





Abstract
Hepatocyte growth factor activator inhibitor (HAI)-2 is an integral membrane Kunitz-type serine protease inhibitor that regulates the proteolysis of matriptase and prostasin in a cell-type selective manner. The cell-type selective nature of HAI-2 function depends largely on whether the inhibitor and potential target enzymes are targeted to locations in close proximity. The N-glycan moiety of HAI-2 can function as a subcellular targeting signal. HAI-2 is synthesized with 1 of 2 different N-glycan modifications: one of oligomannose-type, which largely remains in the endoplasmic reticulum/GA, and another of complex-type, which is targeted toward the apical surface in vesicle-like structures, and could function as an inhibitor of matriptase and prostasin. HAI-2 contains 2 putative N-glycosylation sites, Asn-57 and Asn-94, point mutations of which were generated and characterized in this study. The protein expression profile of the HAI-2 mutants indicates that Asn-57, and not Asn-94, is responsible for the N-glycosylation of both HAI-2 species, suggesting that the form with oligomannose-type N-glycan is the precursor of the form with complex-type N-glycan. Unexpectedly, the vast majority of non-glycosylated HAI-2 is synthesized into multiple disulfide-linked oligomers, which lack protease inhibitory function, likely due to distorted conformations caused by the disarrayed disulfide linkages. Although forced expression of HAI-2 in HAI-2 knockout cells artificially enhances HAI-2 oligomerization, disulfide-linked HAI-2 oligomers can also be observed in unmodified cells. These results suggest that N-glycosylation on Asn-57 is required for folding into a functional HAI-2 with full protease suppressive activity and correct subcellular targeting signal.
Introduction
Hepatocyte growth factor activator inhibitor (HAI)-2 is an integral membrane Kunitz-type serine protease inhibitor that is widely expressed in organ systems containing epithelial elements (Kawaguchi et al. 1997). The structural complementarity between the Kunitz domain 1 of HAI-2 and the serine protease domain of the type 2 transmembrane serine protease matriptase and that of the membrane-associated serine protease prostasin means that HAI-2 is a potent inhibitor of these proteases (Takeuchi et al. 2000; Shipway et al. 2004). More importantly, an enzyme–inhibitor relationship between these proteins has been observed in cellular and physiological settings. For example, HAI-2 complexes with matriptase and prostasin have been purified from human milk (Lai et al. 2016a). HAI-2 complexes with prostasin were also detected in lysate prepared from intestinal tissues (Shiao et al. 2017). HAI-2 has also been identified as the primary protease inhibitor of matriptase in neoplastic B-cells, although HAI-2 is not effective at controlling extracellular matriptase activity due to the fact that majority of the HAI-2 remains intracellular (Chiu et al. 2019). Studies from animal models have further demonstrated that matriptase and prostasin are physiologically relevant target proteases of HAI-2 (Kawaguchi et al. 2019; Szabo et al. 2009, 2012; Szabo and Bugge 2018; Szabo et al. 2019).
Despite the widespread expression of this inhibitor, mutations of the SPINT2 gene, which encodes HAI-2, primarily impact the gastrointestinal tract, while sparing other organs, resulting in the severe diarrhea observed in patients with syndromic congenital sodium diarrhea (SCSD) (Heinz-Erian et al. 2008; Sivagnanam et al. 2010; Slae et al. 2013; Bou Chaaya et al. 2017; Hirabayashi et al. 2018; Holt-Danborg et al. 2019). This organ-selective pathology suggests that there exists some intestine-selective functionality. This hypothesis is supported by the fact that HAI-2 is not responsible for prostasin inhibition in human skin, where HAI-1 serves as the primary inhibitor for prostasin as well as for matriptase (Chen et al. 2010; Lu et al. 2021). In fact, HAI-1-mediated inhibition of matriptase and prostasin has been observed in the vast majority of epithelial/carcinoma cells tested (Tseng et al. 2010; Su et al. 2016). In contrast, HAI-2 regulates matriptase and prostasin proteolysis in a cell-type selective manner (Chang et al. 2015, 2020; Shiao et al. 2017; Lee et al. 2018). The differential regulation of matriptase and prostasin in different contexts has been attributed to differences in their subcellular distribution and localization (Lai et al. 2016b; Kataoka et al. 2000; Oberst et al. 2005; Tseng et al. 2010; Chen et al. 2014; Wu et al. 2014; Chang et al. 2015, 2020; Lee et al. 2018). Although HAI-1 and HAI-2 closely resemble one another with respect to overall domain structure and exhibit similar specificity and potency against proteases (Lai et al. 2015), the subcellular targeting signals embedded within their respective intracellular domain are different (Huang et al. 2022). The plasma membrane export motif in HAI-1, combined with relatively few Arg/Lys-rich endoplasmic reticulum (ER) retention-like motifs facilitates the translocation of more than half of total cellular HAI-1 to the cell surface at the steady state. As a result, HAI-1 has direct access to matriptase and prostasin. In contrast, an incomplete plasma membrane export motif along with multiple Arg/Lys-rich ER retention-like motifs, keeps the majority of HAI-2 inside cells, thereby denying access to matriptase and prostasin (Huang et al. 2022). It appears, however, that in the human intestine cells have evolved to have a subcellular microenvironment, in which HAI-2 and prostasin co-localized near to the terminal web underneath the microvilli of the human enterocytes (Shiao et al. 2017). HAI-2, therefore, gains direct access to prostasin and becomes the primary protease inhibitor of prostasin in enterocytes.
A previous study with a chimeric molecule that contains the HAI-2 extracellular domain and HAI-1 intracellular domain indicated that the HAI-2 extracellular domains also contain a subcellular targeting signal that seems to be responsible for directing HAI-2 to the relatively large vesicle-like structures observed (Huang et al. 2022). This subcellular targeting signal apparently involves the complex-type N-glycan modification of HAI-2. In addition to modification with complex-type N-glycan, a proportion of HAI-2 is synthesized instead with oligomannose-type N-glycan, the vast majority of which is detected primarily in the region of the ER and the Golgi apparatuses (GA). Furthermore, while the 2 HAI-2 species are both expressed at high levels, only the form with the complex-type N-glycan, and not the mannose-type N-glycan form, is responsible for the cellular control of matriptase and prostasin proteolysis (Lai et al. 2015). HAI-2 contains 2 putative N-glycosylation sites and it remains largely unknown how the 2 putative sites are involved in the expression of the 2 HAI-2 species and how the different N-glycan modifications alter the protease suppressive activity of HAI-2. To explore this issue the current study made use of 3 HAI-2 variants with point mutations of the 2 putative N-glycosylation sites that were analyzed and characterized.
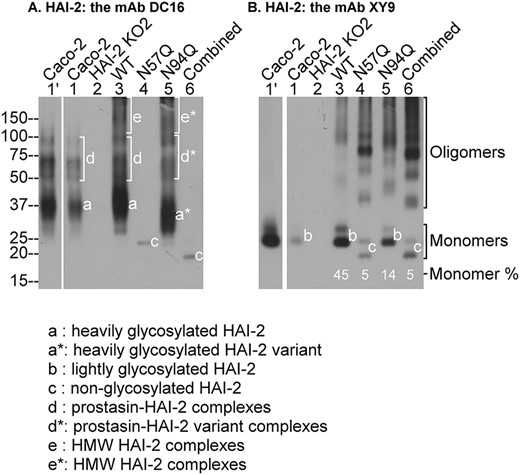
Asn-57, but not Asn-94, is responsible for the N-glycosylation of HAI-2. Wild-type HAI-2 (WT, lanes 3) or mutant HAI-2 variants Asn-57 (N57Q, lanes 4), or Asn-94 (N94Q, lanes 5), or Asn-75 + Asn-94 (combined, lanes 6) expression constructs were transfected into HAI-2 knockout Caco-2 cells. Lysates prepared from these cells, the HAI-2 KO cells (HAI-2 KO2, lanes 2), and parental Caco-2 cells (Caco-2, lanes 1 and lanes 1′ for longer exposure) were analyzed by immunoblot for HAI-2 species using the mAb DC16 (A) and the mAb XY9 (B). The various HAI-2 species are indicated. The ratios of HAI-2 monomer to total HAI-2 recognized by the mAb XY9 are also indicated.
Results
Asn-57, and not Asn-94, is the HAI-2 N-glycosylation site
HAI-2 contains 2 putative N-glycosylation sites, Asn-57 (N-57) and Asn-94 (N-94). Three HAI-2 variants with point mutations of Asn-57 (N57Q), Asn-94 (N94Q), or both (combined) to glutamine were transiently expressed in a HAI-2 deleted variant of Caco-2 human colorectal adenocarcinoma cells. The potential role of N-57 and N-94 as HAI-2 glycosylation sites and the impact of N-glycan loss on HAI-2 protein expression and biochemical function as a protease inhibitor were subsequently studied. Previous studies have shown that HAI-2 is naturally expressed in cells as 1 of 2 different forms: one being heavily glycosylated with complex-type N-glycan and the other being lightly glycosylated with oligomannose-type N-glycan. Two different HAI-2 monoclonal antibodies (mAbs), DC16 and XY9, were developed and characterized as previously described (Chang et al. 2015; Lai et al. 2015). The heavily glycosylated form of HAI-2 can be selectively detected by the mAb DC16 but not XY9. In contrast, the lightly glycosylated HAI-2 form can be detected by the mAb XY9 and likely by DC16 as well when the detection signal is strong (Lai et al. 2015). In the parental Caco-2 cells, the endogenous HAI-2 can be detected using the 2 HAI-2 mAbs as multiple species due to the differential glycosylation of HAI-2 and the presence of stable enzyme–inhibitor complexes of HAI-2 with activated prostasin. The pattern of bands is further complicated by the presence of some degradation products (Shiao et al. 2017).
The presence of multiple HAI-2 species within Caco-2 cells was again observed in this study (Fig. 1 lanes 1 and 1′ for long exposure). Three HAI-2 monomer bands can readily be distinguished: band a that is detected by the mAb DC16, but not XY9 (Fig. 1, lanes 1′ and 1); band b, a doublet, detected primarily by the mAb XY9 (Fig. 1B, lanes 1′ and 1). Band a represents the heavily glycosylated HAI-2 species with complex-type N-glycan modification; band b represents the lightly glycosylated HAI-2 species with oligomannose-type N-glycan modification. In addition to the monomers, the endogenous HAI-2 was also detected in multiple complexed forms. Our previous study showed that the heavily glycosylated but not the lightly glycosylated HAI-2 forms stable enzyme–inhibitor complexes with prostasin (Shiao et al. 2017), and the same result was also observed here (Fig. 1, bands d).
As expected, no HAI-2 signal is observed in the HAI-2 KO Caco-2 cells (Fig. 1, lanes 2). When the wild-type HAI-2 expression construct was transfected into the HAI-2 KO Caco-2 cells, the expression profile of this exogenously expressed wild-type HAI-2 (Fig. 1, lanes 3) was very similar to that of endogenous HAI-2 in the Caco-2 parental cells (Fig. 1, comparing lanes 3 with lanes 1′ and 1). Novel HAI-2 species with a very high molecular weight (HMW) were also detected, not only for heavily glycosylated HAI-2 (Fig. 1A, lane 3, band e), but also for lightly glycosylated HAI-2 (Fig. 1B, lane 3). It appears that the forced expression of HAI-2 could lead to the generation of HMW HAI-2 species. These HMW HAI-2 species of lightly glycosylated HAI-2 are in fact disulfide-linked HAI-2 oligomers as demonstrated later (Fig. 3).
The band profiles in lysates from cells transfected with one of the 3 HAI-2 mutants were next examined. The complete loss of the heavily glycosylated form of HAI-2 in cells transfected with both the N57Q and the combined mutants suggests that Asn-57 is occupied with the complex-type N-glycan (Fig. 1A, lanes 4 and 6). The sizes of N94Q monomer doublets are identical to the sizes observed for lightly glycosylated wild-type HAI-2 suggesting that this putative N-glycosylation site is not involved in N-glycosylation (Fig. 1B, comparing lane 5 to lane 3 for band b). The conclusion is consistent with the apparent lack of N-glycan or other modification on Asn-94, which allowed the amino acid residue to be sequenced by Edman degradation in a previous study (Kawaguchi et al. 1997). Taken together, these data indicate that Asn-57 is the glycosylation site at which HAI-2 is modified in both the heavily and lightly glycosylated forms of HAI-2.
It is worth noting that mutation at Asn-94 does appear to cause a slight change in the apparent size or migration rate of the heavily glycosylated HAI-2 on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 1A, lane 5, band a*) but not the lightly glycosylated HAI-2 form (Fig. 1B, lane 5, band b doublet). Although the mechanism underlying the structural or conformational changes in the HAI-2 variants remains elusive, the different degree of N-glycosylation may represent the likely mechanism.
An unexpected consequence of N57Q mutation was the observation of a marked decrease in the proportion of the monomer relative to the total lightly glycosylated HAI-2, which was determined to be 100% for the endogenous HAI-2 (Fig. 1B, lane 1′), near half for the exogenously expressed wild-type HAI-2, and less than 5% in both the N57Q and the combined mutant forms (Fig. 1B, comparing lanes 4 and 6 with lane 3). This decrease in the ratio of the monomer relative to the total of lightly glycosylated HAI-2 was also observed for N94Q (Fig. 1B, lane 5, 14%). Nevertheless, a significant proportion of the N94Q variant still undergoes extensive N-glycan branching modification and is observed as a monomer (Fig. 1A, lane 5, band a*), which can function as a potent protease inhibitor against prostasin by formation of stable enzyme–inhibitor complex (Fig. 2, lane 5).
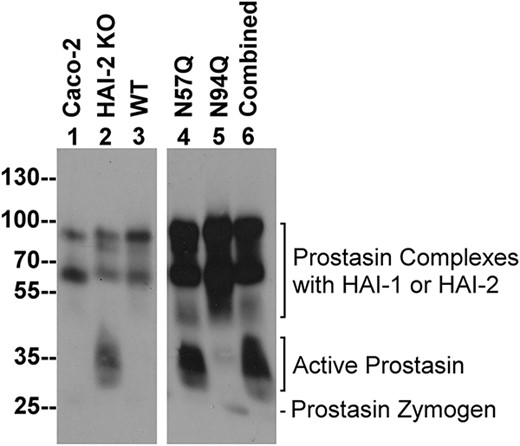
N-glycosylation is required for HAI-2 to reverse the enhanced prostasin proteolysis caused by targeted HAI-2 deletion in Caco-2 cells. Wild-type HAI-2 (WT, lane 3), or HAI-2 mutants, constructs N57Q (lane 4), N94Q (lane 5), or combined (lane 6) were transiently expressed in HAI-2 knockout Caco-2 cells. Lysates prepared from these cells, the HAI-2 KO cells (HAI-2 KO, lane 2), and parental Caco-2 cells (Caco-2, lane 1) were analyzed by immunoblot for prostasin species using the mAb YL11. Prostasin species include prostasin complexes with the HAIs, active prostasin, and prostasin zymogen, as indicated.
The increase in prostasin proteolytic activity caused by HAI-2 loss in Caco-2 cells, cannot be reversed by transfection with HAI-2 variants lacking N-glycosylation
Caco-2 cells express prostasin at high levels but also actively activate the prostasin, particularly when Caco-2 cells have been maintained at relatively high confluence. Despite this high level of zymogen activation, cellular amounts of enzymatically active prostasin are, however, only observed at very low levels due to the presence of HAI-1 and HAI-2 (Barndt et al. 2021). This tight control of prostasin proteolytic activity has been also observed in lysates of human intestinal tissues (Shiao et al. 2017). The vast majority of the prostasin detected in the parental Caco-2 cells was observed in the activated form but in complexes with the HAIs in previous studies (Shiao et al. 2017; Barndt et al. 2021), and in the current study (Fig. 2, lane 1). Targeted deletion of HAI-2 created a severe imbalance favoring prostasin proteolysis with the presence of abundant uncomplexed, enzymatically active prostasin (Fig. 2, comparing lane 2 with lane 1). The imbalance caused by targeted HAI-2 deletion can be reverted by the re-expression of wild-type HAI-2 in the knockout cells, resulting in the disappearance of the enzymatically active prostasin (Fig. 2, comparing lane 3 with lane 2). In contrast, transfection with the N57Q and the combined putative glycosylation site mutants, both of which result in the expression of HAI-2 without modification with N-glycans (Fig. 1), failed to revert the proteolytic imbalance with active prostasin detected at high levels (Fig. 2, comparing lanes 4 and 6 with lane 2). The inability of the N57Q HAI-2 variant to suppress the excess prostasin proteolysis caused by targeted HAI-2 deletion suggests an important role for Asn-57 in the control of prostasin proteolysis in Caco-2 cells. Given that Asn-57 represents the sole N-glycosylation site in HAI-2 (Fig. 1), the prevention of N-glycan modification on Asn-57 is the most likely reason for the inability of the N57Q HAI-2 variant to suppress the increased prostasin proteolysis caused by targeted HAI-2 deletion in Caco-2 cells.
In contrast to the N57Q HAI-2 variant, transfection of the HAI-2 knockout cells with the N94Q mutant, which results in the expression of both the heavily and lightly glycosylated HAI-2 forms (Fig. 1), was able to suppress prostasin proteolytic activity in a similar manner to transfection with wild-type HAI-2 (Fig. 2, comparing lane 5 with lane 3). The results of the N94Q transfection experiment serves also as an experimental control for the result of the N57Q transfection and supports the conclusion that N-glycan modification on Asn-57 is important for the role of HAI-2 in the control of prostasin proteolysis. Although HAI-2 is synthesized as heavily and lightly glycosylated forms, a previous study has shown that heavily glycosylated HAI-2, but not the lightly glycosylated form of HAI-2, is responsible to the control of prostasin proteolysis (Lai et al. 2015; Shiao et al. 2017). In conjunction with the importance of Asn-57 in the expression of both HAI-2 forms identified in the current study (Fig. 1), the N-glycan branching conducted in the GA onto the existing oligomannose-type N-glycan attached via Asn-57 appears to be an important N-glycan modification, which renders heavily glycosylated HAI-2 responsible to the control of prostasin proteolysis. The complex-type N-glycans are, therefore, structurally required for HAI-2 in the cellular control of prostasin proteolysis in Caco-2 cells.
It is worth noting that the higher apparent MW of enzymatically active prostasin than that of its zymogen form is due to the fact that the immunoblot analysis was performed under non-reducing and non-boiled conditions. Under such conditions, the active and zymogen forms of prostasin contain same number of amino acid residues. The slower migration rate of the active prostasin in SDS-PAGE is likely due to the relatively flexible and loose conformation of active prostasin.
The N-glycan modification of HAI-2 is important for protein folding by reducing formation of disulfide linked complexes
The inability of non-glycosylated HAI-2 to revert the imbalance favoring prostasin proteolysis might simply be attributed to the different subcellular localizations of non-glycosylated HAI-2 and prostasin. The differences in the protein profiles observed, particularly the observation that the vast majority of non-glycosylated HAI-2 is present as multiple higher MW complexes indicates, however, that other mechanisms might also contribute to the phenotype. For example, these non-glycosylated HAI-2 complexes may not possess similar protease-inhibitory activity as their monomer counterparts, and indeed, the relative protease-inhibitory activity of non-glycosylated HAI-2 monomer has been not characterized. The nature and structural basis of the protein–protein interactions, which result in HAI-2 complex formation, could provide insights to the anti-protease activity of these complexes of non-glycosylated HAI-2. Thus, wild-type HAI-2 and the 3 mutants were next analyzed by immunoblot under 3 different conditions: (i) non-reducing and non-boiled, under which these non-glycosylated HAI-2 complexes are stable; (ii) non-reducing but boiled, under which non-glycosylated HAI-2 monomers might be dissociated and released if the interactions involved in complex formation are non-covalent; and (iii) reducing and boiled, in which non-glycosylated HAI-2 monomers would be dissociated and released if disulfide linkages are involved. Examination of the immunoblot results revealed that boiling treatment alone did not cause changes in the protein band profiles for these HAI-2 complexes, regardless of their glycosylation status, whereas the wild-type HAI-2 signal slightly increased consistently among the repeated experiments (Fig. 3, comparing lanes 2 to lanes 1). These data indicate that the HAI-2 complexes are involve some kind of covalent linkage and exclude the possibility that the complexes represent enzyme–inhibitor HAI-2 complexes with active protease, which are non-covalent in nature. When a reducing agent was introduced, the signal from the complexes disappeared that was coincided with a dramatical increase in the level of HAI-2 monomer (Fig. 3, lanes 3), indicating the dissociation of the complexes into the monomer form. These data suggest that the HAI-2 complexes are formed via disulfide linkages. Furthermore, when the size of the HAI-2 monomer (Fig. 3, band a) and the apparent size of the complexed bands (Fig. 3, bands b, c, and d) are considered, it seems likely that the complexes are comprised of homo-oligomers of HAI-2 rather than HAI-2 complexes with other proteins. For example, the predicted size of a HAI-2 homodimer is similar to that of protein band b; the trimer to protein band c; and the tetramer to protein band d (Fig. 3, as indicated). In addition, the doublet band profile of the HAI-2 monomer (Fig. 3, band a) can also be observed for the dimer (Fig. 3, band b) and faintly for the trimer (Fig. 3, band d) before the loss of resolution on SDS-PAGE limits separation (Fig. 3). The size difference between the lightly glycosylated HAI-2 and non-glycosylated HAI-2 was also observed for the respective oligomers. Taken together, these data suggest that when HAI-2 overexpression is enforced, HAI-2 can undergo oligomerization. The increase in disulfide-linked HAI-2 complexes observed with the HAI-2 mutants lacking N-glycosylation suggests that the N-glycan modification is important for the folding of HAI-2 by reducing the formation of HAI-2 complexes with intramolecular disulfide linkages.
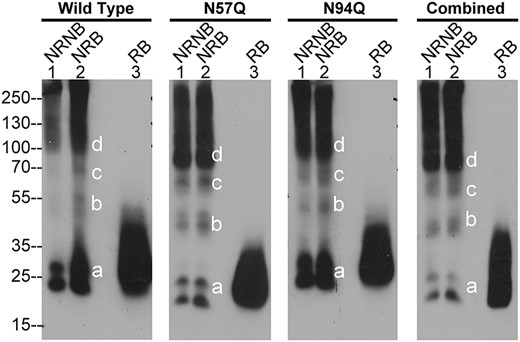
Exogenously expressed HAI-2 tends to form disulfide-linked oligomers, particularly non-glycosylated HAI-2. Wild-type HAI-2 (wild type) and HAI-2 variants, including N57Q mutant (N57Q), N94Q mutant (N94Q), and the combined mutant (combined) were transiently expressed in HAI-2 knockout Caco-2 cells. Lysates prepared from these cells were analyzed by immunoblot for HAI-2 species using the mAb XY9 under 1) non-reducing and non-boiled condition (NRNB, lanes 1), 2) non-reducing and boiled condition (NRB, lanes 2), and reducing and boiled condition (RB, lanes 3). Different HAI-2 species based on their sizes were indicated.
N-glycosylation status affects the binding of HAI-2 to trypsin in solution
Mature HAI-2 (lacking the signal peptide) contains 12 cysteine residues, which form 3 pairs of disulfide linkages within each Kunitz domain for a total of 6 pairs for the entire protease inhibitor. The 3 pairs of disulfide bonds in each Kunitz domain are required for maintenance of the native conformation of the Kunitz domains and are essential for protease inhibitory function (Hedstrom 2002). There are no free cysteine residues, therefore, to form intermolecular disulfide bond for the formation of these HAI-2 oligomers. It is, therefore, plausible that in order for oligomerization, some of these intramolecular disulfide bonds are not correctly formed in the ER. As a result, unpaired cysteine residues are available to participate in intermolecular disulfide linkages allowing the formation of HAI-2 dimers, trimers, and oligomers, with the likely result that the protease inhibitory function would be compromised due to a distorted conformation.
In order to test whether oligomerization contributes to the loss of the ability of the N57Q HAI-2 variant to inhibit active prostasin (Fig. 2), the ability of the non-glycosylated variant HAI-2 to form complexes with active trypsin was next evaluated along with the heavily glycosylated and lightly glycosylated wild-type counterparts (Fig. 4). Lysate containing both the heavily and lightly glycosylated forms of HAI-2 were prepared from Caco-2 parental cell lysates by immune depletion of HAI-1 and prostasin from the lysates. Lysate containing non-glycosylated HAI-2 was prepared from the lysates of N57Q HAI-2 transfected HAI-2 KO Caco-2 cells following immune depletion of HAI-1 and prostasin from the lysates. Samples of the lysate preparations containing the different HAI-2 species were then mixed with increasing amounts of trypsin and incubated at 4°C for 30 min. The incubation time was terminated by the addition of SDS-sample buffer and the formation of HAI-2 complexes with trypsin was analyzed by immunoblot (Fig. 4). Examination of the heavily glycosylated HAI-2 protein band profiles reveals that complex formation at the cost of the HAI-2 monomer occurred in a dose-dependent manner, when the amount of enzymatically active trypsin in the reaction was presumably less than or comparable to the amount of cellular HAI-2 (Fig. 4A, left panel, lanes 1 through 4). Further increases in the amount of trypsin beyond this level resulted in the rapid degradation of the heavily glycosylated HAI-2 complexes (Fig. 4A, left panel, lanes 5 through 7). It is worth noting that 2 different heavily glycosylated HAI-2-trypsin complexes were formed; one with an apparent size of 60 kDa and the other of 100 kDa. The 60 kDa complex appears to be consistent with the formation of a 1:1 trypsin:HAI-2 complex, whereas the 100 Da complex mostly likely represents one HAI-2 molecule in complex with 2 trypsin molecules, consistent with the presence of 2 functional Kunitz domains in HAI-2, both of which possess inhibitory activity against trypsin-like serine proteases. While complex formation with trypsin, and trypsin-mediated degradation, were also observed for the lightly glycosylated form of HAI-2, the level of complex observed was much lower than for the heavily glycosylated counterpart, and was not in proportion with level of the monomer present (Fig. 4A, right panel). Although the presence of multiple oligomers of non-glycosylated HAI-2 could have masked the signals of complex formation somewhat, the fact that similar levels of the individual non-glycosylated HAI-2 species were maintained, along with the trypsin-mediated degradation of these species as the amount of trypsin increased, suggests that complex formation did not occur or to a negligible extent (Fig. 4B). Taken together, these comparisons demonstrate that heavily glycosylated HAI-2 exhibits a much strong binding affinity for trypsin than the lightly glycosylated HAI-2 and, that non-glycosylated HAI-2 appears to have no binding affinity for trypsin. HAI-2 N-glycosylation may play an important role in the correct folding of HAI-2 into a functional protease inhibitor with full suppressive activity against trypsin-like serine proteases. More importantly, the loss of the protease suppressive function of non-glycosylated HAI-2 in solution is consistent with the failure of the N57Q HAI-2 variant to suppress the increase in enzymatically active prostasin observed in HAI-2 knockout cells (Fig. 2). It is worth noting that the inability of N57Q HAI-2 variant to suppress trypsin results from the oligomerization. Our current study did not evaluate if N57Q HAI-2 monomer retains the anti-trypsin activity.
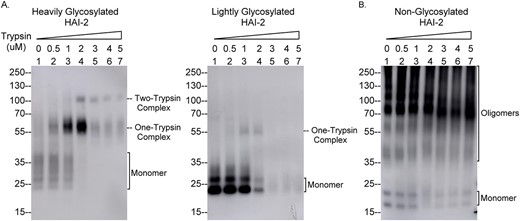
The N-glycosylation status affects the ability of HAI-2 to form enzyme–inhibitor complexes with trypsin in solution. Equal amounts of cellular proteins from lysates of Caco-2 cells (A) and the HAI-2 KO Caco-2 cells (B), in which N57Q HAI-2 variant has been transiently expressed, were incubated with different amounts of a trypsin preparation, as indicated at 4°C for 30 min. The reaction was terminated by the addition of 1X SDS-sample buffer. The samples were analyzed by immunoblot for HAI-2 species and trypsin-HAI-2 complexes using the HAI-2 mAb. HAI-2 monomer and HAI-2 complexes with trypsin are indicated.
HAI-2 monomer represents the predominant reserve of the cellular anti-trypsin activity in human enterocytes
Caco-2 cells express HAI-1, HAI-2, and probably other protease inhibitors that work together to control proteolysis mediated by trypsin-like serine proteases, including prostasin and matriptase. At steady state, in parallel with the zymogen activation of the serine proteases, a proportion of these cellular protease inhibitors are used, and consumed by the formation of enzyme–inhibitor complex with these activated proteases. The remaining protease inhibitors, in their monomer forms, constitute the overall cellular reserve available for the control of serine protease activity. The level of this reserve can be determined by titration using different amounts of trypsin added to a fixed amount of cell lysate, a situation similar to that shown in Fig. 4. An assay was established in which a fixed amount of trypsin (12 pmol trypsin for 20 μg total cell lysate protein) was selected, at which the trypsin activity, as determined by chromogen release from a synthetic chromogenic substrate, N-Benzoyl-Val-Gly-Arg p-nitroanilide hydrochloride, was largely suppressed by the cellular protease inhibitors present in the lysates, as shown in a representative experiment (Fig. 5A, Caco-2). The trypsin activity was determined quantitatively by measuring the average rate of chromogen release from the synthetic substrate in 3 independent experiments (Fig. 5B). Using this predetermined trypsin levels, the relative importance of HAI-1 versus HAI-2 in the control of cellular anti-trypsin activity can be determined. For example, the modest increase in trypsin activity from 1 × 10−3 CU/min in Caco-2 lysate to 1.4 × 10−3 CU/min in the HAI-1 depleted Caco-2 lysate indicates that HAI-1 is not a particularly significant contributor with respect to the cellular anti-trypsin reserve (Fig. 5B, comparing H1 De with Caco-2). In contrast, a robust increase in trypsin activity from 1 × 10−3 CU/min in Caco-2 lysate to 2.8 × 10−3 CU/min was observed in the HAI-2 depleted Caco-2 lysate, suggesting that HAI-2 is much more important than HAI-1 with respect to its contribution to the cellular anti-trypsin reserve. The difference between HAI-1 and HAI-2 can be further estimated in quantitative manner. The combination of HAI-1 and HAI-2 in contributing to the cellular anti-trypsin reserve was estimated to be 2.2 × 10−3 CU/min by subtracting 1 × 10−3 CU/min in Caco-2 lysate from 3.2 × 10−3 C/min in the HAI-depleted lysate (Fig. 5B, H1 + H2 De). As such, HAI-2 represents an approximate 80% versus 20% for HAI-1 as the combined reserve of the HAIs (Fig. 5C). HAI-2, therefore, represents a cellular anti-trypsin reserve that is roughly 4-fold that of HAI-1. HAI-1 has previously been shown to play important role in the control of matriptase and prostasin proteolysis in Caco-2 cells. The minor proportion of HAI-1 in the protease inhibitor reserve may result from the high levels of matriptase and prostasin zymogen activation, with the result that a significant proportion of the available HAI-1 has already been sequestered in the formation of complexes with activated matriptase and prostasin.
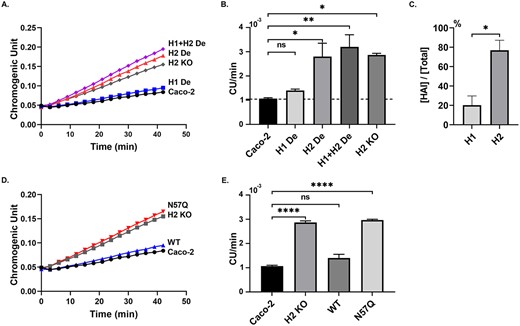
The heavily glycosylated form of HAI-2 represents the predominant cellular anti-trypsin activity of Caco-2 cells. HAI-1 and HAI-2 were immune-depleted from lysates prepared from Caco-2 cells individually (A and B, H1 De and H2 De) or together (A and B, H1 + H2 De). Equal amounts of protein from these samples and lysates of Caco-2 parental cells (A, B, and D, E, Caco-2), HAI-2 KO Caco-2 cells (A, B, D, and E, H2 KO), and the HAI-2 KO cells, in which the N57Q HAI-2 variant was transiently expressed (D, and E, N57Q), were incubated with trypsin at 4°C for 30 min. The residual trypsin activity in these samples was measured using a synthetic, chromogenic substrate, N-Benzoyl-Val-Gly-Arg p-nitroanilide hydrochloride (A and D, chromogenic unit versus time). The residual trypsin activities in these samples were quantitatively determined by the rate of chromogen release (B, and E, chromogen unit per min, CU/min) from 3 independent experiments. The ratio of the individual HAI relative to the combined anti-trypsin activity of the HAIs in Caco-2 cells were calculated and presented (C). CU stands for chromogen unit. Error bar represents SEM. Significant differences are derived from one-way ANOVA (B, E) or Student’s t-test (C). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Exogenous expression of N57Q HAI-2 mutant does not significantly increase the overall anti-trypsin capacity in HAI-2 KO Caco-2 cells
The loss of the ability of non-glycosylated HAI-2 to form enzyme–inhibitor complexes with trypsin (Fig. 4) indicates the loss of the inhibitory function of non-glycosylated HAI-2, and is consistent with its lack of the ability to suppress the increase in prostasin proteolysis, caused by targeted HAI-2 deletion (Fig. 2). Thus, N-glycosylation on Asn-57 seems to be a prerequisite for the protease inhibitory function of HAI-2. The importance of N-glycosylation on Asn-57 to the protease inhibitory function of HAI-2 was further investigated by comparing the ability of the HAI-2 N57Q variant versus wild-type HAI-2 to suppress the increase in the cellular anti-antitrypsin capacity, caused by targeted HAI-2 deletion. When wild-type HAI-2 was expressed exogenously by transfection of the HAI-2 KO Caco-2 cells, the trypsin activity present was robustly suppressed (Fig. 5D and E, comparing WT with H2KO) to a level similar to that in the parental Caco-2 cells (Fig. 5D and E, comparing WT with Caco-2). In contrast, exogenous expression of the N57Q HAI-2 mutant failed to restore the cellular anti-trypsin activity (Fig. 5D and E, N57Q).
HAI-2 oligomerization is not simply a transfection artifact and is observed in LoVo colon cancer cells
The lack of oligomerization of lightly glycosylated HAI-2 in parental Caco-2 cells (Fig. 1B) or in another colorectal adenocarcinoma cells, HT29 (Fig. 6A) raises a concern that the oligomerization is an experimental artifact caused by the forced expression of HAI-2. HAI-2 oligomerization has, however, been observed in at least one unmodified colorectal adenocarcinoma line: LoVo cells (Fig. 6B). In addition to the monomer, lightly glycosylated HAI-2 was also detected as dimers, trimers, and tetramers, which were also found to be thermally stable under non-reducing condition (Fig. 6B, lanes 1 and 2) and dissociated by reducing agents (Fig. 6B, lane 3). Two thermolabile, lightly glycosylated HAI-2 species with apparent masses of 120 and 65 kDa complexes were also observed (Fig. 6B, lane 1, indicated by *), which might represent HAI-2 complexes with as yet unidentified proteases. The 120 kDa complex was also seen in HT29 cells (Fig. 6A, lane 1). The presence of lightly glycosylated HAI-2 oligomers in LoVo cells indicates that HAI-2 oligomerization can occur with endogenously expressed HAI-2.
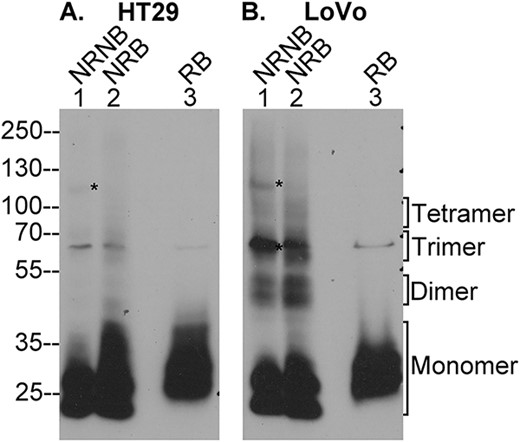
LoVo colon adenocarcinoma cells endogenously express disulfide linked HAI-2 oligomers. Lysates of colorectal adenocarcinoma cells HT29 (A) and LoVo (B) were analyzed by immunoblot for HAI-2 species using the mAb XY9 under 3 different conditions: 1) non-reducing and non-boiled condition (NRNB, lanes 1), 2) non-reducing and boiled condition (NRB, lanes 2), and reducing and boiled condition (RB, lanes 3). The monomer and oligomer forms of HAI-2 are indicated. Two putative enzyme–inhibitor complexes of HAI-2 with an as yet unidentified protease(s) are indicated by *, which disappeared when the samples underwent boiling treatment (lanes 2).
Discussion
Through a systemic analysis of the putative N-glycosylation sites, the current study reveals an interesting and important mechanism governing the protease inhibitory function of HAI-2. While most Kunitz-type serine protease inhibitors are synthesized with their Kunitz domains in a mature correctly folded conformation, several mechanisms have been identified that limit the ability of some Kunitz domains to suppress proteolytic activity of their cognate target proteases. The unusual spatial arrangement of the Kunitz domains in HAI-1 has been shown to result in Kunitz domain 1 preventing access of Kunitz domain 2 to the substrates, even though Kunitz domain 2 possesses the full capacity to suppress protease activity with the expected specificity and potency as has been shown when this domain is expressed and assayed in isolation (Miyata et al. 2007; Liu et al. 2017). Subcellular compartmentalization may deny access of fully functional Kunitz inhibitors to their cognate proteases in some contexts. For example, the intracellular localization of HAI-2 means that it cannot inhibit matriptase on the cell surface of keratinocytes and lymphoma cells, resulting the overall low efficiency of the inhibition of matriptase by HAI-2 in these cells (Lai et al. 2016a; Chiu et al. 2021). The discovery of HAI-2 oligomerization presented here reveals a novel mechanism by which the protease suppressive activity of Kunitz domains becomes completely lost caused by an altered conformation. More importantly, we demonstrate that this the oligomerization can be observed in unmodified cell systems indicating that this is not simply an experimental artifact caused by the enforced expression of HAI-2 by transfection. The mechanism, which causes HAI-2 to undergo oligomerization in LoVo cells, provides an interesting insight, beyond subcellular mistargeting, for a potential mechanism underling the pathogenesis of SCSD, caused by SPINT2 mutations. Although mechanisms other than the loss of the inhibition of target proteases could contribute to development of sodium diarrhea in patients with SDSD, the uncontrolled prostasin proteolysis is likely the direct consequence of SPINT2 mutations and could subsequently cause the sodium diarrhea. The correlation between the degree of N-glycosylation and the extent of oligomerization suggests a role for N-glycosylation in preventing the synthesis of nonfunctional HAI-2 protein. It would be of interest to analyze the HAI-2 N-glycosylation status in LoVo cells and in tissues from SCSD patients in future studies.
Like other membrane and secreted glycoproteins, HAI-2 undergoes a 2-stage N-glycosylation process. In the lumen of the ER, HAI-2 undergoes co-translational modification with the transfer of a 14-monosaccharide Glc3Man9glcNAC2 glycan to Asn-57 of the nascent polypeptide, followed by the trimming of terminal glucoses and/or some mannoses (Suzuki et al. 2022). The lightly glycosylated form of HAI-2 may be the product of this first stage of N-glycosylation due to the fact that the N-glycan of this form can bind to concanavalin A (Con A) but not to wheat germ agglutinin (WGA) and, thus, likely contains the oligomannose type N-glycan. The lightly glycosylated form of HAI-2 exits the ER to enter the GA for the second stage of N-glycosylation. Trimming the existing N-glycan followed by the transfer of N-acetylglucosamine then likely occurs to generate the heavily glycosylated form of HAI-2, which binds to WGA but not to Con A (Lai et al. 2015). The primary function of the first stage of N-glycosylation in the ER is likely involved in ensuring the proper protein folding of HAI-2 prior to exit to the GA. The N-glycan branching modification conducted in the Golgi is to provide the subcellular targeting signal and, unexpectedly, leads to enhancement of the protease inhibitory activity of HAI-2, as shown in the current study. Although protein folding is dictated largely by the primary sequence, proper protein folding also involves the formation of secondary structures, α-helices and β-sheets, the formation of disulfide bonds, and quaternary association via multimerization. The oxidative redox environment in the ER promotes the formation of disulfide bonds. The presence of HAI-2 oligomers in LoVo cells suggests that HAI-2 may also naturally not only be folded to form monomers, but also undergo oligomerization in the ER. This oligomerization, however, causes the loss of the protease inhibitory activity of HAI-2.
Although HAI-2 oligomerization results in the loss of its protease suppressive activity (Figs 4 and 5), it remains largely unclear if HAI-2 oligomers represent a misfolded product that will be degraded. The involvement of disulfide linkages indicates that HAI-2 oligomerization should occur in the ER. If HAI-2 oligomers represent misfolded HAI-2, these HAI-2 molecules would be translocated to the cytoplasm and degraded by the proteasome. Since HAI-2 oligomers are naturally synthesized at a certain level in LoVo cells, and that the oligomers can be synthesized to very high levels in modified cell model with forced HAI-2 protein expression, HAI-2 oligomerization may represent a mechanism by which the HAI-2 protein can pass the quality control for misfolded protein. If this is the case, there could be some cellular and molecular situations, which can induce or facilitate HAI-2 oligomerization. Understanding of the underlying mechanisms could provide important insights to some pathophysiological processes involving HAI-2, such as in patients with SCSD, caused by SPINT2 mutations. The lack of N-glycosylation appears to be the strongest factor that causes HAI-2 to be synthesized almost exclusively in the oligomer form, based on the observation that forced expression of N57Q HAI-2 results in 95% of the non-glycosylated HAI-2 forming oligomers (Fig. 1B). This is in marked contrast to wild-type HAI-2 naturally expressed in un-modified (parental) Caco-2 cells, in which HAI-2 is synthesized as heavily and lightly N-glycosylated forms and HAI-2 oligomers are not detectable (Fig. 1). The lack of HAI-2 oligomers has been also observed in more than 20 other non-modified cell lines (Lai et al. 2015) with an exception of LoVo cells (Fig. 6). When we searched COSMIC (the Catalogue of Somatic Mutations in Cancer), a missense mutation, c.442C > T, of SPINT2 was found in LoVo cells (https://cancer.sanger.ac.uk/cell_lines/mutation/overview?id=102958553). This missense SPINT2 mutation has also been found in some patients with congenital diarrhea (Salomon et al. 2014). Although this somatic mutation causes a substitution of Arg-148 with a Cys residue within Kunitz domain 2 of HAI-2, the level of heavily glycosylated HAI-2 is exceptionally low in LoVo cells (data not shown). In contrast to the “normal” level of lightly glycosylated HAI-2, the low expression of heavily glycosylated HAI-2 indicates that R148C HAI-2 somehow encounters difficulty in N-glycan branching in the GA. There could, therefore, be a correlation between HAI-2 oligomerization and a dysfunctional HAI-2 N-glycan branching in LoVo cells. Given the fact that HAI-2 oligomerization in the ER precedes HAI-2 N-glycan branching in the GA, this inverse correlation could result from mechanisms that are simultaneously present in both the ER and GA and can detect and correspondingly respond to the change in the HAI-2 amino acid sequence and the resultant altered 3D structure caused by the R148C mutation. If this is true, the defect in HAI-2 N-glycan branching has no role in HAI-2 oligomerization. Alternatively, the R148C HAI-2 mutant undergoes normal N-glycosylation in the absence of oligomerization in the ER. Lightly N-glycosylated R148C HAI-2, however, encounters a difficulty in N-glycan branching in the GA and is returned back to the ER for oligomerization. A combination of the 2 possible situations could be also present.
In addition to the 3D structure of HAI-2, the secretory pathway itself could also contribute to HAI-2 oligomerization. In our previous studies, the expression of heavily and lightly N-glycosylated HAI-2 forms have been compared in more than 20 human cell lines (Lai et al. 2015). Although the levels of the HAI-2 proteins vary significantly in these different cell lines, a relative constant ratio of lightly glycosylated HAI-2 to heavily glycosylated HAI-2 is maintained. This constant ratio suggests that there is some intrinsic mechanism by which a relatively constant proportion of lightly glycosylated HAI-2 undergoes N-glycan branching in the GA regardless the overall expression level. This proposed intrinsic mechanism excludes the possibility that the presence of high levels of lightly glycosylated HAI-2 is due to some derangement of the N-glycan branching machinery in the GA. N-glycan branching not only converts the lightly glycosylated HAI-2 into the heavily glycosylated form but also, at the same time, enhances the suppressive activity of HAI-2 (Figs 4 and 5) and provides HAI-2 with the subcellular targeting signal to direct it to the large vesicles (Lai et al. 2015; Huang et al. 2022). Although oligomers of lightly glycosylated HAI-2 have been generated in the model with overexpression of the wild-type HAI-2 construct (Figs 1B and3), the oligomers are not detectable in almost all non-modified cell lines tested with the exception of LoVo cells (Fig. 6). The lack of oligomerization of lightly glycosylated HAI-2 and certainly the heavily glycosylated HAI-2 in non-modified cells in the native (or endogenous) state indicates that the intrinsic mechanism underlying HAI-2 N-glycosylation can effectively prevent the oligomerization. A significant proportion of wild-type HAI-2 (45%) was, however, detected in oligomer forms when HAI-2 was overexpressed expressed. The presence of almost half of the wild-type HAI-2 expressed in oligomer form could simply be attributed to the “artifact” of forced expression in modified cells. This phenomenon, however, suggests that the proposed intrinsic mechanism is apparently delicately and precisely regulated. As such, the modification of cells and the manipulation of gene expression could adversely impact this mechanism and the oligomerization appears as a consequence. As described above, the amino acid sequences and the resultant 3D structure are important for N-glycosylation, particularly N-glycan branching, and the oligomerization of HAI-2. This conclusion is supported by the studies with N94Q HAI-2 (Fig. 1). Although Asn-94 is not the N-glycosylation site, the majority of the lightly glycosylated N94Q HAI-2 variant (86%) is synthesized in oligomers (Fig. 1B) and the heavily glycosylated HAI-2 is synthesized with altered complex-type N-glycan, the size of which is apparently smaller than its wild-type counterpart (Fig. 1A).
In summary, HAI-2 must undergo a series of tightly controlled molecular processes in the secretory pathway for the synthesis and translocation of functional HAI-2 to the apical side of the plasma membrane to perform its physiological function. N-glycosylation plays an integral role in facilitation of these molecular processes (Fig. 7) (Suzuki et al. 2022). In the ER lumen, N-glycosylation serves as a quality control mechanism to ensure the folding of nascent HAI-2 into a state with correct conformation and certain levels of suppressive activity against serine protease (Fig. 7, steps 1 through step 4). Those HAI-2 molecules that remain misfolded will be retrotranslocated to the cytoplasm, following by deglycosylation and degradation by the proteasome (Fig. 7, step 6). Disruption and interference with the initial N-glycosylation could lead to HAI-2 oligomerization involving the formation of intermolecular disulfide bonds (Fig. 7, steps 7 and 8). As a result, a functional conformation cannot be achieved for the Kunitz domains, and as a result the anti-protease activity is impaired. Once the proper conformation is achieved, the newly synthesized HAI-2 with oligomannose-type N-glycan enters the GA (Fig. 7, step 5), where the N-glycan branching takes place to generate HAI-2 species with complex-type N-glycan. Unexpectedly, the N-glycan branching modification appears to enhance the existing suppressive activity of the Kunitz domains against serine proteases. The polysaccharide moiety also serves as the subcellular targeting signal directing HAI-2 into relatively large vesicles, which apparently translocate to the apical plasma membrane at a slow pace due to the unfavorable plasma membrane targeting signals embedded in the intracellular domain. As a result, the majority of the HAI-2 remains inside the cells at steady state. Disruption of the addition of complex-type N-glycan will also misroute the less-active HAI-2 species away from the physiological localization and the target proteases, such as prostasin in enterocytes.
![Schematic summary of the role of N-glycosylation in HAI-2 folding and oligomerization in the ER. The nascent HAI-2 polypeptide undergoes co-translational modification with the transfer of a 14-monosaccharide Glc3Man9GlcNAc2 glycan to Asn-57 of HAI-2 (step 1). Upon removal of 2 glucose, the monoglucosylated N-glycans serve as a quality control to assist protein folding via the interplays with lectin-like chaperone, such as calnexin (CNX) and ER-resident proteins, such as ERp57 (step 2). Correctly folded HAI-2 molecules will exist the ER after the removal of terminal glucose via α-glucosidase II (GII) (steps 3 and 5). The glycosidase and UDP-glucose glycoprotein glucosyltransferase (UGGT) together also facilitate HAI-2 folding via cycling of the removal and addition of the terminal glucose (step 4). HAI-2 glycoproteins remaining in misfolded conformations will be retrotranslocated to the cytoplasm, where the HAI-2 molecules are deglycosylated and degraded by the proteasome (step 6). Some of these HAI-2 species with oligomannose-type N-glycan undergoes oligomerization to escape the proteasome-mediated degradation (step 7). The oligomerization process appears to become a mechanism by which non-glycosylated HAI-2 passes the quality control and escapes proteasome-mediated degradation at the cost of the loss of protease suppressive function (step 8). (Figure modified from Fig. 39.2 in Essentials of Glycobiology [Internet]. 4th edition; Suzuki et al. 2022).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/glycob/33/3/10.1093_glycob_cwad002/1/m_cwad002f7.jpeg?Expires=1750792092&Signature=UPMChbeVze9010XTMMp3loKZxyTjj7xRF6CbmB0fADWgqez0ndscQrVW7fiMg~xjKyqEFDNpvdp-M~wVQVhdpOrwkuCpHapm5QbSm9ezLRL5W0~ecsnC-DL4UK4Q-KHjn3fE1U1iz0rJw87~0dK7aO~AII7RbumOcS2DsGHjWN0MWjgyGapjIA-en4oLfsCUONKHRwtLzDY87HGdhCAl4ghDfbV5MzpyrWAV0LMx-CPE~DXaoE6TAFEfCrU3FtxampKr6~F2PpRJ8SIBCs3yNol6hGEr5CNfwKkFyinRXk2wpVHSLBiJKipC0dXLpYbqYeZr1osY80k5zMQ30bNWow__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Schematic summary of the role of N-glycosylation in HAI-2 folding and oligomerization in the ER. The nascent HAI-2 polypeptide undergoes co-translational modification with the transfer of a 14-monosaccharide Glc3Man9GlcNAc2 glycan to Asn-57 of HAI-2 (step 1). Upon removal of 2 glucose, the monoglucosylated N-glycans serve as a quality control to assist protein folding via the interplays with lectin-like chaperone, such as calnexin (CNX) and ER-resident proteins, such as ERp57 (step 2). Correctly folded HAI-2 molecules will exist the ER after the removal of terminal glucose via α-glucosidase II (GII) (steps 3 and 5). The glycosidase and UDP-glucose glycoprotein glucosyltransferase (UGGT) together also facilitate HAI-2 folding via cycling of the removal and addition of the terminal glucose (step 4). HAI-2 glycoproteins remaining in misfolded conformations will be retrotranslocated to the cytoplasm, where the HAI-2 molecules are deglycosylated and degraded by the proteasome (step 6). Some of these HAI-2 species with oligomannose-type N-glycan undergoes oligomerization to escape the proteasome-mediated degradation (step 7). The oligomerization process appears to become a mechanism by which non-glycosylated HAI-2 passes the quality control and escapes proteasome-mediated degradation at the cost of the loss of protease suppressive function (step 8). (Figure modified from Fig. 39.2 in Essentials of Glycobiology [Internet]. 4th edition; Suzuki et al. 2022).
Materials and methods
Cell culture
Caco-2 human colorectal adenocarcinoma cells were obtained as a gift from Dr Toni Antalis of the University of Maryland Baltimore. The preparation and characterization of a Caco-2 variant with targeted HAI-2 deletion made using clustered regularly interspaced short palindromic repeats technology has been described in a previous study (Barndt et al. 2021). HT29 and Lovo human colorectal adenocarcinoma cells were obtained from the Tissue Culture and Biobanking Shared Resource of the Georgetown Lombardi Comprehensive Cancer Center. The cells were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere with 5% CO2.
Plasmid preparation
Wild-type (WT) HAI-2 plasmid (pCMV-HAI2) was purchased from OriGene Technologies, Inc. (CAT#: RC202044) and used after removal of the Myc and DDK tags, as previously described Huang et al. 2022).
Site-directed mutations of the putative N-glycosylation sites N57 and N94 to glutamine, individually and in combination on the modified pCMV-HAI2 plasmid were generated using QuikChange XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instruction. The sequences of the primers used are list below.
N57Q: 5′-aggatccgtcagtgacctggtaccaccacctaggc-3′
5′-gcctaggtggtggtaccaggtcactgacggatcct-3′
N94Q: 5′-caggtcacccgtggcctgctctgtgacagtggc-3′
5′-gccactgtcacagagcaggccacgggtgacctg-3′
Transfection
Transfection was performed when the cells reach approximately 30% confluency. For a 100 mm dish, 3000 ng of HAI-2 plasmid was used. Transfection was performed with TransIT-LT1 Transfection Reagent (Prod. No. MIR 2300, Mirus Bio LLC, Madison WI) at a 3:1 ratio to the DNA according to the manufacturer’s instruction. The cells were harvested for analyses 48 h post-transfection.
Western blotting and antibodies
The cells were lysed in radioimmunoprecipitation assay buffer, containing 1 mM 5,5-dithio-bis-(2-nitrobenzoic acid). After removal of insoluble debris by centrifugation, the protein concentration in the supernatant was determined using Bio-Rad Protein Assay Dye Reagent (Bio-Rad Laboratories, Hercules, CA, USA) and a Wallac 1420 Victor 2 microplate reader. Samples of cell lysate were diluted in 5× SDS sample buffer containing no reducing agent, unless indicated, and incubated at room temperature for 5 min prior to loading onto the gels. For reducing condition, 1 μl 2-mercaptoethanol was added to 25 μl sample preparation with SDS-sample buffer, followed by incubation at 100°C (boiling) for 5 min. Proteins were separated by 10% SDS-PAGE, transferred to 0.45-μm nitrocellulose membranes, and probed with the indicated primary mAbs. The binding of mAbs was detected by incubation with horseradish peroxidase-conjugated secondary antibodies (Kirkegaard & Perry Laboratories) and visualized on X-ray films using Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences). The mAbs used were for prostasin, mAb YL11, for HAI-1, mAb M19, and for HAI-2, mAbs XY9 and DC16. The generation and characterization of these mAbs can be found in our previous publications (Lin et al. 1999; Lai et al. 2015).
Immunodepletion
For immunodepletion, protein samples (200 μl) were mixed with 15 μl mAb-conjugated Sepharose beads. The suspensions were mixed by end-over-end rotation in a cold room for 2 h. The supernatant containing the unbound proteins was separated from the agarose beads by centrifugation and collected. The protein concentration of the supernatant was determined.
Trypsin complex formation with HAI-2 and trypsin proteolytic activity assay
Lysates containing 20 μg cellular proteins were incubated on ice with 4 μl of trypsin preparations, containing different concentrations of trypsin, for 30 min. The lysate–trypsin mixtures were then analyzed by western blot for the formation of enzyme–inhibitor complex and by amidolytic assay for trypsin-like protease activity. The amidolytic assay was performed in 96-well microtiter plates using a chromogenic oligopeptide substrate, N-Benzoyl-Val-Gly-Arg p-nitroanilide hydrochloride (CAT# sc-215440, Santa Cruz Biotechnology, Inc.). The reaction buffer contained 100 mM Tris buffer, pH 8.5, and 125 μM of the chromogenic substrate in a total volume of 0.1 ml. The trypsin activity was determined by measuring the release of chromogen by 405 nm absorbance using a Wallac 1420 Victor 2 microplate reader for 45 min with an interval of 3 min. All experiments have been repeated 3 times.
Con A: Concanavalin A
ER: Endoplasmic reticulum
GA: Golgi apparatuses
HAI: Hepatocyte growth factor activator inhibitor
HMW: High molecular weight
KO: Knockout
PAGE: Polyacrylamide gel electrophoresis
SCSD: Syndromic congenital sodium diarrhea
SDS: Sodium dodecyl sulfate
WGA: Wheat germ agglutinin
WT: Wild-type
Funding
This study was supported by National Cancer Institute (NCI) Grant R01 CA 123223 (to MDJ and CYL), Grants (MAB-109-042 and MND-MAB-110-045) from the Ministry of National Defense Medical Affairs Bureau, Taiwan (to JKW), and Grants (TSGH-E-109211, TSGH-E-110190, and TSGH-E-111203) from Tri-Service General Hospital, Taipei, Taiwan (to PYC). We also acknowledge the assistance provided by the Tissue Culture Shared Resource, which is supported in part by the Lombardi Comprehensive Cancer Center support grant (NIH/NCI grant P30-CA051008). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest statement. There is no conflict of interest for the authors.
Data Availability Statement
The data underlying this article are available in the article.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}