficient mice and human patients with homozygous ATOH7 mutations described in this study are that the mice failed to demonstrate complete retinal detachment and any obvious anterior segment abnormalities were absent (7,8). The disparity in retinal detachment between both species most likely reflects differences between the relative volume of the lens and vitreous chamber, the retinal architecture and timing of neuronal and vascular development. The anterior segment discrepancies reflect the dissimilarities in the posterior segment, which show a milder phenotype in the mutant mice. Nevertheless, it is interesting to note that dominant mutations in the master eye control gene PAX6 (paired box 6), which is an upstream regulator of ATOH7 (6,41), also causes both anterior segment and retinal abnormalities (42,43). However, unlike ATOH7 which is restricted to developing neuronal cells and is required for RGC development (6–10), PAX6 is not only involved in mediating retinal progenitor cell fate (44) but also has a role in very early development in the migration of neural crest cells to form the lens placode (45,46). Our data on the human ATOH7 mutant phenotype extends the primarily retinal observations noted previously to include both anterior segment and vascular abnormalities. Despite these observations, given that retinal neurogenesis precedes the development of the vasculature during early development (47–50) and the previously reported findings in transgenic animal models (7–9), it is likely that the absence of RGCs may be the primary cause of disease pathogenesis in the patients described in this study.
In summary, we have identified homozygous mutations in ATOH7 as a cause of global eye developmental defects in human patients. This finding will benefit patients with ATOH7 mutations and their families by facilitating accurate diagnosis and counselling as well as help in the development of future therapies. Furthermore, in addition to the established function of ATOH7 in retinal progenitor cell specification, these findings reveal wider roles in the regression of the hyaloid vasculature, the development of the retinal vasculature as well as the anterior segment structures of the eye.
MATERIALS AND METHODS
Subjects
This study was performed using a process approved by the Pakistan Medical Foundation in Lahore and the Leeds Teaching Hospitals Trust Research Ethics Committee. Informed consent was obtained from all participants in accordance with the tenets of the Declaration of Helsinki. Clinical examination involved a detailed eye inspection and ultrasound examination in selected patients. In addition, affected members of NE1 underwent orbital MRI and cranial CT imaging. Peripheral blood was taken from available family members and genomic DNA was extracted according to standard procedures. Control subjects were normal individuals who were recruited as siblings of patients subject to genetic testing. None of the families involved had any member with an inherited eye abnormality and all the individuals were of Asian subcontinent extraction.
Autozygosity mapping
DNA from the affected individuals was genotyped using SNP arrays (Affymetrix 6.0, AROS Applied Biotechnologies, Aarhus, Denmark) and regions of homozygosity highlighted using IBD finder software as described before (51). Linkage was confirmed using polymorphic microsatellite markers on a genetic analyser according to the manufacturer's instructions (3130xl Genetic Analyser, Applied Biosystems, Warrington, UK).
Next generation sequencing
A customized liquid-phase ‘SureSelect Target Enrichment’ biotinylated cRNA bait was designed using the Agilent Technologies eArray platform (Agilent Technologies Ltd., Edinburgh, UK). This reagent was designed against all the UCSC Genome Browser coding exons and flanking 20 bp from the known genes within the critical interval. Three micrograms of patient DNA was sheared by sonication and ligated onto adaptors using standard procedures. This mixture was hybridized against the customized baits and eluted to capture the enriched DNA library which was sequenced for 80 bp single-end reads on a single lane of the Illumina Genome Analyzer GAIIx (Illumina Inc., CA, USA).
The raw data files were processed by the Illumina pipeline (version 1.3.4) and the sequencing reads were aligned to the human reference sequence (hg19/GRCh37) with Novoalign software (Novocraft Technologies, Selangor, Malaysia). Alignments were processed in the SAM/BAM format (52) using Picard and The Genome Analysis Toolkit (GATK) (53) java programs in order to correct alignments around indel sites and mark and remove potential PCR duplicates. Variants for the coding regions were reported in the VCF format using the Unified Genotyper and DINDEL (54) functions of GATK, before filtering against variants recorded in dbSNP131.
Direct DNA sequencing
Primers for the amplification of ATOH7, PBLD and FRMPD2 are depicted in Supplementary Data. For all primers pairs, amplification was performed using HotShot Diamond PCRTM mastermix according to the manufacturer's instructions (Clent Life Science, Stourbridge, UK). PCR products were digested using ExoSAP-IT (USB Corporation, Cleveland, OH, USA) and sequenced using the Big Dye terminator cycle sequencing Kit v.3.1 (Applied Biosystems) on the ABI 3130XL DNA analyser (Applied Biosystems). The results were analysed using SeqScape version 2.5 software (Applied Biosystems).
Bioinformatic analysis
For the prediction of the impact of an amino acid substitution on the structure and function of a protein, PolyPhen-2 (Polymorphism Phenotyping v.2) scores were calculated at http://genetics.bwh.harvard.edu/pph2/ under a HumVar model (55). For evolutionary conservation, the protein sequences were downloaded from the NCBI (http://www.ncbi.nlm.nih.gov/protein) and the alignments calculated using ClustalW (http://www.ebi.ac.uk/clustalw2/).
Protein modelling
The structural model of the mutant and wild-type bHLH domain of human ATOH7 (accession number NP_660161) was derived by homology modelling using the M4T server (56,57). The crystal structure of the mouse Neurogenic differentiation factor 1 bound to DNA (PDB code 2ql2), which shares 61% sequence identity to the bHLH domain of human ATOH7, was used as template. The quality and stereochemistry of the model was assessed using ProSA-II (https://prosa.services.came.sbg.ac.at/prosa.php) and PROCHECK (http://www.ebi.ac.uk/thornton-srv/software/PROCHECK/), respectively, and the figures were generated using PYMOL (http://pymol.sourceforge.net/).
SUPPLEMENTARY MATERIAL
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the Wellcome Trust (grant number 090224; K.K. is a Clinical Research Training Fellow), Yorkshire Eye Research (grant number 022), The Royal Society (C.T. is a URF) and The Sir Jules Thorn Charitable Trust (grant number 09/JTA). Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
ACKNOWLEDGEMENTS
The authors would like to thank the patients and their families for their help with this study. We would also like to thank Professors Bart Leroy and Elfride de Baere (Department of Ophthalmology and Centre for Medical Genetics, Ghent University, Ghent, Belgium) for sending DNA samples from patients with retinal dysplasia for mutation screening.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors and the last three authors should be regarded as having contributed equally to this work.

{kind=link}
{kind=link}