





Abstract
The challenges of breeding autotetraploid potato (Solanum tuberosum) have motivated the development of alternative breeding strategies. A common approach is to obtain uniparental dihaploids from a tetraploid of interest through pollination with S. tuberosum Andigenum Group (formerly S. phureja) cultivars. The mechanism underlying haploid formation of these crosses is unclear, and questions regarding the frequency of paternal DNA transmission remain. Previous reports have described aneuploid and euploid progeny that, in some cases, displayed genetic markers from the haploid inducer (HI). Here, we surveyed a population of 167 presumed dihaploids for large-scale structural variation that would underlie chromosomal addition from the HI, and for small-scale introgression of genetic markers. In 19 progeny, we detected 10 of the 12 possible trisomies and, in all cases, demonstrated the noninducer parent origin of the additional chromosome. Deep sequencing indicated that occasional, short-tract signals appearing to be of HI origin were better explained as technical artifacts. Leveraging recurring copy number variation patterns, we documented subchromosomal dosage variation indicating segregation of polymorphic maternal haplotypes. Collectively, 52% of the assayed chromosomal loci were classified as dosage variable. Our findings help elucidate the genomic consequences of potato haploid induction and suggest that most potato dihaploids will be free of residual pollinator DNA.
HIGHLY prized in plant breeding and research, haploid plants can be obtained through culture of immature gametophytes or, more conveniently, through inter- or intraspecific crosses in which the genome of one parent, the haploid inducer (HI), does not appear in the progeny (Forster et al. 2007; Ishii et al. 2016). Having been documented in 74 crosses between monocotyledonous species and 35 involving dicotyledonous species, this phenomenon is not uncommon [reviewed in Ishii et al. (2016)], and is widely exploited for the rapid generation of inbred lines, as well as genetic mapping and germplasm base expansion. However, the genetic properties that make an HI are largely unknown, with a couple of exceptions. Artificial manipulation of centromeric histone H3 can result in an HI (Ravi and Chan 2010; Karimi-Ashtiyani et al. 2015; Kuppu et al. 2015; Maheshwari et al. 2015; Kelliher et al. 2016). Furthermore, natural maize HIs depend on inactivation of the phospholipase encoded by the matrilineal locus (Gilles et al. 2017; Kelliher et al. 2017; Liu et al. 2017).
In potato (Solanum tuberosum L.), the world’s fourth most important crop in terms of calories consumed per person per day (http://www.fao.org/faostat/en/#compare), haploid seed can be routinely obtained via pollination with select HI varieties from the diploid S. tuberosum Andigenum Group [formerly S. tuberosum Phureja Group or S. phureja (Spooner et al. 2014)]. Such crosses with tetraploid potato (2n = 4x = 48) produce 2n = 2x = 24 dihaploids that can be used for genetic mapping (Kotch et al. 1992; Pineda et al. 1993; Ercolano et al. 2004; Velásquez et al. 2007; Mihovilovich et al. 2014; Bartkiewicz et al. 2018). Additionally, these crosses produce hybrids that can be either triploid or tetraploid (Wagenvoort and Lange 1975; Hanneman and Ruhde 1978), and can be identified as seed because they express a purple embryo spot, a dominant anthocyanin marker encoded by the HIs that is expected to be absent in the dihaploids (Figure 1) (Hermsen and Verdenius 1973). In embryo spot-negative dihaploid populations, 3.5–11.0% of aneuploids are commonly found, exhibiting 2n = 2x + 1 = 25 and, rarely, 2n = 2x + 2 = 26 karyotypes (Wagenvoort and Lange 1975).
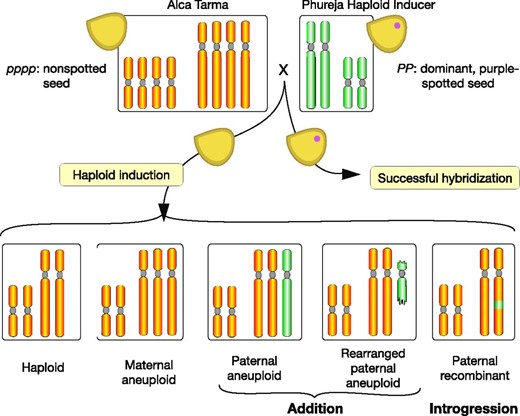
Production of the dihaploid population and expected types. Haploid inducers IVP101 or PL4, both diploids, were used to pollinate tetraploid cultivar Alca Tarma. For simplicity, the potato genome is represented by two chromosome types. The haploid inducer is homozygous for the dominant seed purple spot marker. Normal fertilization and development resulting in hybrids with spotted seed, defined as successful hybridization, results in biparental triploids or tetraploids, depending on the ploidy of the male gamete. Maternal dihaploids are expected among plants germinated from nonspotted seeds. Plants displaying more than eight stomatal chloroplasts (an indication of increased nuclear content) or unusual phenotypes potentially consistent with aneuploidy were discarded (Velásquez et al. 2007; Mihovilovich et al. 2014). Genetic haploid inducers can act through either parthenogenesis (Forster et al. 2007) (development of an unfertilized egg) or genome elimination (Ishii et al. 2016) (rejection of the haploid inducer genome). Addition or introgression of residual haploid inducer DNA indicates the second mode of action.
An ongoing question regarding haploid induction in potato is the cytogenetic mechanism by which it occurs. Two mechanisms have been proposed. The first mechanism is parthenogenesis, in which HI pollen triggers the development of unfertilized egg cells without making a genetic contribution to the embryo. This is supported by three lines of evidence: (i) endosperms from 4x by 2x potato haploid induction crosses are usually hexaploid instead of the expected pentaploid, suggesting abnormal pollen (von Wangenheim et al. 1960); (ii) HIs frequently produce 24-chromosome restitution sperm nuclei, thought to be a consequence of failed pollen mitosis II; and (iii) colchicine-treated pollen of noninducer S. tarjinse also exhibit restitution sperm nuclei and can induce haploids (Montelongo-Escobedo and Rowe 1969). Based on these observations, it was speculated that a 2x restitution sperm fertilizes the central cell, leaving no sperm to fertilize the egg. The second mechanism is genome elimination, in which HI chromosomes are eliminated from the embryo after fertilization. This alternative hypothesis is supported by the presence of inducer-specific AFLP, RFLP, or isozyme markers in presumably dihaploid progeny. Often, progeny exhibiting genetic markers from the HI are also aneuploid, suggesting inheritance of an entire chromosome from the HI (Clulow et al. 1991, 1993; Waugh et al. 1992; Wilkinson et al. 1995; Allainguillaume et al. 1997; Clulow and Rousselle-Bourgeois 1997; Straadt and Rasmussen 2003; Ercolano et al. 2004). These results are consistent with haploid induction crosses in maize (Riera-Lizarazu et al. 1996; Zhao et al. 2013), Arabidopsis (Kuppu et al. 2015; Maheshwari et al. 2015; Tan et al. 2015), and oat–maize hybrids (Riera-Lizarazu et al. 1996) in which one or more HI chromosomes persist in otherwise haploid plants.
Recently, widespread and ubiquitous introgression of very short DNA regions (> 100 bp) from the HI genome into potato dihaploids has been reported by SNP genotyping (Bartkiewicz et al. 2018) and by whole-genome sequencing (Pham et al. 2019). In the latter case, depending on the progeny, 25,000–300,000 translocation events were inferred, suggesting a massive contribution from the transient HI genome to the maternally contributed genome. Genetic information in short segments of HI DNA could persist through three mechanisms: (i) nonhomologous recombination leading, for example, to insertion; (ii) homologous recombination leading, for example, to gene conversion; and (iii) autonomous replication. To clarify the underlying molecular arrangement, we use the term “addition” to indicate the presence in the dihaploid genome of an additional copy derived from the HI. We use the term “introgression” to indicate that the DNA from the HI has recombined with the donor genome. If recombination is homologous, this could result in copy-neutral transfer of information. If confirmed, such widespread recombination would require rethinking of both breeding and biotechnology experimental strategies to either avoid or exploit it, depending on the context.
In light of these observations, we resequenced a population of 167 primary dihaploids derived from tetraploid Andigenum Group cultivar Alca Tarma to address three questions: First, does a curated set of phenotypically normal primary dihaploids display aneuploidy? If so, which parent contributes the additional chromosome(s)? Second, are single chromosomes or large chromosome segments from the HI added to otherwise dihaploid progeny? Third, are shorter segments of the HI genome introgressed or added to the dihaploids? If so, on what scale? We considered two hypotheses: first, that occasional failure to eliminate the entire HI chromosome set could result in the persistence of an additional chromosome, whole or fragmentary, in an otherwise dihaploid potato. We have previously demonstrated that events involving entire chromosomes or chromosome segments are readily detectable with low-coverage whole-genome sequencing, and chromosome dosage analyses, in Arabidopsis (Henry et al. 2010; Kuppu et al. 2015; Maheshwari et al. 2015; Tan et al. 2015) and poplar (Henry et al. 2015; Zinkgraf et al. 2016). In potato, extensive copy number variation (CNV), which has been described in numerous cytological and genomic surveys of diploid and tetraploid cytotypes, is a potentially confounding factor that should be taken into account (Iovene et al. 2013; de Boer et al. 2015; Hardigan et al. 2016; Hardigan et al. 2017; Pham et al. 2017). Second, smaller-scale introgression described could be detected by deeper sequencing of selected individuals. Our genomic analysis did reveal frequent whole-chromosome aneuploidy and widespread segmental dosage variation, but these were never attributable to the HI. Notwithstanding the ability of dihaploids to tolerate chromosomal dosage imbalance, we found no evidence that HIs contributed large chromosomal segments to the progeny. Further, by a set of standard criteria, we found no short segmental introgression either.
Materials and Methods
Plant material
A population of 167 putative dihaploids described in Velásquez et al. (2007) and Mihovilovich et al. (2014) was raised from the progeny of tetraploid Andigenum Group landrace cultivar Alca Tarma and one of two HI genotypes: IVP-101 or CIP596131.4. For convenience, we refer to CIP596131.4 as PL4 throughout. Both HIs are homozygous for a dominant embryo spot marker that results in anthocyanin accumulation at the base of the cotyledons visible through the seed coat. Only seeds lacking the embryo spot marker were planted, and seedlings exhibiting more than an average of eight chloroplasts per guard cell (minimum 10 measured cells) were discarded. Seedlings were germinated on soil and maintained as in vitro cuttings thereafter.
Genomic DNA library preparation, sequencing, and preprocessing
Approximately 750 ng of genomic DNA extracted from leaf tissue as previously described (Ghislain et al., 1999) was sheared to an average size of 300 bp using a Covaris E-220 sonicator in a 50-µl reaction volume using the following settings: 175 W peak power, 10% duty factor, 200 cycles per burst, 50-sec treatment time, 4° minimum temperature, and 9° maximum temperature. Genomic libraries were constructed using 375 ng of sheared DNA and a KAPA Hyper Prep Kit (catalog number KK8504) with half-scale reactions, custom 8-bp dual-indexed adapters, and library amplification cycles as specified in Supplemental Material, Table S1. Libraries were sequenced on an Illumina HiSeq 4000 in either 50-nt single-end or 150-nt paired-end mode by the University of California, Davis DNA Technologies Core and Vincent Coates Genome Sequencing Laboratory.
Libraries were demultiplexed using a custom Python script available on our laboratory website (allprep-12.py; http://comailab.genomecenter.ucdavis.edu/index.php/Barcoded_data_preparation_tools).
Variant calling
For paired-end sequencing, sequence reads were processed with Cutadapt (v.1.15) to remove low-quality (< Q10) bases, adapter sequences, and reads ≤ 40 nt after trimming. The DM1-3 v4.04 genome assembly, as well as DM1-3 chloroplast and mitochondrion sequences, were retrieved from (http://solanaceae.plantbiology.msu.edu/pgsc_download.shtml), concatenated, and used as the reference sequence for paired-end read alignment with BWA MEM (v.0.7.12-r1039) (Li 2013), with mismatch penalty six and all other parameters left at the program default. PCR duplicates were removed using Picard (v.2.14) MarkDuplicates, and only reads with mates mapping to the same chromosome were retained. For reads with overlapping mates, one of the two reads was soft-clipped in the overlap region using bamUtil::clipOverlap (Jun et al. 2015). Variants were called on processed alignment files using freebayes (v.1.1.0) (Garrison and Marth 2012) with minimum read mapping quality 41, minimum base quality 20, population priors not considered, and ploidy specified for each sample as a CNV map. To remove low-quality variants, the following site filters were applied in RStudio (v.3.4.0): NUMALT == 1, CIGAR == 1X, MQM ≥ 50, MQMR ≥ 50, |MQM - MQMR| < 10, RPPR ≤ 20, RPP ≤ 20, EPPR ≤ 20, EPP ≤ 20, SAP ≤ 20, SRP ≤ 20, and DP ≤ 344. Only sites with called homozygous Alca Tarma genotypes without reads matching HI alleles were retained. Sites that were called homozygous for the Alca Tarma allele in either HI were removed. Several additional quality filters were applied on each sample: depth ≥ 10 in all three parents, ≤ 5% Alca Tarma allele representation at called homozygous HI loci, and 40–60% Alca Tarma allele representation at called heterozygous HI loci. After filtering, 798,468 SNPs were retained for analysis. Putative introgression loci were identified via heterozygous genotype calls in any of the three high-coverage dihaploids.
Dosage analysis
Single-end reads were aligned to the DM1-3 reference genome as described above, and only reads with mapping quality ≥ Q10 were retained. Standardized coverage values were derived by taking the fraction of mapped reads that aligned to a given bin for that sample, dividing it by the corresponding fraction from the same bin for tetraploid Alca Tarma, and doubling the resulting value to indicate the expected diploid state. To mitigate mapping bias due to read type and length, Alca Tarma forward mates were hard trimmed to 50 nt and remapped. When all chromosomes were treated as equivalent, the distribution of per-chromosome standardized coverage values approximated a Gaussian distribution (quantile-quantile (Q-Q) plots not shown), satisfying the assumption of a Z-score analysis. Aneuploidy was then called if chromosomal standardized coverage exceeded the all-chromosome population by ≥ 3 SD. We identified local dosage variants by clustering standardized coverage values of nonoverlapping 250-kb bins using the R package MeanShift (CRAN - Package MeanShift). The clustering bandwidth parameter was set to the 50th percentile of interpoint distances in each 250-kb bin.
Parental origin analyses of trisomy and dosage variants were carried out as previously described (Henry et al. 2015). Briefly, reads with mapping quality ≥ Q20 and base calls ≥ Q20 were used to compute allele-specific read depth at the subset of 800,384 SNP loci identified above, located on chromosomes 1–12 of the DM1-3 v4.04 assembly. The percentage of reads supporting the HI allele reads among all reads at loci within a nonoverlapping 1-Mb bin was then reported. As a biological positive control was not available for SNP analysis, we empirically evaluated limitations of the SNP dosage assay by comparing dihaploids with simulated triploid hybrids expected to resemble an introgressed HI chromosome segment at all tested genomic loci. To construct triploid hybrids in silico, pseudorandom subsets of exactly 2,015,413 and 1,007,706 forward mates were drawn 100 times from raw sequencing reads of Alca Tarma (SRA ID SRR6123032) and IVP101 (SRA ID SRR6123183), respectively. The number of parental reads was chosen such that parental reads would be present in a 2:1 ratio expected for a triploid, and that the number of raw reads for each in silico hybrid would match the fifth percentile of raw read counts in the dihaploid sequencing data set. Similarly, 100 in silico hybrids were constructed using Alca Tarma and PL4 reads. As a negative control, pseudorandom subsets of 3,023,119 reads were drawn 100 times from Alca Tarma. Raw reads from all simulated hybrids were hard trimmed to 50 nt, and then processed using the SNP dosage pipeline described above with nonoverlapping 1-Mb bins. For each bin, if the ranges of HI allele percentages from either in silico hybrid group overlapped with the corresponding range of the negative control, the bin was withheld from analysis. Using this approach, 608 of 730 bins (83%) were considered in this analysis. Similarly, to determine whether unique dosage variants could be genotyped confidently, we compared the distributions of % HI allele values between simulated hybrid and negative control groups in the affected interval.
To estimate absolute copy number, per-position read was calculated using samtools depth, using only reads with mapping quality ≥ Q20. Median read depth in nonoverlapping 10-kb windows was then determined using custom Python software available from (https://github.com/kramundson/LOP_manuscript). We observed a positive correlation of window median read depth and GC content, suggesting that PCR amplification bias was introduced during sequencing library construction (Benjamini and Speed 2012). For each 10-kb bin, this bias was corrected by dividing median read depth by GC content for that bin. The resulting values were clustered using the MeanShift package in R Ciollaro and Wang (2016). The centroid of the largest cluster was designated as the copy number corresponding to the expected ploidy (four for Alca Tarma and two for dihaploids), and multiples of the centroid were used to designate the remaining copy number states.
Cytological analysis
Chromosome spreads were prepared from root tips as previously described (Watanabe and Orrillo 1993), with minor modifications. Commercial permethrin was used at a concentration of 75 ppm as a pretreatment to induce chromosome condensation. Roots were kept in an ice-cold water bath for 24 hr before hydrolysis in 1M HCl for 10–15 min, then stained with lacto-propionic acid and squashed.
Data availability
Sequence data have been deposited in the National Center for Biotechnology Information Sequence Read Archive: BioProject identifier PRJNA408137. Analysis code has been deposited at https://github.com/kramundson/LOP_manuscript. Supplemental material available at figshare: https://figshare.com/projects/Genomic_Outcomes_of_Haploid_Induction_Crosses_in_Potato_Solanum_tuberosum_L_/70793.
Results
Induction, selection, and sequencing of dihaploids
A population of 167 primary dihaploids was generated from Alca Tarma via pollination with HIs IVP101 or PL4 (Velásquez et al. 2007; Mihovilovich et al. 2014) over the course of two previous studies (Velásquez et al. 2007; Mihovilovich et al. 2014). The dihaploids in this population lacked the homozygous dominant embryo spot seed marker present in both His, and displayed the expected count of guard cell chloroplast counts and root cell chromosomes (Velásquez et al. 2007; Mihovilovich et al. 2014) (Figure 1). We carried out genome resequencing to identify dosage variation and aneuploidy among the population. Alca Tarma, IVP101, PL4, and three selected haploids were sequenced to 40–56× coverage. For the remaining dihaploids, we generated an average of 3.88 million reads per individual (Table S1).
Maternally derived trisomy
We hypothesized that introgressions into the host genome could derive from at least three types of events, each associated with specific predictions: (i) nonhomologous transposition of HI segments to the host genome, resulting in three copies of the corresponding region with a SNP ratio of 1:2 HI:host; (ii) homologous recombination leading to replacement of a segment, either interstitial or terminal, resulting in no copy number change of the affected region and a SNP ratio of 1:1 HI:host; and (iii) gene conversion from a noncrossover event producing a very short (25–50-bp) conversion tract and resulting mostly in a single SNP with a 1:1 ratio. Formally, homologous recombination could also result in duplication and resemble case (i) (Das et al. 1991).
We first screened the population for whole-chromosome aneuploidy. Sequencing reads were aligned to the DM1-3 reference genome and read counts per chromosome were normalized to those of the tetraploid parent, such that values near 2.0 were obtained for chromosomes present in two copies and values deviating from 2.0 indicated aneuploidy. In 19 individuals, standardized coverage was significantly elevated for a single chromosome, suggesting a primary trisomy (Figure 2A). Root tip chromosome spreads were evaluated for 15 of the 19 putative trisomics, confirming the 2n = 2x + 1 = 25 karyotype in all cases (Figure 2, B and C and Figure S1). In total, six trisomics of chromosome 2, two of chromosomes 4, 5, 7, and 8, and one of chromosomes 1, 3, 6, 10, and 12 were detected in the population (Figure 2A and Figure S1). To determine the parental origin of each trisomy, 382,967 SNPs homozygous for the same allele in both HIs, but homozygous for an alternate allele in Alca Tarma, were identified from sequencing the parental genomes (Supplemental Data set S1). The fraction of HI-specific allele calls along all chromosomes was then calculated for each trisomic individual. Using this measurement, a trisomy from either HI was expected to exhibit ∼33% HI allele across the affected chromosome. To empirically validate this expectation, we evaluated 200 simulated low-coverage hybrids each consisting of 2 million reads from Alca Tarma, and 1 million reads from either IVP101 or PL4 (see Materials and Methods). In each trisomic dihaploid, HI alleles were nearly absent from the trisomic chromosome (Figure 2D), indicating inheritance from the noninducer parent.
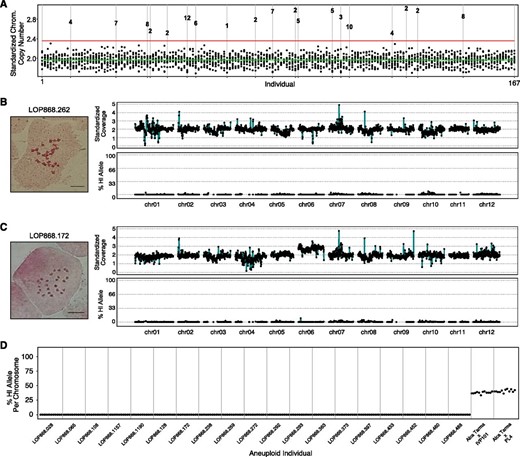
Aneuploidy detection in 167 primary dihaploids by genome sequencing. (A) Chromosome copy number for each individual in the population. Each individual of the population is displayed along the x-axis, with the stack of dots at each x coordinate corresponding to standardized copy number for each chromosome type, with a value of 2.0 representing the expected diploid state. The green line corresponds to the mean chromosome copy number among the population and the red line indicates 3 standard deviations greater than the mean. Outliers in this distribution correspond to the affected chromosome in each trisomic and are numbered according to the chromosome present in excess. (B) Cytogenetic and in silico karyotype of a representative euploid dihaploid LOP868.262. Left: Root tip somatic metaphase karyotype. Top right: copy number plot; individual data points represent read depth in nonoverlapping 250-kb bins standardized to tetraploid Alca Tarma counts such that the expected diploid state corresponds to copy number 2.0. Bottom right: haploid inducer SNP allele plot; black points correspond to the fraction of base calls supporting haploid inducer alleles at all informative sites in nonoverlapping 1-Mb bins. (C) Cytogenetic and in silico karyotype of chromosome 6 trisomic LOP868.172, illustrating that trisomy of chromosome 6 was not derived from either IVP101 or PL4. See Figure S1 for dosage plots of the remaining trisomics. (D) SNP plot showing the percentage of haploid inducer allele present in the trisomics identified in panel (A). For each individual, the 12 points correspond to the 12 chromosomes displayed in order (i.e., the first dot is chromosome 1, the second chromosome 2, etc.). For each individual, points near 0% for the affected chromosome and all others indicate a maternal trisomy. Observed % haploid inducer allele values from two representative simulated hybrid controls, one Alca Tarma × IVP101 and the other Alca Tarma × PL4, are shown on the right. Chrom., chromosome; HI, haploid inducer.
Determining which SNP bins are informative
To survey the population for HI chromosome addition or introgression, the SNP dosage analysis described above was repeated using nonoverlapping 1-Mb bins. To account for a low density of homozygous parental SNP markers in some regions, we included an additional set of SNPs that did not fit the optimal criteria of the original set (Supplemental Data set S2). Of the added SNPs, 170,273 were heterozygous in one HI and homozygous in the other, while 247,144 were heterozygous in both HIs. A consequence of including these additional SNP markers is that an HI allele contribution could be lower than the expected 33%. Therefore, we empirically determined the expected percentages for each bin by comparing the percentages obtained from low-coverage in silico triploid hybrids and negative controls. Any bin in which the distributions of observed HI allele percentages of the hybrid and negative control groups exhibited any overlap was withheld from consideration. To call an introgression, we required at least three adjacent bins to exhibit an HI allele percentage that overlapped with the empirical thresholds determined from the in silico hybrid analysis. Among 1-Mb bins that were considered in this analysis, no such events were found (Figure S2). Notably, a recombination event that substitutes an Alca Tarma chromosome segment by the corresponding segment from either HI would produce a higher HI allele percentage than an addition event (50 vs. 33%), suggesting that neither addition nor introgression of an HI chromosome segment occurred in the dihaploid population.
CNV analysis
To complement the approach described above, we also asked whether rare structural variants existed in the population and, if so, from which parent they were derived. The dosage analysis was repeated as described above for nonoverlapping 250-kb bins of the reference genome. As before, a value of 2 indicates the expected diploid complement and values deviating from 2 indicate structural variation. Inferred karyotypes of a representative 2n = 2x = 24 dihaploid and a 2n = 2x + 1 = 25 maternal trisomy are shown in Figure 2, C and D, respectively. By overlaying dosage plots for each dihaploid, it became evident that many CNVs are recurrent in the population, with copy number gains and losses of the same locus among the dihaploids (Figure 3, A and B and Figure S3). This behavior is evident in the pericentromeric heterochromatic region of all chromosomes, while the euchromatic, gene-rich arms are more uniform (Figure 3, A–G). Chromosomes 2 and 4 are exceptions in that they display recurring dosage variation in their short arms (Figure 3B and Figure S3).
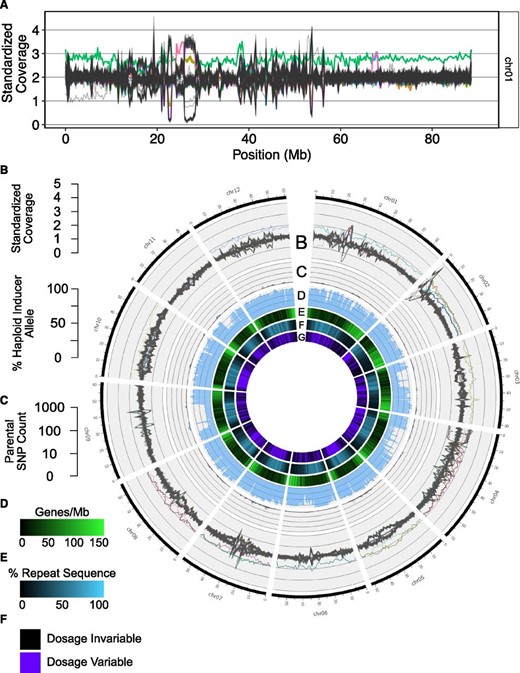
Chromosomal distributions of dosage variation and parent-informative markers in the dihaploid population. (A) Read depth standardized to tetraploid Alca Tarma in nonoverlapping 250-kb bins of chromosome 1. Each individual in the dihaploid population is represented by a single line. Dihaploids that are trisomic for any chromosome are represented by colored lines. For example, chromosome 1 trisomic LOP868.238 is displayed as a green line. The plot also displays unique segmental dosage variants, one starting at 24 Mb and involving a trisomic of eight (pink line), a second one at 67 Mb and involving two trisomics of two (overlapping magenta and blue lines), and a terminal deletion of the short arm in an otherwise euploid line (gray line). (B–F) Circos plots for the 12 chromosomes. For each chromosome, 167 lines are shown. Aneuploids of any chromosome are uniquely colored and all other individuals are colored gray. (B) Standardized read-depth plots of all chromosomes smoothed to 1-Mb bins. (C) Percent haploid inducer allele at all parent-informative marker loci in nonoverlapping 1-Mb bins. Refer to Figure S3 for expected haploid inducer allele percentages derived from simulated hybrid analyses. (D) Log10-scaled counts of parent-informative markers in nonoverlapping 1-Mb bins. (E) Genes per 1-Mb sliding window and 200-kb step. (F) Percent repeat sequence per 1-Mb sliding window and 200-kb step. Chr, chromosome.
To define structurally polymorphic loci and alleles at these loci, we clustered relative coverage values separately for each 250-kb bin (Figure S4). Structural variation was widespread, with multiple clusters detected in 48% of 250-kb bins (Figure 3G). From joint consideration of the number of individuals in a cluster and read depth of high-coverage samples, we inferred that most dosage variants represented segregating polymorphism among Alca Tarma haplotypes, as exemplified by a 1-Mb block of chromosome 6 that is present on one chromosome and absent on the other three (Figure 4 and Figure S5). Among 14 duplications that were ≥ 750 kb and present in < 5% of dihaploids, 5 were clearly derived from Alca Tarma (Table S2). Based on comparison of simulated hybrids, SNP marker density was too low to conclusively resolve the parental origin of the remaining 9, but the fraction of HI SNPs in power analyses classified them as low-probability outliers (Figure S6).
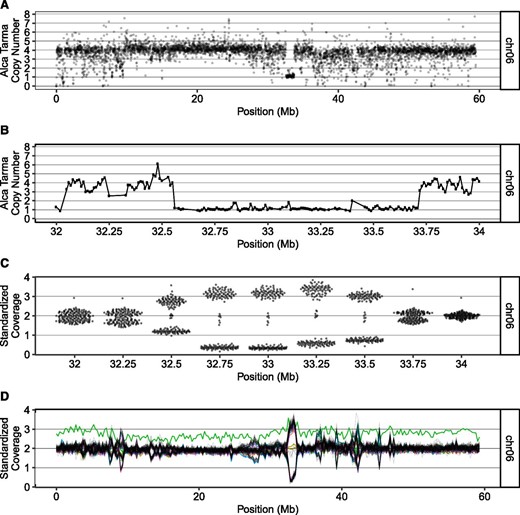
Example of segregating copy number variation among dihaploid population. (A) GC-normalized median read depth of nonoverlapping 10-kb bins for tetraploid Alca Tarma (see Materials and Methods). Chromosome 6 is shown. Each dot is plotted with partial transparency to emphasize overplotting. (B) GC-normalized median read depth, zoomed in on 1-Mb region of chromosome 6. (C) Swarm plots of standardized coverage values in bins affected by 1-Mb deletion. The population segregates the high- and low-dosage states in an ∼1:1 ratio, consistent with random chromosome segregation of a deletion in triplex allele dosage. (D) Population standardized values for chromosome 6. Each dihaploid is displayed as a single contiguous line; green line: chromosome 6 trisomic. The region corresponding to the large deletion shown in panel (A) exhibits segregating dosage variation in the dihaploid population. Chr, chromosome.
SNP loci consistent with addition or introgression were rare, and dispersed
To search for addition and introgression events at higher resolution, three dihaploids were sequenced to 19–30× depth with Illumina paired-end sequencing. To minimize spurious genotype calls, SNP loci that passed our quality filtering steps were further filtered to exclude sites with even a single Alca Tarma read that matched an HI allele. In total, 800,384 loci were assayed, with 725,952–745,535 loci assayed in each of the three dihaploids. The fraction of loci with heterozygous genotypes consistent with addition or introgression was very low (0.157–0.195%). Among heterozygous sites in the dihaploids, HI alleles tended to be underrepresented relative to expectations for either addition (∼33%) or introgression (50%) of HI DNA (Figure S7), and read depth was lower in both Alca Tarma and the dihaploid at hand (Figure S8). Upon observing the low read depth and underrepresentation of HI alleles at many putative introgression loci, we applied additional filters (minimum HI allele depth ≥ 5 and minimum allele depth representation 10%) to investigate the subset of possible introgression events with the best read support in our data set. Applying these filters reduced the number of putative introgression loci from 1217 to 266 in LOP868.004, from 1457 to 358 in LOP868.064, and from 1124 to 198 in LOP868.305 (Supplemental Data set S3).
We further investigated the distribution of all putative introgression loci with respect to the reference genome. As loci that were heterozygous in the two HIs were also included in this analysis and the phase was unknown, introgression of an HI segment may appear discontinuous. Therefore, we estimated a lower bound of the number of introgression events using a seed-and-extend approach: starting at SNP loci with inducer-specific allele introgression events, putative introgressions were extended in both directions until a parent-homozygous SNP locus with no evidence of HI alleles in the dihaploid was encountered. The total number of SNP markers, as well as markers exhibiting HI alleles, were tallied for each event. Dihaploids LOP868.004, LOP868.064, and LOP868.305 exhibited 1037, 1191, and 806 events, respectively. Approximately one-half (49.6%) of putative introgression events consisted of a singleton SNP, i.e., a single locus exhibiting HI alleles flanked by homozygous parental loci that did not support introgression in the dihaploid (mean distance between loci flanking a singleton = 9110 bp and median distance = 735 bp). Among nonsingleton events, very few markers exhibited HI alleles for each event (Figure S9).
Finally, read alignments were manually inspected for the introgression event with the highest number of parental SNP loci exhibiting HI alleles. This event spans a ∼2-Mb region of chromosome 5 in dihaploid LOP868.305, including 5712 parental SNP loci, of which only 43 exhibited HI alleles. Specifically, we looked for phased variants on the same read consistent with introgression of a contiguous HI haplotype. Remarkably, no reads supporting HI introgression at each of these 43 loci matched a local haplotype present in either HI (Figure S10 and Supplemental Data set 4).
Taken together, these analyses revealed rare maternally associated chromosome remodeling among a backdrop of widespread structural heterogeneity (Figure 3A), including several large and novel variants detected in the genome of the tetraploid parent. Robust evidence for chromosomal introgression from the HI was not detected in any case, and a detailed survey of three dihaploid genomes revealed sites that, while superficially consistent with introgression of HI DNA, resemble sequencing or alignment artifacts.
Discussion
We analyzed the genomes of 167 primary dihaploids produced by pollinating the S. tuberosum Andigenum Group cultivar Alca Tarma with HIs IVP101 and PL4. This population is representative of a typical dihaploid progeny set used for breeding in that, during its development, selection has been applied against individuals with DNA content differing from the dihaploid state, against individuals carrying the genetic color marker from the HI (Figure 1), and against severe abnormality. Using low-pass sequencing, we derived karyotypes of each progeny, identifying primary trisomy in 11.4% of individuals. By comparing parental SNP genotypes, we established that whole-chromosome aneuploidy was maternally inherited. Widespread variation in DNA dosage consistent with segregation of maternal structural variation was already evident at 1-Mb resolution (Figure 3). This analysis does not consider small structural events at the genic or transposable element scales. Using randomly downsampled data to simulate a triploid hybrid, we empirically show that our low-pass sequencing approach provided an effective and affordable method to decipher the complexity of populations with highly variable genomic structure, such as is often employed in breeding (Barrell et al. 2013; Hirsch et al. 2013). Our findings lead to several conclusions.
Genetic contribution from HI was not detected, despite frequent aneuploidy and widespread dosage variation. Genetic HIs act by either stimulating parthenogenesis in the female, or chromosome instability in the embryo resulting in missegregation and loss of one parental chromosome set. In potato, evidence in support of parthenogenesis has been reported (von Wangenheim et al. 1960; Montelongo-Escobedo and Rowe 1969; Peloquin et al. 1996). At the same time, genome elimination is supported by the detection of genetic markers from the HI in euploids and aneuploids arising from haploid induction crosses (Clulow et al. 1991, 1993; Waugh et al. 1992; Wilkinson et al. 1995; Allainguillaume et al. 1997; Clulow and Rousselle-Bourgeois 1997; Straadt and Rasmussen 2003; Ercolano et al. 2004). Whole-genome sequencing provides a more informative and reliable method to assess the genetic contribution of the HI. In Arabidopsis haploids, DNA from the HI can be identified readily from low-pass sequencing (Tan et al. 2015). It consists of whole chromosomes or segmental subsets of a single chromosome, consistent with the incomplete elimination of certain chromosomes, which persist autonomously, whole or rearranged.
We employed a similar approach with the dihaploids derived from Alca Tarma. Chromosomal addition or introgression comparable to the described cases should be evident by the appearance of whole chromosomes or large segments containing an HI centromere (Riera-Lizarazu et al. 1996; Zhao et al. 2013; Tan et al. 2015). Although aneuploidy was common, convincing evidence of chromosomal contribution from the HIs was not observed, consistent with AFLP analysis on this set (Velásquez et al. 2007) and analysis of another dihaploid population (Samitsu and Hosaka 2002). The capability of our method to identify long chromosomal segments that diverge in SNP or copy number is validated by in silico reconstructions (Materials and Methods) and its effectiveness in comparable systems (Tan et al. 2015; Henry et al. 2015), indicating that the transfer of large segments of HI DNA (Riera-Lizarazu et al. 1996; Zhao et al. 2013; Kuppu et al. 2015; Tan et al. 2015) did not take place.
Assessment of small introgressions is more challenging. Recent genotyping (Bartkiewicz et al. 2018) or sequencing (Pham et al. 2019) of other dihaploid potato populations found that ∼1% of SNP loci displayed the presence of HI DNA in very small tracts, and with lower than expected allelic ratios. As in these reports, using high-coverage sequence data from three dihaploids, we detected many, widely dispersed SNPs represented by proportionally fewer aligned reads than expected for the addition of an HI DNA segment, or alternatively, replacement of a non-HI haplotype by homologous recombination. To address these observations, Pham et al. have suggested that tens of thousands of recombination events took place in each dihaploid during early growth before the HI genome was eliminated. Further, they have proposed that low allelic frequency could be explained by tissue chimerism. Mechanistically, this type of short introgression could be explained by somatic recombination caused by double-stranded DNA breaks followed by synthesis-dependent strand annealing or dsDNA break repair (Pâques and Haber 1999). Notably, while recombination of ectopic sequences has been demonstrated in plants (Puchta 1999; Čermák et al. 2017; Filler Hayut et al. 2017), these events are infrequent and require careful interpretation (Puchta and Hohn 2012). In this case, the scale of these changes ranged from ∼30,000 to 300,000 per sequenced haploid and affected all examined haploids (Pham et al. 2019), implying extremely high efficiency of recombination. The hypothesis of autonomous replication of HI DNA segments, for which there are precedents (Cohen et al. 2008; Shibata et al. 2012), could relieve the need for recombination. Nevertheless, it would also require high-efficiency propagation of extrachromosomal elements. These problems suggest a conservative interpretation of our data: these signals are artifactual and could have causes comparable to those identified in other gene conversion studies (Wijnker et al. 2013; Qi et al. 2014).
We conclude that, for the cross between HIs IVP101 or PL4 and tetraploid Alca Tarma, either the mechanism of haploid induction did not involve egg fertilization or genome elimination resulted in the loss of all HI chromosomes before the plants were evaluated. Events resulting in chromosome addition or introgression may be infrequent. For this reason, it may be premature to rule out genome elimination until more dihaploids derived from different parental combinations are evaluated. If haploid induction acts via genome elimination, both the addition of large DNA segments in the form of chromosomes and the rare introgression of small segments via recombination could be identified, and used for manipulation of the potato genome.
Our findings suggest that after tuberosum × phureja crosses, plants derived from seeds that did not express the purple spot marker and that display 2x genome content by flow cytometry are likely to be clean dihaploids (free of pollinator genome). That 11.4% of the dihaploid population evidently escaped initial screening against aneuploidy based on chloroplast counts, visual phenotyping, and chromosome counts underscores the difficulty of identifying aneuploids in highly variable dihaploid progeny. Their occurrence is consistent with the high frequency of aneuploid gametes in autotetraploids (Comai 2005) and with the ability of certain genotypes to tolerate imbalance (Rick and Notani 1961; Henry et al. 2010). The frequency of chromosome 2 trisomy, which carries the nucleolar organizing region of potato, may be explained if increasing the ribosomal RNA gene copy number offsets the disadvantage of linked dosage imbalance. Alternatively, ribosomal gene transcription (or some other unknown feature) could interfere with segregation (Tomson et al. 2006).
Conclusions
We undertook this investigation with the objective of assessing large-scale structural variation and its causes in dihaploids produced through genetic induction. Using cost-effective low-pass sequencing, we documented extensive, large-scale structural variation affecting > 52% of the genome. We found that 11% of the dihaploids were trisomic, frequently for the chromosome that carries the nucleolar-organizing region. In spite of multiple previous reports of genomic contamination by the HI used as the pollinator, we did not detect any large-scale introgression of the HI genome.
Acknowledgments
The authors thank Awais Khan for encouraging and coordinating the early phase of this collaboration; Michell Feldmann, Jordan Weibel, Jeanine Montano, and Helen Tsai for constructive comments on writing and data analysis; and the CGIAR Collaborative Program on Roots Tubers and Bananas for supporting the development of the LOP population used here. This work was supported by the National Science Foundation Plant Genome Integrative Organismal Systems (IOS) Grant 1444612 (Rapid and Targeted Introgression of Traits via Genome Elimination) to L.C.
Author contributions: K.R.A., E.M., E.H.T., M.B., I.M.H., and L.C. designed experiments. M.S. performed cytological analysis. K.R.A., B.O., and E.H.T. performed experiments. K.R.A. and L.C. analyzed data and wrote the manuscript with input from all authors.
Footnotes
Supplemental material available at figshare: https://figshare.com/projects/Genomic_Outcomes_of_Haploid_Induction_Crosses_in_Potato_Solanum_tuberosum_L_/70793.
Communicating editor: J. Birchler
Literature Cited
Author notes
Present Address: Duquesa Business Centre, P. O. Box 157, Manilva, Malaga, Spain 29692.

{kind=link}
{kind=link}
{kind=link}
{kind=link}