





Abstract
The evolution of genomic incompatibilities causing postzygotic barriers to hybridization is a key step in species divergence. Incompatibilities take 2 general forms—structural divergence between chromosomes leading to severe hybrid sterility in F1 hybrids and epistatic interactions between genes causing reduced fitness of hybrid gametes or zygotes (Dobzhansky–Muller incompatibilities). Despite substantial recent progress in understanding the molecular mechanisms and evolutionary origins of both types of incompatibility, how each behaves across multiple generations of hybridization remains relatively unexplored. Here, we use genetic mapping in F2 and recombinant inbred line (RIL) hybrid populations between the phenotypically divergent but naturally hybridizing monkeyflowers Mimulus cardinalis and M. parishii to characterize the genetic basis of hybrid incompatibility and examine its changing effects over multiple generations of experimental hybridization. In F2s, we found severe hybrid pollen inviability (<50% reduction vs parental genotypes) and pseudolinkage caused by a reciprocal translocation between Chromosomes 6 and 7 in the parental species. RILs retained excess heterozygosity around the translocation breakpoints, which caused substantial pollen inviability when interstitial crossovers had not created compatible heterokaryotypic configurations. Strong transmission ratio distortion and interchromosomal linkage disequilibrium in both F2s and RILs identified a novel 2-locus genic incompatibility causing sex-independent gametophytic (haploid) lethality. The latter interaction eliminated 3 of the expected 9 F2 genotypic classes via F1 gamete loss without detectable effects on the pollen number or viability of F2 double heterozygotes. Along with the mapping of numerous milder incompatibilities, these key findings illuminate the complex genetics of plant hybrid breakdown and are an important step toward understanding the genomic consequences of natural hybridization in this model system.
Introduction
The evolution of postzygotic reproductive barriers, caused by incompatibilities between interacting genes and/or meiotic dysfunction in chromosomally divergent hybrids, is a key component of speciation (Coyne and Orr 2004; Fishman and Sweigart 2018; Coughlan and Matute 2020). Experimental hybridization of closely related species provides a window into the nature and origins of postzygotic barriers, revealing both the genetic mechanisms of genomic incompatibility and potential magnitude of barriers to gene flow upon secondary contact. The characterization of both early- and late-generation experimental hybrid populations is a particularly promising approach (Matute et al. 2020). Multigeneration approaches can capture extrinsic incompatibilities dependent on environmental context (Walter et al. 2020), replicate natural hybrid zones (Pritchard and Edmands 2012), and, under controlled growth conditions, allow comparison of early and late hybrid generations. Additional rounds of recombination and distinct assemblages of hybrid genotypes in later generations provide increased power to detect and localize incompatibilities (Moyle and Nakazato 2008). Furthermore, comparing patterns of hybrid breakdown across experimental generations can reveal how initial selection against incompatible genotypes shapes hybrid genomes and whether genetic and chromosomal incompatibilities quickly resolve (e.g. sort to parental genotypes) after initial hybridization or are maintained as potentially costly polymorphisms.
Fitness breakdown in experimental hybrids can be characterized in 2 complementary ways: by direct measurement of fertility, viability, and other fitness metrics and by characterizing deviations from expected Mendelian allele or genotype frequencies (transmission ratio distortion—TRD). TRD is a common feature of experimental hybrid populations, and interspecific mapping populations often exhibit significant distortion across a large fraction of their chromosomes (Fishman and McIntosh 2019). In plants, TRD at individual loci has many documented sources, including environmental selection (Yin et al. 2004), meiotic drive by chromosomes (Fishman and Saunders 2008), exposure of suppressed gamete killers (Koide, Ikenaga, et al. 2008), pollen competition (Fishman et al. 2008), and Dobzhansky–Muller incompatibilities that kill gametes or zygotes (Leppälä et al. 2008; Kerwin and Sweigart 2017). In some cases, strong Dobzhansky–Muller incompatibilities (e.g. gametic or gametophytic lethals) can generate both local distortion and linkage disequilibrium between physically unlinked loci if they kill a given multilocus genotypic class (Colomé-Tatché and Johannes 2016). Although differentiating among multiple mechanisms of hybrid TRD can be challenging, joint mapping of fitness traits and TRD in multiple generations increases power and precision to characterize intrinsic postzygotic barriers.
Along with genic Dobzhansky–Muller incompatibilities, chromosomal rearrangements can play important direct and indirect roles in postzygotic breakdown. Inversions often do not cause direct underdominant effects on hybrid fertility in plants (Fishman and Sweigart 2018; Huang and Rieseberg 2020; Zhang et al. 2021), but their suppression of recombination across large genomic regions in hybrids can link multiple incompatibility loci and extend their barrier effects (Noor et al. 2001; Rieseberg 2001; Livingstone and Rieseberg 2004). Reciprocal translocations tend not to be as common as inversions within species but often distinguish sister species of flowering plants (Grant 1971; Fishman et al. 2013; Ostevik et al. 2020). In contrast to inversions, translocations can directly cause meiotic dysfunction and sterility in F1 hybrids (Stathos and Fishman 2014); with or without crossovers, segregation of paired chromosomes with reciprocal translocations often produces at least 50% unbalanced gametes (Burnham 1956). Because novel translocations should be strongly disfavored by selection until reaching >50% frequency (Lande 1984), their initial establishment and spread in diverging populations have been a long-standing puzzle in evolutionary biology. However, some plant species maintain permanent translocation heterozygosity (Golczyk et al. 2014) and/or avoid adjacent segregation, so populations segregating for translocations may rapidly shift (genetically or epigenetically) to minimize the deleterious effects of heterokaryotypy. Thus, understanding how translocations contribute to postzygotic breakdown and genomic transmission after initial hybridization (both directly and indirectly) is an important step in understanding the processes that maintain plant species barriers.
Here, we investigate the patterns and underlying mechanisms of hybrid breakdown in multiple generations of hybrids between closely related monkeyflowers [Phin Mimulus section Erythranthe, Mimulus cardinalis, and M. parishii (also known as Erythranthe cardinalis and E. parishii)]. Despite dramatic differences in floral morphology (Beardsley et al. 2003; Fishman et al. 2015; Liang et al. 2022), hummingbird-pollinated M. cardinalis and selfer M. parishii hybridize naturally and show signatures of recent introgression where their ranges overlap in southern California (Nelson, Stathos, et al. 2021). The Erythranthe group, which also includes bee-pollinated M. lewisii, has been a long-standing model for understanding speciation by pollinator shifts and other ecological factors (Hiesey et al. 1971; Bradshaw et al. 1995; Bradshaw and Schemske 2003; Angert et al. 2008) and has recently emerged as a model system for studying Asteridae floral development (Yuan et al. 2016; Yuan 2019; Liang et al. 2022). Because phylogenomic analyses reveal substantial genomic reticulation among all taxa despite rapid morphological and structural divergence (Nelson, Stathos, et al. 2021), Erythranthe is a particularly rich system for investigating the origins, maintenance, and interactions of both pre- and postzygotic species barriers.
As a platform for investigations of both postzygotic barriers (this study) and dramatic floral, life history, and vegetative divergence, we generate dense genetic linkage maps of M. parishii × M. cardinalis F2 and recombinant inbred line (RIL) hybrids using genome-wide double-digest restriction-site associated DNA sequencing (ddRADseq) anchored in a new M. cardinalis genome assembly (mimubase.org). Previous coarse mapping in hybrids of each focal species with M. lewisii (Fishman et al. 2013, 2015), plus genotyping of breakpoint markers in a small number of M parishii × M. cardinalis hybrids (Stathos and Fishman 2014), suggest that these naturally hybridizing taxa are distinguished by at least one reciprocal translocation. High-resolution genome-wide mapping of male fertility (pollen viability and number) and TRD in each generation allows us to address specific hypotheses about putative barriers, identify major genic and chromosomal incompatibilities genome-wide, and track their evolution and influence from F2 to advanced-generation hybrids.
Specifically, we ask: are patterns of postzygotic breakdown (hybrid sterility and TRD) consistent in multiple environments (i.e. between different F2 populations) and through multiple generations of self-fertilization (F2 hybrids vs RILs)? In particular, are epistatic genic incompatibilities causing hybrid sterility rapidly lost during RIL construction, resulting in restored fertility but loss of incompatible genotypic combinations? Do chromosomes with reciprocal translocation sort to parental or other meiotically compatible karyotypes in advanced-generation hybrids, or does chromosomal underdominance persist? Does TRD and cross-chromosome linkage disequilibrium due to other sources dissipate (as might be the case with some postmating prezygotic barriers such as pollen competition) or amplify (as might occur with some recessive incompatibilities or directional selection) with multiple generations of selfing? Are major incompatibility loci shared with other Mimulus section Erythranthe hybrids, or are there novel postmating reproductive barriers unique to this florally divergent cross?
Methods
Study system and plant lines
The monkeyflowers of the M. cardinalis species complex (Mimulus section Erythranthe; Phrymaceae) are a well-established model system for understanding the genetics of floral evolution and speciation (Yuan 2019). Hummingbird-pollinated M. cardinalis has a broad latitudinal range in the western United States with distinct Arizonan, Southern Californian, and Sierran clades (Nelson, Stathos, et al. 2021). M. cardinalis is parapatric in the Sierras with bee-pollinated high-elevation specialist M. lewisii, where reproductive isolation is maintained by local adaptation to divergent habitats and pollinator preferences (Hiesey et al. 1971; Bradshaw and Schemske 2003; Ramsey et al. 2003), as well as 2 reciprocal translocations causing underdominant F1 hybrid sterility (Fishman et al. 2013; Stathos and Fishman 2014). M. parishii is a small-flowered annual selfer restricted to Southern California, where it cooccurs with M. cardinalis in ephemerally wet desert washes. Hybrids between M. cardinalis and M. parishii have been observed in the field, and local sharing of identical organellar genomes (as well as nuclear reticulation) indicates an extensive history of mating and introgression despite extreme differences in floral morphology (Nelson, Stathos, et al. 2021).
The plants in this study were all derived from 2 highly (>10 generations) inbred lines of Sierran M. cardinalis (CE10) and M. parishii (PAR), which were also used in previous investigations of species barriers (Bradshaw et al. 1998; Schemske and Bradshaw 1999; Bradshaw and Schemske 2003; Ramsey et al. 2003; Fishman et al. 2013, 2015; Nelson, Muir et al. 2021). We generated PAR × CE10 F1 hybrids by hand-pollination (with prior emasculation of the PAR seed parent in the bud) and F2 hybrids by self-pollination of F1 hybrids. The F2 hybrids were grown in 2 separate greenhouse common gardens at the University of Montana (UM-F2; total N = 524) and the University of Connecticut (UC-F2 N = 253), along with parental control lines, and were phenotyped for numerous floral and vegetative traits including the pollen fertility traits presented here. RILs were generated by single-seed-descent from additional F2 individuals grown at the University of Georgia and California State Polytechnic University, Pomona; a total of 167 RILs were formed through 3–6 generations of self-fertilization.
DNA extraction and sequencing
Genomic DNA was extracted from bud and leaf tissue of the greenhouse-grown F2 and RIL mapping populations using a cetyltrimethylammonium bromide (CTAB) and chloroform protocol modified for 96-well plates (dx.doi.org/10.17504/protocols.io.bgv6jw9e). We used a ddRADSeq protocol to generate genome-wide sequence clusters (tags), following the BestRAD library preparation protocol (dx.doi.org/10.17504/protocols.io.6awhafe), using restriction enzymes PstI and BfaI (New England Biolabs, Ipswich, MA, USA). Postdigestion, half plates of individual DNAs were labeled by ligation of 48 unique in-line barcoded adapters and then pooled for size selection. Libraries were prepared using NEBNext Ultra II library preparation kits for Illumina (New England BioLabs). Each pool was indexed with a unique NEBNext i7 adapter and an i5 adapter containing a degenerate barcode and PCR amplified with 12 cycles. The F2 libraries were size-selected to 200–700 bp using BluePippin 2% agarose cassettes (Sage Science, Beverly, MA, USA) and sequenced (150-bp paired-end reads) in a partial lane of an Illumina HiSeq4000 sequencer at GC3F, the University of Oregon Genomics Core Facility. The RIL library was sequenced (150-bp paired-ends) without size-selection on an Illumina HiSeq4000 at Genewiz (South Plainfield, NJ, USA).
Sequence processing and linkage mapping
After sequencing, two separate ddRAD data sets were analyzed: one with samples from both F2 populations (N = 283 UM-F2 hybrids with 3 M. parishii and 2 M. cardinalis controls, and 253 UC-F2 hybrids, with 3 each F1, M. parishii and M. cardinalis controls) and one with samples from the RIL population (N = 167). Samples from both data sets were demultiplexed using a custom Python script (dx.doi.org/10.17504/protocols.io.bjnbkman), trimmed using Trimmomatic (Bolger et al. 2014), mapped to the M. cardinalis CE10 v2.0 reference genome (http://mimubase.org/FTP/Genomes/CE10g_v2.0) using the Burrows-Wheeler Aligner maximal exact match (bwa-mem) algorithm and indexed using SAMtools (Li et al. 2009). The RIL data set was also filtered in SAMtools using a mapping quality ≥29. We called SNPs in both data sets using HaplotypeCaller in GATK v3.3 in F2s, v4.1.8.1 in RILs (McKenna et al. 2010).
Next, we performed a series of filtering steps to generate sets of high-quality SNPs. In the F2 dataset, we filtered using vcftools (Danecek et al. 2011), retaining sites with read depth ≥5, mapping quality ≥10, and <40% missing data. We also filtered out loci deviating from Hardy–Weinberg Equilibrium at P < 0.00005; this threshold was empirically chosen to only remove rare clusters of “bad” SNPs (generally entire tags with high excess heterozygosity due to cross-mapping of reads) deviating strongly from the overall trends in transmission distortion across chromosomes (Supplementary Fig. 1). In the RIL data set, we filtered a combined GVCF file using default quality control parameters in GATK, retaining sites with read depth ≥4 × N (with N = number of RIL samples) and <10% missing genotypes. For both data sets, we used custom scripts to remove sites that were not polymorphic in the parents and heterozygous in the F1 hybrids (F2: https://github.com/bergcolette/F2_genotype_processing). We excluded individuals from the F2 data set with >10% missing data and from the RIL dataset with low coverage, high missingness, or excessive heterozygosity (>50%, indicating line contamination). These filtering steps produced an F2 data set with 18,119 SNPs (N = 252 UM-F2 and 253 UC-F2) and an RIL data set with 47,851 SNPs (N = 145).
To produce sets of high-quality marker genotypes for mapping, we binned each data set into 18-SNP windows using custom Python and R scripts (provided at github links above), requiring ≥8 sites to have SNP genotype calls to assign a windowed genotype. In the F2 binning script, M. cardinalis homozygotes were coded as 2, M. parishii homozygotes as 0, and heterozygotes as 1. We called windows with mean values <0.2 as parishii homozygotes, >1.8 as cardinalis homozygotes, and between 0.8 and 1.2 as heterozygotes. Windows with means outside of these ranges were coded as missing genotypes. For the RILs, we required ≥88% of SNP calls to match each other to assign the window homozygous genotype (e.g. 16/18 sites must be called M. parishii homozygotes to assign that window as M. parishii; the same is true for heterozygotes and M. cardinalis homozygotes; https://github.com/vasotola/GenomicsScripts).
We generated linkage maps for each data set using Lep-MAP3 (Rastas 2017). First, we used the SeparateChromosomes2 module to assign markers to linkage groups (F2: LodLimit = 25, theta = 3, RIL: LodLimit = 28, theta = 0.2). In the RIL data set, 10 markers were assigned to linkage groups inconsistent with the reference genome assembly; we manually reassigned these markers to linkage groups corresponding to their reference assembly chromosomes. Next, we performed iterative ordering using the OrderMarkers2 module (Kosambi mapping function; 6 iterations/per linkage group in the F2s, 10 in the RILs); the order with the highest likelihood for each linkage group was chosen. This resulted in an F2 map with 997 markers in 7 linkage groups and an RIL map with 2,535 markers in 8 linkage groups. In the RIL data set, the genotype matrix output by Lep-MAP3 differed in 2 important respects from the input file. First, due to stringent thresholds for calling windowed genotypes, our input file includes a high percentage of missing data (23% of genotypes are coded as “no call”), whereas the output file contains no missing data (Lep-MAP3 converts each “no call” genotype to a called genotype). Second, the Lep-MAP3 output file contains more heterozygous genotype calls than the input file. The reason for this increase in heterozygosity is that Lep-MAP3 disproportionately converts “no call” genotypes to heterozygotes: relative to the input file, the output genotype matrix includes 115% more heterozygotes, compared to only 18% more M. cardinalis homozygotes and 20% more M. parishii homozygotes. Notably, Lep-MAP3 frequently converted “no call” genotypes to heterozygotes when they occur at single markers between recombination breakpoints. Because most recombinational switches in this RIL population are between alternative homozygotes, any window that contains an actual breakpoint will carry a mixture of M. cardinalis and M. parishii homozygotes at the 18 SNPs (and thus be coded as “no call” in our windowed genotype matrix). To circumvent these problems, for all downstream analyses, we used a modified version of the genotype matrix output from Lep-MAP3 in which genotypes were recoded as “no call” as in the input file.
TRD and linkage disequilibrium
Genotype frequencies were calculated, plotted, and tested for significant deviation from Mendelian expectations (X2 with 2 df) separately for the 2 F2 populations. TRD due solely to extrinsic factors (e.g. selection on parental alleles affecting germination under different conditions, environmentally dependent hybrid incompatibilities) may be distinct between the F2 growouts (Fishman and McIntosh 2019), whereas TRD due to intrinsic genic or chromosomal incompatibilities may be more likely to be shared. For the RILs, we conducted parallel tests with expectations for alternate homozygotes set to 0.47 and heterozygotes to 0.06, which is the expectation given our final composition of RILs (9 individuals with 3 generations of selfing, 116 with 4, 19 with 5, and 1 individual with 6 generations of selfing). For all sets, we used both uncorrected (a = 0.01; critical value = 6.635) and stringent (Bonferonni-corrected; F2 critical value = 16.442, RIL critical value = 18.217) chi-squared tests to assess the significance of distortion.
We used the package pegas (Paradis 2010) in the program R (R Core Team 2021) to calculate pairwise linkage disequilibrium (r) between all markers within the UM-F2 and RIL mapping populations. To minimize false positives driven by the large number of pairwise comparisons, we plotted only those r values greater than the 95% quantile after bootstrapping 1,000 times. All plots were visualized with R.
QTL mapping of pollen traits
In the UM-F2 and RIL populations, we directly assessed male fertility by collecting all 4 anthers of the first flower from each plant into 50 mL of lactophenol-aniline blue dye. We counted viable (darkly stained) and inviable (unstained) pollen grains using a hemocytometer (≥100 grains/flower). We estimated total pollen grains per flower (count per mL × 50) and pollen viability as viable grains/total counted. For a handful of individuals with <100 pollen grains counted, pollen viability was scored as missing data. Phenotyping in the RIL population was performed on siblings or selfed progeny of the individuals genotyped. In a few cases in the RILs (N = 12), the first flower had <100 pollen grains, so both traits were instead collected from the second flower.
We mapped pollen quantitative trait loci (QTLs) in Windows QTL Cartographer 2.5 (Wang et al. 2012) using composite interval mapping (CIM; Zeng 1993, 1994), with forward–backward stepwise regression, a window size of 10 cM, 5 background markers, and a 1-cM walk speed. We used permutations (N = 1000) to set genome-wide significance thresholds for QTL peaks and calculated 1.5-LOD drops to determine confidence intervals for QTL locations. Because pollen viability QTLs exhibited complex interactions in F2s (see Results), we directly estimated QTL effects (at each peak marker) and interactions using the Generalized Linear Model module in JMP16 (SAS Institute, Cary, NC, USA, starting with a full factorial model of all QTLs and two-way interactions and removing nonsignificant (P > 0.05) interactions. To test for pollen viability signatures of a 2-locus gametic incompatibility (LG4–LG8) detected from TRD and linkage disequilibrium (LD) patterns, we contrasted Least Squared Means for each extant F2 class from an ANOVA also including the large-effect translocation breakpoint marker at 60.55 Mb on Chr 6.
QTL mapping of underdominant effects in RILs has low power due to (relatively) few heterozygotes, plus most RIL QTL-mapping algorithms (including those in WinQTLCart) exclude nonhomozygous genotypes. To directly test for persistent underdominant effects of the Chr 6–7 translocation in RILs, we screened for genotype-pollen viability associations at uniquely positioned markers with a joint sample size >70 across the breakpoint region (57.07–61.08 Mb of Chr6 and 7.51–13.05 of Chr 7; n = 42), using t-tests in the response screening module in JMP16 (SAS Institute, Cary, NC, USA). However, we caution that because RIL hybrid male sterility was measured on siblings or descendants of the genotyped individuals, underdominant effects will likely be underestimated. That is, regions genotyped as heterozygous might actually be homozygous in phenotyped individuals, potentially explaining why a few of them are highly fertile. We conservatively controlled for multiple tests by using an false discovery rate (FDR)-corrected P-value of 0.05.
Results
Comparative linkage mapping
Across most of the genome, the F2 and RIL genetic maps are highly collinear, with linkage groups and marker order largely reconstructing the physical order of the 8 chromosomes of the M. cardinalis reference genome (Supplementary Figs. 2 and 3). The notable exception to this pattern involves markers on Chromosomes (Chr) 6 and 7, where previous work suggested an M. cardinalis-specific reciprocal translocation vs both M. parishii and M. lewisii (Fishman et al. 2013, 2015; Stathos and Fishman 2014). There is no single linear order of markers in heterozygotes for a reciprocal translocation (Livingstone et al. 2000), which generates linkage across the entire involved chromosomes during F1 meiosis and a continuous LG 6&7 in the F2 hybrids. In the RILs, the portion of Chr 6 distal to the translocation forms a distinct linkage group, while tight linkage between markers on the end of Chr 6 and the first 8 Mb of Chr 7 in M. cardinalis (purple and green/red segments, respectively, in Fig. 1) generates a second composite linkage group. The RIL map is ∼35% longer than the F2 map (total length = 892.95 vs 661.37 cM), consistent with the additional generations of recombination. In both maps, recombination rates per Mb are dramatically lower (near zero) across the central 20–40 Mb of each chromosome (Supplementary Fig. 2). This pattern is also evident for all chromosomes other than Chr 7 in an intraspecific M. cardinalis map (Nelson, Muir, et al. 2021; Nelson, Stathos, et al. 2021), consistent with a primarily metacentric chromosomal structure in Erythranthe species.
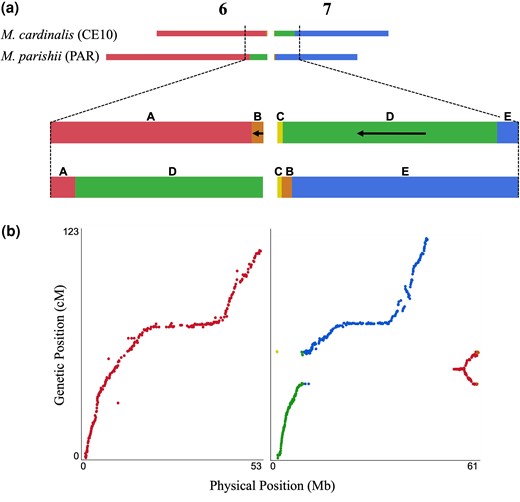
Mimulus cardinalis (CE10) and M. parishii (PAR) exhibit a reciprocal translocation between Chromosomes (Chr) 6 and 7. a) Schematic showing the affected chromosomes and regions with the CE10 order labeled A to E from left to right: a distal region of Chr 6 in PAR has moved to Chr 7 in CE10 (segment D) and a smaller region of Chr 7 in PAR has moved to the end of Chr 6 in CE10 (segment B). Black arrows indicate blocks where the CE10 order is inverted relative to the ancestral PAR order. Genome coordinates for the blocks are given in Table S1. b) Plot of physical position (Mb, CE10g_v2.0) by genetic position for RIL linkage groups (LGs) 6 and 6&7. SNP markers (dots) are colored by the locations defined in (a). RIL LG6 includes only markers from the collinear region of Chr 6. RIL LG6&7 includes all markers from the translocated regions, but the genetic order reflects multiple distinct linkages associated with the divergent parental chromosomes (e.g., markers near the translocation breakpoint at ∼60 Mb in CE10 are tightly linked to markers in all other segments).
Genome-wide patterns of TRD
All linkage groups, with the exception of LG2 in the UM-F2 mapping population, exhibited strong TRD in all 3 maps (Fig. 2; Supplementary Fig. 4, Supplementary Tables 2 and 3). Patterns of TRD were very similar between the 2 F2 populations, suggesting that TRD primarily reflects shared aspects of the hybrid context (e.g. pollen competition or incompatibilities) vs local environmental selection (e.g. germination conditions different between the 2 growouts). The 2 F2 growouts shared regions of excess M. cardinalis homozygosity on LGs 1, 4, 5, and 6 (distal to translocation), excess M. parishii on LG8, and excess heterozygosity on LG3 and in the translocation region on LG6&7. The UC-F2 also exhibited excess heterozygosity and M. cardinalis alleles on LG2. Additional generations of recombination and selection in the RIL population strengthened and refined the locations of shared TRD peaks on LGs 1, 2, 3, 6&7, and 8. However, RILs exhibited a novel region of very strong M. parishii excess on one arm of LG5; this signal was absent or opposite in the F2 hybrids, suggesting an advantage of M. parishii alleles in this region specific to either the environment or the increasingly homozygous genetic background of RIL formation. In addition to local peaks of TRD, the RIL population exhibited slightly higher residual heterozygosity across the genome (9.3%) than the Mendelian expectation [6% (Fig. 2)]. While some of this overall excess is due to major local peaks in heterozygosity shared with the F2 hybrids (e.g. on LG3 and in the LG6&7 breakpoint region, Fig. 2), the remainder may reflect genome-wide selection against weakly incompatible parental homozygous combinations in the RILs (Thompson et al. 2022).
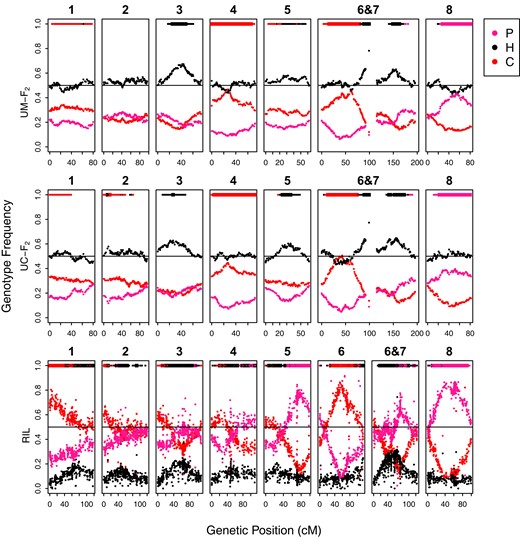
Genotype frequencies across each linkage group in the F2 and RIL populations. Red dots indicate homozygous M. cardinalis, pink is homozygous M. parishii, and black is heterozygous. The bars at the top of each plot indicate regions with significant transmission ratio distortion by Χ2 tests at α= 0.01 (thin lines, critical value = 6.635) and at a more stringent, Bonferonni-corrected level (thick lines, F2 critical value = 16.442, RIL critical value = 18.217).
Tests of hybrid incompatibilities as the source of TRD
To investigate whether peaks of TRD were caused by hybrid incompatibilities (e.g. gametic or zygotic lethals), we characterized pairwise LD between all markers in the UM-F2 and RIL populations (Fig. 3; Supplementary Tables 4 and 5). In the F2 hybrids, there was significant off-diagonal LD over most of LG6 and 7 (due to the translocation), between LG4 and the center of LG8, and between the distal ends of LG2 and LG8. In the RILs, parallel TRD on different chromosomes (e.g. near fixation of M. parishii alleles on both LG5 and LG8) can cause high interchromosome LD even without epistatic selection during RIL formation. Thus, it is not surprising that, in addition to sharing the F2 LD regions, the RILs exhibit more abundant and diffuse LD genome-wide (Fig. 2, Supplementary Table 5). However, some of these regions may house multilocus incompatibilities that are too mild and/or late-acting to be statistically detected in the F2 generation (e.g. F2 sporophytic hybrid sterility).
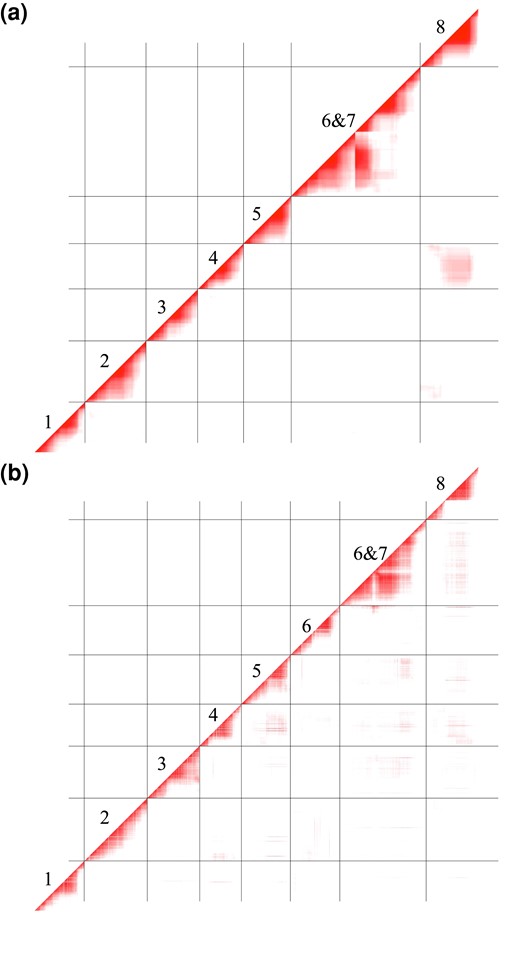
Linkage disequilibrium heatmaps for M. parishii × M. cardinalis UM-F2 a) and RIL b) populations, ordered by linkage group. Data shown are r values greater than the 95% quantile after bootstrapping (95% F2: 0.176; 95% RIL: 0.305). Values range from 0 (white) to 1 (dark).
Strong and opposite TRD on LG4 (excess M. cardinalis) and LG8 (excess M. parishii) in the F2 hybrids (Fig. 2), as well as high and persistent LD between these regions (Fig. 3), suggested a major 2-locus incompatibility. To investigate further, we tallied all 2-locus genotype combinations at TRD-peak markers (6.8 Mb on Chr 4, 40 Mb on Chr 8; Fig. 2; Supplementary Fig. 4) in both the UM-F2 and RIL populations (Table 1). These regions clearly deviate from the Mendelian expectation of independent segregation, with 3 of the expected 9 genotypic classes entirely absent from both populations (Table 1). To test for the mechanism, we calculated the expected genotype frequencies under 2 different scenarios: (1) a gametic incompatibility in which all haploid gametes (both pollen and ovules) are inviable if they carry the M. parishii–M. cardinalis allelic combination at LG4 and LG8 (i.e. P;C gametes missing), and (2) a 2-locus dominant zygotic incompatibility in which hybrids die if they carry at least one M. parishii allele at LG4 and at least one M. cardinalis allele at LG8 (P_; C_ zygotes die). In the F2 population, genotype frequencies best fit the sex-independent gametic incompatibility model, which distinctively predicts the numerous double heterozygotes observed (Table 1). The RIL genotypes are also most consistent with a sex-independent gametophytic mechanism; however, the observed excess of M. parishii transmission on LG8 exceeds that expected from a gametic incompatibility alone.
Two-locus segregation pattern for loci on LG4 (6.75–7.65 Mb) and LG8 (12.87–40.08 Mb) showing opposite single-locus TRD and strong LD in M. parishii × M. cardinalis F2 hybrids and RILs.
| UM-F2 population | RIL population | |||||||
|---|---|---|---|---|---|---|---|---|
| Genotype (LG4;LG8) | Observed | Mendelian | P;C gametes inviable | P_;C_ zygotes inviable | Observed | Mendelian | P;C gametes inviable | P_;C_ zygotes inviable |
| C;C | 24 | 16 | 29 | 37 | 18 | 31 | 42 | 42 |
| H;C | 0 | 32 | 0 | 0 | 0 | 2 | 0 | 0 |
| P;C | 0 | 16 | 0 | 0 | 0 | 31 | 0 | 0 |
| C;H | 61 | 32 | 57 | 73 | 6 | 2 | 4 | 6 |
| H;H | 53 | 64 | 57 | 0 | 6 | 4 | 4 | 0 |
| P;H | 0 | 32 | 0 | 0 | 0 | 2 | 0 | 0 |
| C;P | 27 | 16 | 29 | 37 | 40 | 31 | 42 | 42 |
| H;P | 58 | 32 | 57 | 73 | 21 | 2 | 4 | 6 |
| P;P | 34 | 16 | 29 | 37 | 47 | 31 | 42 | 42 |
| UM-F2 population | RIL population | |||||||
|---|---|---|---|---|---|---|---|---|
| Genotype (LG4;LG8) | Observed | Mendelian | P;C gametes inviable | P_;C_ zygotes inviable | Observed | Mendelian | P;C gametes inviable | P_;C_ zygotes inviable |
| C;C | 24 | 16 | 29 | 37 | 18 | 31 | 42 | 42 |
| H;C | 0 | 32 | 0 | 0 | 0 | 2 | 0 | 0 |
| P;C | 0 | 16 | 0 | 0 | 0 | 31 | 0 | 0 |
| C;H | 61 | 32 | 57 | 73 | 6 | 2 | 4 | 6 |
| H;H | 53 | 64 | 57 | 0 | 6 | 4 | 4 | 0 |
| P;H | 0 | 32 | 0 | 0 | 0 | 2 | 0 | 0 |
| C;P | 27 | 16 | 29 | 37 | 40 | 31 | 42 | 42 |
| H;P | 58 | 32 | 57 | 73 | 21 | 2 | 4 | 6 |
| P;P | 34 | 16 | 29 | 37 | 47 | 31 | 42 | 42 |
For each population, observed counts of individuals with each of the 9 possible genotypes (C = M. cardinalis homozygote, P = M. parishii homozygote, and H = heterozygote) are compared with expected counts under normal Mendelian inheritance (Mendelian), expected counts under a model of sex-independent gametic inviability (P;C gametes inviable), and expected counts under a model of dominant zygote inviability (P_;C_ zygotes inviable). Counts are shown rounded to the nearest integer. See Supplementary Table 6 for unrounded counts and frequencies.
Two-locus segregation pattern for loci on LG4 (6.75–7.65 Mb) and LG8 (12.87–40.08 Mb) showing opposite single-locus TRD and strong LD in M. parishii × M. cardinalis F2 hybrids and RILs.
| UM-F2 population | RIL population | |||||||
|---|---|---|---|---|---|---|---|---|
| Genotype (LG4;LG8) | Observed | Mendelian | P;C gametes inviable | P_;C_ zygotes inviable | Observed | Mendelian | P;C gametes inviable | P_;C_ zygotes inviable |
| C;C | 24 | 16 | 29 | 37 | 18 | 31 | 42 | 42 |
| H;C | 0 | 32 | 0 | 0 | 0 | 2 | 0 | 0 |
| P;C | 0 | 16 | 0 | 0 | 0 | 31 | 0 | 0 |
| C;H | 61 | 32 | 57 | 73 | 6 | 2 | 4 | 6 |
| H;H | 53 | 64 | 57 | 0 | 6 | 4 | 4 | 0 |
| P;H | 0 | 32 | 0 | 0 | 0 | 2 | 0 | 0 |
| C;P | 27 | 16 | 29 | 37 | 40 | 31 | 42 | 42 |
| H;P | 58 | 32 | 57 | 73 | 21 | 2 | 4 | 6 |
| P;P | 34 | 16 | 29 | 37 | 47 | 31 | 42 | 42 |
| UM-F2 population | RIL population | |||||||
|---|---|---|---|---|---|---|---|---|
| Genotype (LG4;LG8) | Observed | Mendelian | P;C gametes inviable | P_;C_ zygotes inviable | Observed | Mendelian | P;C gametes inviable | P_;C_ zygotes inviable |
| C;C | 24 | 16 | 29 | 37 | 18 | 31 | 42 | 42 |
| H;C | 0 | 32 | 0 | 0 | 0 | 2 | 0 | 0 |
| P;C | 0 | 16 | 0 | 0 | 0 | 31 | 0 | 0 |
| C;H | 61 | 32 | 57 | 73 | 6 | 2 | 4 | 6 |
| H;H | 53 | 64 | 57 | 0 | 6 | 4 | 4 | 0 |
| P;H | 0 | 32 | 0 | 0 | 0 | 2 | 0 | 0 |
| C;P | 27 | 16 | 29 | 37 | 40 | 31 | 42 | 42 |
| H;P | 58 | 32 | 57 | 73 | 21 | 2 | 4 | 6 |
| P;P | 34 | 16 | 29 | 37 | 47 | 31 | 42 | 42 |
For each population, observed counts of individuals with each of the 9 possible genotypes (C = M. cardinalis homozygote, P = M. parishii homozygote, and H = heterozygote) are compared with expected counts under normal Mendelian inheritance (Mendelian), expected counts under a model of sex-independent gametic inviability (P;C gametes inviable), and expected counts under a model of dominant zygote inviability (P_;C_ zygotes inviable). Counts are shown rounded to the nearest integer. See Supplementary Table 6 for unrounded counts and frequencies.
Genetics of male fertility traits in hybrids
F2 Hybrids (UM-F2) exhibited substantial variation for pollen viability (mean = 0.48, but significantly nonnormal by Shapiro–Wilks test, P < 0.0001; Supplementary Fig. 5), whereas both parental lines were relatively fertile (mean = 0.73 for CE10 and 0.84 for PAR, N = 2 and 8, respectively). A major underdominant pollen viability QTL mapped to the translocation breakpoints on LG6&7, along with smaller QTLs on LGs 2, 3, and 4 (Fig. 4; Table 2). To investigate the contribution of epistasis to the observed pollen inviability and test for unlinked modifiers of the translocation-associated underdominant sterility, we conducted a model selection analysis starting with a factorial model of the 4 peak markers and all 2-way interactions. Only interactions including LG6&7 were significant at P = 0.05 (along with all 4 single QTLs), so we interpret QTL effects under that reduced model (r2 = 0.42). The LG6&7 QTL was strongly underdominant (Table 2), while the other 3 pollen viability loci exhibited at least partial dominance of the M. parishii allele, with M. cardinalis homozygotes significantly more fertile than heterozygotes and M. parishii on LG4, significantly less fertile on LG3, and heterozygotes most fertile (but not significantly different from M. cardinalis) on LG2. The interactions involving the QTL on LG6&7 did not appear to modify the low fertility of translocation heterozygotes, which was consistent across genetic backgrounds (i.e. LG6&7 heterozygotes with alternative genotypes elsewhere were indistinguishable by Tukey's Honest Significant Difference tests; Supplementary Fig. 6). Instead, the interactions involved idiosyncratic combinations of other genotypes, generally with low sample sizes; for example, F2 hybrids with M. parishii genotype at QTL PV3 and M. cardinalis at PV6&7 were about twice as fertile [least squared mean (LSM) = 0.86 ± 0.11, N = 2] as those with the opposite homozygous combination (LSM = 0.45 ± 0.06, N = 8).
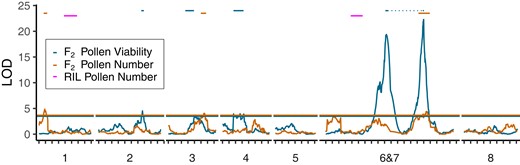
QTLs for pollen viability (blue) and pollen number (orange) in M. parishii × M. cardinalis F2 hybrids, as well as the locations of 2 pollen number QTLs in descendant RILs (pink, aligned by the physical position of markers). Horizontal lines denote the permutation-derived (N = 1000) LOD significance thresholds for each F2 trait, and the bars indicate QTL confidence intervals (1.5 LOD drop). The single major underdominant pollen viability QTL associated with the LG6&7 translocation maps to markers near both breakpoints (QTL locations connected by light dotted line), which cannot be linearly resolved in F2 hybrids. Because the RIL QTL-mapping algorithm excluded heterozygotes, no RIL pollen viability QTLs were detected despite the negative effects of retained heterozygosity (see text).
Male fertility QTL locations and effects in M. parishii × M. cardinalis UM-F2 and RIL populations.
| Pop. | Trait | LG/Chrom | cM position (1.5 LOD) | Mb position (1.5 LOD) | r2 | a | d | Summary |
|---|---|---|---|---|---|---|---|---|
| F2 | Pollen viability | 2 | 67.02 (65.3–69.1) | 43.96 (43.28–45.27) | 0.076 | 0.022 | 0.111 | H+ |
| 3 | 40.59 (39.23–47.66) | 13.84 (12.30–42.72) | 0.062 | −0.007 | 0.105 | H+ | ||
| 4 | 26.93 (16.91–33.22) | 7.65 (3.44–13.26) | 0.071 | −0.073 | −0.040 | P−, dom. | ||
| 6&7* | 92.88 (91.39–94.88) + 149.95 (148.58–150.15) | 6_60.55 (59.99–60.55) + 7_9.92 (6.73–10.10) | 0.315 | −0.027 | −0.231 | H− | ||
| Pollen count | 1 | 8.95 (7.3–12.59) | 1.42 (1.32–2.48) | 0.076 | −8044 | −6254 | P−, dom. | |
| 3 | 54.64 (49.84–57.78) | 46.39 (43.46–47.12) | 0.061 | −6764 | 8769 | P−, rec. | ||
| 6&7 | 155.55 (141.9–159.28) | 7_15.10 (4.13–21.30) | 0.064 | 1863 | 13,059 | H + | ||
| RIL | Pollen count | 1 | 50.37 (38.29–58.30) | 8.29 (5.52–10.96) | 0.111 | −5819 | P− | |
| 6 | 45.88 (38.64–56.53) | 7.54 (4.83–11.24) | 0.374 | −6830 | P− |
| Pop. | Trait | LG/Chrom | cM position (1.5 LOD) | Mb position (1.5 LOD) | r2 | a | d | Summary |
|---|---|---|---|---|---|---|---|---|
| F2 | Pollen viability | 2 | 67.02 (65.3–69.1) | 43.96 (43.28–45.27) | 0.076 | 0.022 | 0.111 | H+ |
| 3 | 40.59 (39.23–47.66) | 13.84 (12.30–42.72) | 0.062 | −0.007 | 0.105 | H+ | ||
| 4 | 26.93 (16.91–33.22) | 7.65 (3.44–13.26) | 0.071 | −0.073 | −0.040 | P−, dom. | ||
| 6&7* | 92.88 (91.39–94.88) + 149.95 (148.58–150.15) | 6_60.55 (59.99–60.55) + 7_9.92 (6.73–10.10) | 0.315 | −0.027 | −0.231 | H− | ||
| Pollen count | 1 | 8.95 (7.3–12.59) | 1.42 (1.32–2.48) | 0.076 | −8044 | −6254 | P−, dom. | |
| 3 | 54.64 (49.84–57.78) | 46.39 (43.46–47.12) | 0.061 | −6764 | 8769 | P−, rec. | ||
| 6&7 | 155.55 (141.9–159.28) | 7_15.10 (4.13–21.30) | 0.064 | 1863 | 13,059 | H + | ||
| RIL | Pollen count | 1 | 50.37 (38.29–58.30) | 8.29 (5.52–10.96) | 0.111 | −5819 | P− | |
| 6 | 45.88 (38.64–56.53) | 7.54 (4.83–11.24) | 0.374 | −6830 | P− |
Additive effects (a) and dominances (d) are negative when the M. parishii allele has a lower value. The summary column indicates the overall pattern of inheritance from a comparison of 2a vs a + d (H+ and H− mean overdominance and underdominance, respectively, while P− means the PAR allele reduces the trait value, and dom. and rec. (if present) indicates the overall dominance or recessivity, respectively, of the PAR allele. For the translocation-associated pollen viability QTL (6&7*) with a split location, effects are given for the higher peak. Dominance was not calculated in the RIL QTL model, which excluded heterozygotes.
Male fertility QTL locations and effects in M. parishii × M. cardinalis UM-F2 and RIL populations.
| Pop. | Trait | LG/Chrom | cM position (1.5 LOD) | Mb position (1.5 LOD) | r2 | a | d | Summary |
|---|---|---|---|---|---|---|---|---|
| F2 | Pollen viability | 2 | 67.02 (65.3–69.1) | 43.96 (43.28–45.27) | 0.076 | 0.022 | 0.111 | H+ |
| 3 | 40.59 (39.23–47.66) | 13.84 (12.30–42.72) | 0.062 | −0.007 | 0.105 | H+ | ||
| 4 | 26.93 (16.91–33.22) | 7.65 (3.44–13.26) | 0.071 | −0.073 | −0.040 | P−, dom. | ||
| 6&7* | 92.88 (91.39–94.88) + 149.95 (148.58–150.15) | 6_60.55 (59.99–60.55) + 7_9.92 (6.73–10.10) | 0.315 | −0.027 | −0.231 | H− | ||
| Pollen count | 1 | 8.95 (7.3–12.59) | 1.42 (1.32–2.48) | 0.076 | −8044 | −6254 | P−, dom. | |
| 3 | 54.64 (49.84–57.78) | 46.39 (43.46–47.12) | 0.061 | −6764 | 8769 | P−, rec. | ||
| 6&7 | 155.55 (141.9–159.28) | 7_15.10 (4.13–21.30) | 0.064 | 1863 | 13,059 | H + | ||
| RIL | Pollen count | 1 | 50.37 (38.29–58.30) | 8.29 (5.52–10.96) | 0.111 | −5819 | P− | |
| 6 | 45.88 (38.64–56.53) | 7.54 (4.83–11.24) | 0.374 | −6830 | P− |
| Pop. | Trait | LG/Chrom | cM position (1.5 LOD) | Mb position (1.5 LOD) | r2 | a | d | Summary |
|---|---|---|---|---|---|---|---|---|
| F2 | Pollen viability | 2 | 67.02 (65.3–69.1) | 43.96 (43.28–45.27) | 0.076 | 0.022 | 0.111 | H+ |
| 3 | 40.59 (39.23–47.66) | 13.84 (12.30–42.72) | 0.062 | −0.007 | 0.105 | H+ | ||
| 4 | 26.93 (16.91–33.22) | 7.65 (3.44–13.26) | 0.071 | −0.073 | −0.040 | P−, dom. | ||
| 6&7* | 92.88 (91.39–94.88) + 149.95 (148.58–150.15) | 6_60.55 (59.99–60.55) + 7_9.92 (6.73–10.10) | 0.315 | −0.027 | −0.231 | H− | ||
| Pollen count | 1 | 8.95 (7.3–12.59) | 1.42 (1.32–2.48) | 0.076 | −8044 | −6254 | P−, dom. | |
| 3 | 54.64 (49.84–57.78) | 46.39 (43.46–47.12) | 0.061 | −6764 | 8769 | P−, rec. | ||
| 6&7 | 155.55 (141.9–159.28) | 7_15.10 (4.13–21.30) | 0.064 | 1863 | 13,059 | H + | ||
| RIL | Pollen count | 1 | 50.37 (38.29–58.30) | 8.29 (5.52–10.96) | 0.111 | −5819 | P− | |
| 6 | 45.88 (38.64–56.53) | 7.54 (4.83–11.24) | 0.374 | −6830 | P− |
Additive effects (a) and dominances (d) are negative when the M. parishii allele has a lower value. The summary column indicates the overall pattern of inheritance from a comparison of 2a vs a + d (H+ and H− mean overdominance and underdominance, respectively, while P− means the PAR allele reduces the trait value, and dom. and rec. (if present) indicates the overall dominance or recessivity, respectively, of the PAR allele. For the translocation-associated pollen viability QTL (6&7*) with a split location, effects are given for the higher peak. Dominance was not calculated in the RIL QTL model, which excluded heterozygotes.
The RILs were, on average, highly fertile compared to the F2 hybrids (mean pollen viability = 0.76; Supplementary Fig. 5), and we detected no significant RIL pollen viability QTLs in WinQTLCart (Supplementary Fig. 7). However, marker genotypes across the translocation region exhibited significant associations with pollen viability (26/42 significant at uncorrected P < 0.05, 18/42 at FDR-corrected P < 0.05). For example, at the peak marker close to the Chr7 breakpoint (Chr7_10.47), heterozygotes were ∼25% less pollen-fertile, on average, than the parental genotypes (H: 0.64 ± 0.04, n = 18; C = 0.79 ± 0.04, n = 18, P = 0.79 ± 0.03, n = 32). Markers on Chr6 showed parallel patterns, though they showed less distortion against M. cardinalis homozygotes. Although some RILs genotyped as heterozygous for LG6&7 markers were highly fertile, consistent with segregation to homozygosity between the genotyped and phenotyped generation (see Methods) and/or stabilization of compatible chromosomal configurations, the underdominant fertility effects of the translocation persist.
Consistent with the parental species' difference in mating system, pollen number in the UM-F2 experiment was nearly seven-fold higher in M. cardinalis than in M. parishii, though the sample size was too small to confirm this statistically (mean pollen number: CE10 = 50.38 × 103, N = 2; PAR = 7.62 × 103, N = 8). Pollen number in the F2 hybrids was intermediate (mean =38.94 × 103, N = 245; Supplementary Fig. 5), and we mapped 3 QTLs that together explain only ∼20% of the F2 variance (Fig. 4; Table 2), consistent with a polygenic basis to quantitative divergence in pollen production. For 2 of the 3 F2 QTLs, M. parishii homozygotes had the lowest pollen counts, while the other (on LG6&7) appeared overdominant (heterozygotes producing most pollen), and there were no significant interactions among QTLs in pairwise tests In the RIL grow-out, pollen counts were overall higher but followed the same pattern, with M. cardinalis ∼7× as pollen-productive and hybrids generally intermediate (mean pollen number: RILs = 19.02 × 103, N = 100; CE10 = 32.26 × 103 N = 17; PAR = 4.39 × 103, N = 11; Supplementary Fig. 5). QTLs on LG1 and LG6 together explain ∼48% of the RIL variance in pollen production, with M. parishii homozygotes at each producing less pollen and no evidence of interactions (Table 2; Supplementary Fig. 7). The genetic bases of variation in pollen number and viability were largely independent, with no significant phenotypic correlation and, with the exception of the region of suppressed recombination near translocation breakpoints, no QTL coincidence in the F2 hybrids (Table 2; Fig. 4).
Notably, F2 pollen viability (or pollen number) QTLs cannot account for the LG4-LG8 gametic incompatibility. Although the LG4 pollen viability QTL is coincident with the LG4 gametic incompatibility locus at ∼6.8 Mb, individuals carrying at least one M. parishii allele have reduced pollen viability regardless of their LG8 genotype, producing a M. parishii-dominant effect (Table 2; Supplementary Fig. 6b). Due to the lack of F1-recombinant P;C gametes (and P;C, H:C, and P;H F2s), the H;H F2 class is the only one capable of forming the incompatible gamete combination (i.e. one-fourth should be missing or sterile). Instead, P;P individuals (all gametes compatible) were just as male-fertile (mean pollen viability = 0.48 ± 0.03 SE, N = 24) as the H:H class (mean = 0.49 ± 0.02, N = 53), consistent with an alternative cause.
Discussion
M. parishii × M. cardinalis genetic maps and RILs: a resource for understanding extreme floral divergence and speciation
The monkeyflowers of Mimulus section Erythranthe have been a model system for understanding plant speciation for over 5 decades, with a primary focus on the floral and elevational adaptation of parapatric M. lewisii and M. cardinalis. Here, we develop foundational genetic maps and RIL resources for the far more florally divergent, yet sympatric and hybridizing, pair of M. parishii and M. cardinalis. Despite structural divergence and substantial hybrid incompatibilities, we constructed consistent and high-quality ddRAD-based genetic maps to guide future investigations of the genetic and molecular mechanisms of floral, mating system, and life history evolution. Previous genetic maps in the Erythranthe group have used sparse PCR-based (Bradshaw et al. 1998; Fishman et al. 2013, 2015) or targeted capture markers; successful extension of this high-density gene-centric ddRAD genotyping technique from M. guttatus (Kolis et al. 2022) to the larger genomes of M. cardinalis and M. parishii holds promise for mapping and population genomics across Mimulus. The mapped RILs are now a permanent resource for research on adaptation and speciation, particularly the study of fitness-relevant traits that benefit from the replication of recombinant genotypes under different biotic or abiotic conditions. Further, the extreme phenotypic diversity of these recombinant plants, their ease of culture, and the growing knowledge base of this flagship system make the RILs valuable for course-based undergraduate research experiences (CUREs) in genetics and evolution.
Overall, patterns of TRD, pseudolinkage, and hybrid sterility reveal substantial genic and chromosomal incompatibilities between this pair of naturally hybridizing taxa. Furthermore, with the exception of the Chromosome 6–7 translocation and consistent M. cardinalis excess in the YUP supergene region of Chromosome 4 (Liang et al. 2023), the specific loci and patterns of hybrid sterility and TRD appeared mostly idiosyncratic to this M. parishii × M. cardinalis cross, rather than shared with either M. lewisii × M. cardinalis (Fishman et al. 2013) or M. parishii × M. lewisii hybrids (Fishman et al. 2015). Because hybrids between M. cardinalis and M. lewisii segregate for an additional translocation involving Chr 1 and Chr 8, severe underdominant pollen sterility likely overshadows other factors influencing the proportion of viable pollen (as each grain can only be sterile once) as well as patterns of marker TRD on Chromosome 8. However, we can conclude that at least 2 major postzygotic incompatibilities are not shared: the LG4–LG8 sex-independent gametophytic lethal detected here by TRD and LD (see more below) and the absent-here cytoplasmic male sterility (CMS) causing anther sterility and zero pollen production in a fraction of M. parishii × M. lewisii hybrids (Fishman et al. 2015). Here, the small-effect pollen number QTLs all have allelic effects consistent with M. parishii (also the organelle-donating grandparent, as in the M. parishii × M. lewisii CMS-producing cross) evolving lower pollen number as part of the selfing syndrome (Sicard and Lenhard 2011). In addition, although there is abundant and consistent TRD in M. parishii × M cardinalis hybrids, we did not recapitulate strong gametophytic TRD favoring M. cardinalis alleles on LG3 in M. lewisii × M. cardinalis hybrids (Fishman et al. 2013). In that cross, patterns of TRD in additional backcrosses revealed the mechanism to be single-locus and strictly male-gametophytic, suggesting a locus involved in the evolution of faster pollen tube growth in the longer-styled hummingbird-pollinated species (Fishman et al. 2013). The absence of a parallel pattern here, where the parental difference in pollen tube growth should (if anything) be accentuated by greater divergence in flower size and mating system, suggests that the previously reported M. cardinalis pollen growth advantage against M. lewisii was dependent on the stylar or genetic context. Similarly, novel strong TRD favoring M. parishii homozygotes on Chr5 in our RILs (vs the opposite pattern in F2 hybrids and in M. parishii × M. lewisii hybrids) suggests specific selection generated by either the greenhouse environment at UGA or the process of RIL formation. Further targeted comparisons of such barrier loci across generations, as well as among the hybrids of these 3 closely related Erythranthe taxa, hold great promise for peeling away the complex layers of genetic interactions that shape hybrid genomes and phenotypes.
Chromosomal translocations: a major and remarkably persistent cause of hybrid sterility
Consistent with previous coarse mapping in M. lewisii × M. cardinalis and newly available genomes (Fig. 1), our results demonstrate that M. cardinalis carries a unique and strongly underdominant reciprocal translocation relative to closely related taxa. Inversions and translocations were among the first posited mechanisms of postzygotic reproductive isolation, as they provide cytogenetically visible evidence of hybrid genomic incompatibility (Stebbins 1958; White 1968; Grant 1971; King 1993). Although empirical studies in plant hybrids often find species-polymorphic and species-diagnostic inversions without underdominant effects on fertility (Zhang et al. 2021), translocations have been consistently associated with severe underdominant sterility (Lai et al. 2005; Fishman et al. 2013). Because the underdominant effect of translocations is a direct effect of chromosomal pairing in heterozygotes (Stathos and Fishman 2014), their initial spread postmutation should be strongly opposed by selection (Lande 1984, 1985) and remains paradoxical. Notably, both hummingbird-pollinated M. cardinalis (Chr 6 and 7) and bee-pollinated M. lewisii (Chr 1 and 8) each have at least one derived reciprocal translocation, while selfer M. parishii carries no novel major rearrangements; thus, strong drift likely does not explain the origin of translocations in this system. Furthermore, this study shows that enforced selfing after hybridization does not eliminate this chromosomal incompatibility and its fitness effects. Thus, rather than quickly sorting to parental chromosomes, translocations may have complex and persistent effects on fertility, recombination, and introgression upon secondary contact between chromosomally divergent plants.
The M. cardinalis Chromosome 6–7 translocation generates complex (nonlinear) linkage relationships across much of both chromosomes in F2 hybrids with M. parishii (Fig. 1), exhibits elevated heterozygosity near the breakpoints (Fig. 2), and causes severe underdominant F2 pollen inviability (Fig. 4). Substantial excess heterozygosity was retained around the translocation breakpoints (Fig. 2), and while some translocation-heterozygous RILs appear to have escaped to compatible chromosomal configurations, many of them exhibited the low pollen viability of their F2 ancestors. Importantly, variation among heterozygous RILs did not appear to reflect the segregation of unlinked modifiers (i.e. one or the other parental genotype substantially conferring enhanced LG6&7 heterozygote fertility by specifying alternate segregation). None of the epistatic interactions between LG6&7 and each other sterility locus had the property of mitigating or exacerbating its underdominant pollen viability effects (Supplementary Fig. 6). This contrasts with previous work in M. lewisii × M. cardinalis hybrids, where M. cardinalis alleles on LG2 specifically accentuated the underdominant effects of the M. cardinalis translocation (Fishman et al. 2013).
This result suggests that translocations may have surprisingly persistent effects on fertility (as well as extended effects on patterns of genomic diversity and introgression) when selfing follows hybridization between structurally divergent incipient species. Population genomics shows that M. parishii has recently captured a Southern Californian M. cardinalis chloroplast, as well as nuclear regions (Nelson, Stathos, et al. 2021), suggesting a history of hybridization involving initial F1 formation with M. cardinalis as the seed parent and then recurrent backcrossing to M. parishii and/or selfing (plus possibly selection for M. parishii-like phenotypes). Since the CE10 chromosomal structure appears widespread throughout the M. cardinalis range (Fishman et al. 2013), we would predict that this genomic region may exhibit elevated diversity and/or unusual patterns of introgression where M. parishii hybridizes naturally with its hummingbird-pollinated relative. This scenario contrasts with the sorting expected for a typical barrier locus and suggests that translocations may behave differently from inversions associated with ecological barriers during speciation and secondary contact, at least in self-compatible plant species. Now that the interchange breakpoints between these naturally hybridizing taxa have been genetically and physically localized, and such predictions are testable with dense sampling in areas of sympatry between M. cardinalis and structurally divergent species.
More broadly, the strong and persistent effects of the M. cardinalis–M. parishii reciprocal translocation on linkage disequilibrium, heterozygosity, and sterility suggest that such incompatible chromosomal rearrangements deserve more theoretical and empirical study. Although the initial spread and fixation of species-diagnostic translocations are puzzling without extreme drift or meiotic drive (Lande 1984, 1985), they are an empirically major cause of hybrid breakdown in plants (Grant 1971). Nonetheless, inversions have garnered more focus recently as causes of adaptive differentiation and species barriers, though they are rarely direct causes of hybrid sterility in plants. In mammals, on the other hand, Robertsonian translocations (centric fission/fusions) are common even within species (Garagna et al. 2014), but do not singly cause the kind of underdominant sterility seen with this Mimulus partial-arm translocation. Now that long-read sequencing technologies make the assembly of near-gapless chromosomes possible across a diversity of systems, revisiting the genomic and molecular causes of organismally distinct patterns of chromosomal evolution and their direct and indirect effects on speciation is newly feasible. Plant systems where diverse chromosomal rearrangements contribute to adaptation and speciation from population to genus-wide scales, such as Helianthus (sunflowers [Ostevik et al. 2020; Owens et al. 2023]) and Mimulus, provide a promising forum for such work.
A novel sex-independent gametophytic incompatibility
Patterns of TRD in both F2 hybrids and RILs indicate absolute nontransmission of both male and female gametes with both M. cardinalis alleles on Chr 8 and M. parishii alleles on Chr 4 (Fig. 2; Supplementary Tables 1 and 6). This extreme pattern of distortion is consistent with a two-locus sex-independent gametophytic incompatibility. In land plants, which have a life cycle with 2 multicellular stages (a.k.a. alternation of generations), incompatibilities may act in the haploid gametophyte or in the diploid sporophyte to cause sterility. Unlike in animal sperm or egg cells (Braun et al. 1989), a substantial proportion of the genome is haploid-expressed in plant gametophytes (Honys and Twell 2004; Wuest et al. 2010; Rutley and Twell 2015). Reflecting this expression activity, most hybrid sterility loci in rice are gametophytic (Ouyang and Zhang 2013), and genome-wide patterns of TRD in multiple F2 populations of Arabidopsis lyrata also seem to be caused by gametic incompatibilities (Leppälä et al. 2013). Abundant gametophytic incompatibilities in plants might be due to the relatively large mutational target of haploid-expressed genes, to rapid divergent evolution of gametophytic genes under efficient sex-specific selection (Gossmann et al. 2013, 2016) or other natural selection (Immler and Otto 2018; Beaudry et al. 2020), or simply to the greater opportunity for exposure of negative allelic interactions in haploid hybrid tissues (similar to hemizygosity as an explanation for Haldane's rule).
The sex independence of the new M. parishii–M. cardinalis incompatibility implies a shared molecular mechanism for the loss of both male and female gamete function—but what could that be? Meiotic and developmental programs resulting in embryo sacs (female) and pollen (male) occur in distinct sexual organs of the flower, and sex-specificity of hybrid sterility loci is predominant in taxa that have been systematically studied, such as rice (Myint and Koide 2022). Nevertheless, sex-independent incompatibility loci have been identified in tomato (Rick 1966), rice (Sano 1990; Koide, Onishi, et al. 2008), lettuce (Giesbers et al. 2018), and yellow monkeyflowers (Kerwin and Sweigart 2017). Because core cellular processes such as meiosis and mitosis are critical for gametogenesis in both sexes, many of the same genes are essential in both sexes (Drews and Yadegari 2002; Ma et al. 2021; Yu et al. 2022; Zhou et al. 2022). Thus, the breakdown of these processes in hybrids could cause sex-independent incompatibility. However, because the LG4–LG8 gametic incompatibility does not colocalize with QTLs for either pollen number or viability, it implies a nonmitotic or meiotic mechanism that does not induce pollen abortion or substantially impair pollen development (at least in a way that could be assessed by our starch stain for viability). This contrasts with the hms1–hms2 system in yellow monkeyflowers (Kerwin and Sweigart 2017), which causes both sex-independent TRD and pollen sterility, but parallels a sex-independent, 2-locus gametophytic barrier between wild and cultivated lettuce(Giesbers et al. 2018). The lack of a pollen-staining effect in both this study and lettuce intriguingly suggests the involvement of genes required specifically for the functioning of mature male and female gametophytes (e.g. genes involved in both female gametogenesis and pollen tube growth/guidance) (Leszczuk et al. 2019; Chen et al. 2020). Like a lethal incompatibility causing albinism in some hybrids between yellow monkeyflowers M. nasutus and M. guttatus (Zuellig and Sweigart 2018), the completeness of gamete failure might suggest duplication and reciprocal deletion of a haploid-essential gene as a potential mechanism. While the completely nontransmitted region on Chromosome 8 contains ∼1,000 genes flanking a putatively centromeric region of low recombination, the interacting locus on Chromosome 4 is ∼1 Mb containing only 80 genes. Given that parallels to other gametic lethals suggest a finite pool of functional candidate genes, the genetic and molecular mechanisms are thus amenable to further dissection.
Conclusions
Overall, our results underline the magnitude, complexity, and diversity of postmating barriers (including postzygotic) that can evolve between closely related plant species. A novel 2-locus sex-independent gametic Dobzhansky–Muller incompatibility and a reciprocal translocation each profoundly affect patterns of transmission and linkage disequilibrium across entire chromosomes. The genic incompatibility completely eliminates gametes recombinant between the 2 involved loci, thus acting as a strong barrier that should shape natural hybridization for whole genomic regions. In contrast, the underdominant reciprocal translocation causes retention of heterozygosity despite enforced selfing, suggesting that it may not act as a typical barrier gene flow despite its major fertility costs. Understanding the mechanisms and origins of these major incompatibilities is key for reconstructing the process of speciation in this model system, as well as predicting patterns of gene flow in areas of contact. Additional regions of consistent F2 TRD amplified across generations to even more strongly skew RIL composition, providing indirect evidence of substantial postmating or postzygotic barriers not detectable from measures of hybrid fertility alone. Beyond illustrating the rapid buildup of multiple postzygotic barriers and providing essential context for investigating the striking floral and life history divergence between these taxa, this work raises new questions about the evolutionary origins of such strong postzygotic barriers and their effects in natural hybrid zones.
Data availability
Raw sequence data from F2 and RIL mapping populations have been archived on the NCBI Sequence Read Archive under PRJNA1003462 and PRJNA948041, respectively. The genotype and phenotype data for linkage, transmission ratio distortion, and QTL mapping are archived on Dryad (doi:10.5061/dryad.v6wwpzh1m).
Supplemental material available at GENETICS online.
Acknowledgments
We thank T. C. Nelson, K. Baesen, and A. Demaree for lab assistance and O. Moynahan of the UM ECOR plant growth facility, M. Moriarty, M. Opel, and C. Liu of the UConn Botanical Conservatory, and Mike Boyd and Greg Cousins of the UGA Plant Sciences Greenhouse for assistance with plant care. We are especially indebted to S. McCann for his help in generating the RILs.
Funding
This work was supported by National Science Foundation grants DEB-1457763 and OIA-1736249 to LF, DEB-1350935 to ALS, and IOS-1827645 to ALS and YWY.
Literature cited
Author notes
Conflicts of interest The authors declare no conflict of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}