





Abstract
DNA–protein cross-links (DPCs) are frequently occurring lesions that provoke continual threats to the integrity of the genome by interference with replication and transcription. Reactive aldehydes generated from endogenous metabolic processes or produced in the environment are sources that trigger cross-linking of DNA with associated proteins. DNA repair pathways in place for removing DPCs, or for bypassing them to enable completion of replication, include homologous recombination (HR) and replication fork remodeling (FR) systems. Here, we surveyed a set of mutants defective in known HR and FR components to determine their contribution toward maintaining resistance to chronic formaldehyde (FA) exposure in Ustilago maydis, a fungus that relies on the BRCA2-family member Brh2 as the principal Rad51 mediator in repair of DNA strand breaks. We found that, in addition to Brh2, Rad52 was also vital for resistance to FA. Deleting the gene for Rec8, a kleisin subunit of cohesin, eliminated the requirement for Brh2, but not Rad52, in FA resistance. The Rad51K133R mutant variant that is able to bind DNA but unable to dissociate from it was able to support resistance to FA. These findings suggest a model for DPC repair and tolerance that features a specialized role for Rad52, enabling Rad51 to access DNA in its noncanonical capacity of replication fork protection rather than DNA strand transfer.
PROTEINS covalently cross-linked to DNA pose a significant threat to genome integrity as such adducts have the potential to hinder transcription and block DNA replication. DNA–protein cross-links (DPCs) are commonly found, highly abundant lesions and can arise from exposure to environmental agents such as aldehydes, certain metals, and ionizing radiation (Barker et al. 2005), and from physiological processes involving DNA and protein oxidation (O’Brien et al. 2005) such as reaction of abasic sites in DNA with nucleosomal core histones (Sczepanski et al. 2013). Reactive aldehydes can arise metabolically (O’Brien et al. 2005; Rosado et al. 2011; Pontel et al. 2015) from lipid peroxidation byproducts, breakdown of ethanol to acetaldehyde, and from enzymatic oxidative demethylation reactions, i.e., histone, RNA, and DNA demethylations, which produce formaldehyde (FA) in immediate proximity to DNA (Kooistra and Helin 2012; Walport et al. 2012). Continual exposure to DPC-inducing agents from environmental pollutants and endogenous sources presents an unrelenting challenge to the cellular systems dedicated to genomic stability (Swenberg et al. 2013).
From molecular and genetic studies performed in microbial as well as metazoan systems it has been found that different repair pathways converge to act in eliminating or tolerating DPCs induced by FA (de Graaf et al. 2009; Ide et al. 2011; Grogan and Jinks-Robertson 2012; Stingele and Jentsch 2015; Vaz et al. 2017). These include the nucleotide excision repair (NER) pathway, DPC-targeted proteolysis (DPC-PR) coupled with translesion bypass synthesis, fork remodeling (FR), and the homologous recombination (HR) system. Emerging models (Stingele and Jentsch 2015; Vaz et al. 2017) envision deployment of the NER system to eliminate short oligonucleotide stretches with cross-linked protein adducts from the DNA (see Figure 1A). DPCs escaping removal would be targeted for degradation by a specific DPC-protease, the action of which is to break down the bulk of the cross-linked protein adduct. Any remnant peptide still tethered to the DNA backbone could still pose a barrier to replicative DNA polymerases, but the site could be traversed by translesion polymerases. The HR and/or FR systems could be brought into play to circumvent the adducted site by switching templates to enable recombinational bypass, to promote remodeling of forks to bypass lesions with or without recombinational involvement to restart replication, and/or to rejoin any fragmented DNA arm broken off by endonucleolytic cleavage of reversed forks. Despite the overall general understanding, many questions concerning mechanisms remain unanswered. Sequential order of action of the various pathways is not yet clear and the hierarchy in the deployment and the contributions of the different repair systems to the overall process appears to differ depending on the organism (Nakano et al. 2007, 2009; Ridpath et al. 2007; de Graaf et al. 2009).
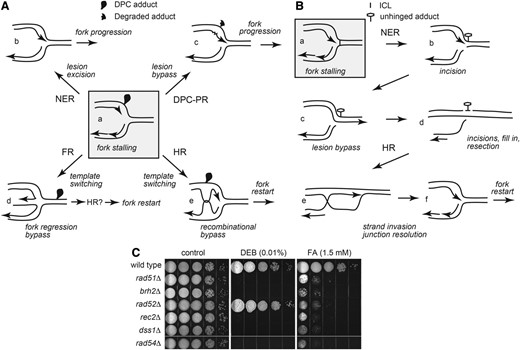
Pathways for resuming replication after fork stalling at cross-links. (A) Repair and tolerance of a DPC. Shaded box (a) adduct on a leading strand template stops replication. (b) NER excision of a tract of nucleotides containing the cross-linked protein adduct. (c) DPC-PR degrades the protein allowing a translesion polymerase to synthesize (squiggle) past the lesion. (d) FR enables regression followed by HR or reversal to restart replication. (e) HR enables template switching of nascent strands to bypass the lesion. (B) Repair of an ICL. A single fork is shown for simplicity in this illustration, but emerging evidence indicates convergence of two forks is required for ICL repair (Zhang et al. 2015). Shaded box (a) replication fork stalling at an ICL. (b) NER structure specific nucleases incise the backbone of one strand unhinging the lesion but creating a single-ended DNA DSB on the other chromatid arm. (c) Fill-in synthesis bypasses the lesion. (d) NER removal of the lesion from the complementary strand and end processing in preparation for HR. (e) HR strand invasion rejoins the broken arm. (f) Resolution of the recombination intermediate to reform the replication fork. Arrowheads in the forks indicate 3′ ends of extending strands. (C) Contribution of HR components to cross-link repair and tolerance. Mutant strains deleted of HR core components were tested for sensitivity to chronic exposure to FA and DEB as described in Materials and Methods. Results shown here and in subsequent figures are representative of multiple determinations. At least three independent determinations were performed for each strain.
In principle, repair of DPCs appears to proceed by a paradigm (see Figure 1B) with features in common with that envisioned for repair of DNA interstrand cross-links (ICLs) (Zhang and Walter 2014; Stingele et al. 2017; Vaz et al. 2017). Here, mounting evidence indicates replication fork progression blocked by an ICL triggers recruitment of NER-associated and other structure-specific nucleases to incise the DNA backbone of one strand on both sides of the adduct unhooking it (Niedernhofer et al. 2004; Huang and D’Andrea 2010; Hodskinson et al. 2014). Translesion polymerases are then enlisted to bypass the adduct-linked nucleotide residue, and HR components are brought in to restore the fork by HR. In metazoans, an additional set of proteins is required for ICL repair (Moldovan and D’Andrea 2009; Kottemann and Smogorzewska 2013; Zhang and Walter 2014). This set, comprising the Fanconi anemia core complex, has ubiquitin ligase activity that promotes monoubiquitination of FANCD2 and FANCI and is essential for ICL repair, possibly by structurally altering components and preparing chromatin in the locale of the lesion for repair processing. Emerging evidence from studies in chicken (Ridpath et al. 2007; Rosado et al. 2011), mammalian (Noda et al. 2011; Pontel et al. 2015), and amphibian systems (Duxin et al. 2014) indicates a role for these factors in DPC repair as well.
HR involves a number of distinct steps (Kowalczykowski 2015). These include preparation of DNA broken ends to reveal single-stranded tracts, assembly of the Rad51-DNA nucleoprotein filament, DNA strand invasion of a homologous sequence and formation of a joint-molecule intermediate, associated repair synthesis, and resolution of DNA structural intermediates. Processing of ICLs is associated with formation of DNA double-strand breaks (DSBs) at sites of stalled replication forks (Moldovan and D’Andrea 2009; Kottemann and Smogorzewska 2013), so it would be expected that the components of the canonical HR system would be brought into play to repair DNA damaged by exposure to diepoxybutane (DEB), which is a well-established bifunctional cross-linking reagent targeting DNA that is used as a diagnostic reagent in testing for Fanconi anemia (Auerbach 2015). Repair of DPCs, on the other hand, does not necessarily proceed through formation of DNA DSBs, so the requirement for HR in the process might not be as absolute as in ICL repair. There have been conflicting reports on the contribution of HR factors in DPC repair in vertebrate cells (e.g., Ridpath et al. 2007; Noda et al. 2011; Rosado et al. 2011), which might be a reflection of a more flexible dependence on HR or differences in assay systems. In view of the incompletely understood role for HR in repair of DPCs, we were interested in investigating the requirement for HR factors in survival during chronic exposure of Ustilago maydis cells to FA. U. maydis is a nonmainstream, but well-characterized yeast with a DNA DSB repair system closer in certain ways to that of human compared with conventional microbes. For example, HR depends on a BRCA2 ortholog, which is not present in the mainstream yeast model systems (Kojic et al. 2002), rather than Rad52, and the nonhomologous endjoining Ku proteins are essential in human, contrary to findings in the mainstream yeasts (de Sena-Tomás et al. 2015).
We were curious about the role of HR factors in DPC repair in U. maydis and wondered if the same components required for processing DNA ICLs would be brought into play in handling DPCs. Therefore, we investigated the requirement for HR components in resistance to FA compared with DEB. We found there is overlap in the requirements for HR factors in repair of DPCs and ICLs, but not complete coincidence. Of particular interest was the finding that the Rad51 mediator requirement for resistance to FA switched from Brh2 to Rad52 depending on the status of the Rec8 kleisin subunit of cohesin.
Materials and Methods
U. maydis genetic methods
Manipulations with U. maydis, culture methods, transformation, gene transfer and disruption procedures, and survival after DNA damage have been described previously [see Kojic et al. (2008) and references therein]. Strains used are tabulated (Supplemental Material, Table S1). Characterization of a number of DNA repair genes in this study has been reported previously. New genes in the study were identified from the U. maydis genome sequence in the annotated Munich Information Center for Protein Sequences (MIPS) database (http://pedant.helmholtz-muenchen.de/) by sequence alignment through BLAST analysis. Genes were disrupted by replacing the open reading frame with cassettes expressing resistance to hygromycin B (HygR), nourseothricin (NatR), or geneticin (G418R) by standard methodology (Kojic et al. 2002). Deletion mutants were verified by PCR analysis using a set of primers internal to the deletion interval to show removal of the open reading frame. Spot assays were performed by making serial 10-fold dilutions from the initial cell suspension of 2–4 × 107/ml then spotting 10 µl aliquots of each dilution in sequence on solid medium containing FA or DEB. Plates were incubated for 3–5 days at 28° for colonies to develop. Cell counting was performed with a microscope using a hemocytometer. Solid medium (1% yeast extract, 2% peptone, 2% sucrose) containing FA was prepared by mixing a freshly prepared 1 M aqueous stock of FA with autoclaved medium cooled to 65°, then allowed to solidify and used within 1 day. Results presented are representative of multiple determinations. At least three independent isolates and measurements were performed for each strain.
Sister chromatid recombination assay
A reporter gene cassette for measuring unequal sister chromatid recombination (SCR) was prepared by joining in tandem two fragments of a gene expressing resistance to hygromycin. Starting with pCM54 as a source of the hygromycin resistance marker (hsp70 promoter-hph gene fusion) contained on a 2.0 kb XhoI–SalI fragment (Kojic and Holloman 2000), a three-way ligation was performed by inserting a 1.5 kb XhoI–SacII fragment containing the proximal region of the phsp70-hph gene and a 1.0 kb EcoRI–SalI fragment containing the distal region of the phsp70-hph gene into pBluescript II opened with EcoRI–SacII. This duplicates a 0.5 kb internal fragment of the hph gene and reverses the configuration of the promoter and terminator sequences. The 2.5 kb fusion was then moved as a HindIII–SacI fragment into pCM691 (Kojic and Holloman 2000) opened with HindIII–SacI. The resulting plasmid was cut with AgeI and targeted to the sdh2 locus in different U. maydis strains by selection for carboxin resistance. SCR was measured by seeding 105 cells containing the SCR reporter into 2 ml YEPS medium with or without 0.75 mM FA or 0.002% DEB, then expanding the cultures for 10 generations. Aliquots were then plated on YEPS medium to determine viability or else on medium containing 100 µg/ml hygromycin to select for recombinants.
Expression of Rad51 and Rad52 variants
Rad51 variants were constructed by site-directed mutagenesis using a QuikChange II kit (Agilent) according to manufacturer instructions. Gene variants were moved into self-replicating plasmid pCM619 (Kojic and Holloman 2000) with selectable carboxin resistance marker (CbxR) for expression under control of the glyceraldehyde 3-phosphate dehydrogenase promoter as described previously (Zhou et al. 2007). Likewise, genes encoding Rad52, the N-terminal fragment encompassing residues 1–359 (Rad52NT), and the C-terminal fragment encompassing residues 359–722 (Rad52CT) were expressed in pCM619 as described previously (Kojic et al. 2008, 2011).
Data availability
The authors state that all data necessary for confirming the conclusions presented in the manuscript are represented fully within the manuscript. Supplemental material available at Figshare: https://doi.org/10.25386/genetics.6887282.
Results
Contribution of HR components to repair of DPCs
We have been interested in understanding the mechanism of repair of DPCs in U. maydis, particularly with regard to involvement of the HR system, and in learning how this compares to repair of ICLs. As these types of lesions stall replication fork progression, but impose structurally different obstacles, we were curious about the deployment of HR and associated factors in coping with them. We used a genetic approach to ask which HR components and associated factors could be involved in DPC repair and how these compared with ICL repair. We focused specifically on mutants defective in HR core components and FR factors, but chose not to examine mutants in substrate preparation and intermediate processing, or in DNA-damage signaling. For experimental purposes, we took resistance to chronic exposure to FA as a measure of DPC repair proficiency and resistance to DEB as a measure of ICL repair, although with the caveat that the specificity of both these agents is by no means strictly confined to these types of lesions (McGhee and von Hippel 1975; Michaelson-Richie et al. 2010; Millard et al. 2012; Hoffman et al. 2015). Both are highly reactive chemicals with broad specificities and with overlap in their respective capabilities in cross-link formation. However, the cytotoxic effects of FA are generally attributed to DPCs (Solomon and Varshavsky 1985; Barker et al. 2005; Lu et al. 2010; Stingele et al. 2014) while those of DEB are attributed to DNA interstrand cross-links (Hartley et al. 1999; Park and Tretyakova 2004; Wadugu et al. 2010). Resistance was assessed as colony forming ability on agar medium containing the cross-linking agent after incubation for several days.
In U. maydis, the primary mediator for Rad51 in DNA DSB repair is the BRCA2 ortholog Brh2. In earlier studies it was found that the brh2∆ mutant phenocopied rad51∆ in loss of resistance to DNA clastogens, in deficiency in gap repair and allelic recombination, and in failure to complete meiosis (Kojic et al. 2002). In accord with that work, we found that Brh2 was required for resistance to both DEB and FA to a similar degree as Rad51 (Figure 1C). These findings reinforced the notion of Brh2 primacy in Rad51 mediator function. However, a surprising exception to this seemingly supreme role of Brh2 as Rad51 mediator came when we examined the rad52∆ mutant. As with previous studies in testing other chemical genotoxins, we found that Rad52 was expendable for DEB resistance, as expected. Unexpected, however, was the finding that Rad52 was required for survival in FA.
Rad52 has had an enigmatic role in DNA repair in U. maydis. As in higher eukaryotes, removal of the gene for Rad52 in U. maydis results in little to no effect on HR or repair of damage to DNA caused by a range of clastogens, including UV or ionizing radiation, or reactive chemicals such as methylmethane sulfonate or cisplatin (Kojic et al. 2008). This stands in contrast to the situation in budding yeast (which lacks a BRCA2 ortholog), where Rad52 governs all aspects of HR and its loss results in profound sensitivity to ionizing radiation and numerous genotoxins including FA (de Graaf et al. 2009). It has been speculated that Rad52 serves as a backup in the absence of BRCA2, but the dual requirement for both Brh2 and Rad52 in FA resistance suggests independent, rather than redundant, functions for these two mediators. Although we have reported previously (Kojic et al. 2008) that loss of Rad52 has a synthetic DNA repair phenotype in the absence of Mph1, Ercc1, or the Rad51 paralog Rec2, this newly documented role for Rad52 in FA resistance is the first instance in our investigations of U. maydis where we have observed a single, stand-alone gene requirement for Rad52 in any DNA repair process.
Other factors involved in the strand invasion step of HR and already established as important contributors to DNA repair proficiency were also examined for involvement in DPC repair. Dss1, an essential Brh2 cofactor (Kojic et al. 2003), and Rec2, a Rad51 paralog (Rubin et al. 1994), were important in both DPC and ICL repair. Rad54, which interacts physically with Rad51 and functions as a motor to drive branch migration (Bugreev et al. 2006) and to pump heteroduplex (Wright and Heyer 2014), was also crucial. These observations indicate that Rad51-promoted homologous pairing and DNA strand invasion likely comprise important steps in the DPC repair pathway, but as Brh2 is the primary Rad51 mediator during HR (Yang et al. 2005) it is by no means clear from this initial survey how Rad52 might be contributing to DPC repair or tolerance.
FR in DPC repair and tolerance
FR provides an important protective mechanism that allows replication forks to reverse course upon encountering an obstacle, to bypass the block by switching templates, then to restore the fork structure so that synthesis can resume (Sidorova 2017; Bhat and Cortez 2018). The process is thought to proceed by coordinated pairing of the two newly synthesized strands and reannealing of the parental strands such that the three-way fork regresses into a four-way structure resembling a Holliday junction (Figure 2A). The regressed arm is then imagined to be subject to additional processing—gap filling synthesis followed by branch migration and fork restoration, or 5′ end resectioning to expose a 3′ protruding single strand suitable as a substrate for Rad51-promoted strand invasion. By either means, the blocking lesion can be bypassed and the fork reconfigured for restarting.
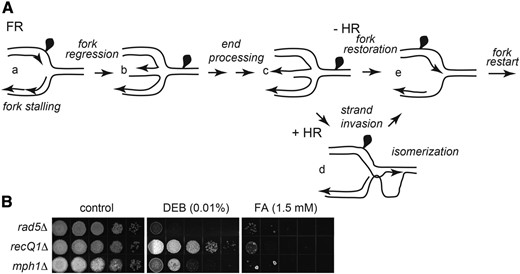
Pathway for bypass of a DPC by FR. (A) Schematic with arbitrary placement of DPC. (a) Protein adduct on a leading strand template stops replication. (b) Fork regression produces an additional DNA branch with a recessed 3′ end. (c) After fill-in synthesis and 5′-end resection a protruding 3′ single-stranded end is revealed. (d) HR promoted 3′-end strand invasion of the newly duplicated tract with the homologous sequence produces a D-loop intermediate. (e) Reversal of the regressed extended fork structure in (c) or isomerization of the HR intermediate in d resets the replication fork junction past the lesion. (B) Contribution of FR components to cross-link repair and tolerance. Mutant strains deleted of FR components were tested for sensitivity to chronic exposure to FA and DEB as in the legend to Figure 1.
Multiple factors that appear capable of FR have been identified in eukaryotic systems. It has been shown in vitro that fork reversal can be catalyzed in yeast by the Rad5 translocase and the Mph1 helicase, and in human by a variety of translocases and helicases that includes the Rad5 and Mph1 orthologs along with other factors not evident in the genomes of lower eukaryotes (Neelsen and Lopes 2015; Quinet et al. 2017; Sidorova 2017; Bhat and Cortez 2018). In addition, the translocase RAD54 in cooperation with RAD51 has been noted to drive fork reversal and promote fork restoration (Bugreev et al. 2011). The multiplicity of factors might indicate redundant roles or different structural specificities for fork regression. To restart replication, the three-way fork needs to be restored. Human RECQ1 was identified as a DNA helicase specialized in restoring reversed forks after topoisomerase I inhibition or genotoxin treatment (Berti et al. 2013; Thangavel et al. 2015; Zellweger et al. 2015).
Orthologs of Rad5, Mph1, and RecQ1 are evident in U. maydis. Rad5 is an ATP-dependent DNA translocase in the SWI/SNF2 family (Unk et al. 2010) for which there is genetic as well as biochemical evidence for a role in fork regression (Torres-Ramos et al. 2002; Blastyák et al. 2007) and replication restart following fork stalling (Minca and Kowalski 2010). Similar to human HTLF, Rad5 contains a HIRAN domain, which is a 3′ ssDNA end binding module demonstrated to be important for fork reversal and proper fork progression following replication stress, but lacks DNA unwinding activity (Kile et al. 2015). The U. maydis rad5∆ mutant was highly sensitive to both FA and DEB (Figure 2B), indicating a likely prominent role of FR in DPC repair. Mph1, an ortholog of the FANCM protein, is a 3′-5′ helicase in the SF2 helicase superfamily (Prakash et al. 2005) shown to dissociate recombination intermediates in vitro (Prakash et al. 2009; Silva et al. 2016), and to play an important role in break-induced replication (Stafa et al. 2014). RecQ1 is the second RecQ-family DNA helicase besides Blm that is present in U. maydis. We found that Mph1 and RecQ1 were both strongly required for resistance to FA. Mph1 was also required for resistance to DEB, but RecQ1 was not.
To summarize, the sensitivities of the various HR and FR mutants to FA are in line with a general model as represented in Figure 1A that features multiple systems in place in U. maydis to help cells repair or tolerate damage caused by formation of DPCs. The findings are consistent with a model in which replication fork stalling caused by introduction of DPCs is defended and overcome in part by a set of HR factors that overlap with those dedicated to repair of ICLs, but that are not completely coincident. The sensitivity to FA resulting from loss of FR factors in addition to HR factors suggests that FR operating in conjunction with the related HR system is a preferred cellular mode for overcoming obstacles to replication caused by DPCs. The most provocative finding among these, from our perspective as proponents of HR mechanisms, is the requirement for Rad52 in FA resistance. Among many different genotoxins examined (Kojic et al. 2008), FA appears unique in eliciting a specific response from Rad52 as a required repair component. The involvement of Rad52 in FA resistance suggests that it does not serve as a backup function for Brh2, but rather is dedicated to repair of a specific type of challenge.
An unorthodox role for Rec8 in recombination associated with DPCs and ICLs
A clue to the role of Rad52 in DPC repair came from a chance observation. In a parallel, but unrelated, study on Rec8, the meiotic kleisin subunit of cohesin, we observed that deletion of the gene led to profound disturbance of meiosis, as was expected based on findings in other systems. Unanticipated, however, was the finding that deletion of the gene also had an effect in mitotic cells—namely a defect in SCR, although there was no sign of any DNA repair defect as evidenced by no loss of resistance of the rec8∆ mutant to DNA clastogens (Figure S1). SCR was measured by an assay similar in design to one developed originally in yeast (Fasullo and Davis 1987). It was based on reconstruction of a drug resistance marker hph (hygromycin phosphotransferase) from two overlapping fragments of the gene reconfigured such that a duplicated internal sequence was arranged in a tandem array (Figure 3A). Hygromycin resistance in haploid cells with an integrated copy of the reporter cassette would be presumed to arise from unequal sister chromatid exchange or conversion events to reconstruct the hph gene. Using this assay, we noted a 10-fold reduction in the spontaneous level of SCR in the rec8∆ mutant compared with wild type, and a decreased level of induction compared with wild type in cells grown in the presence of FA or DEB (Figure 3B).
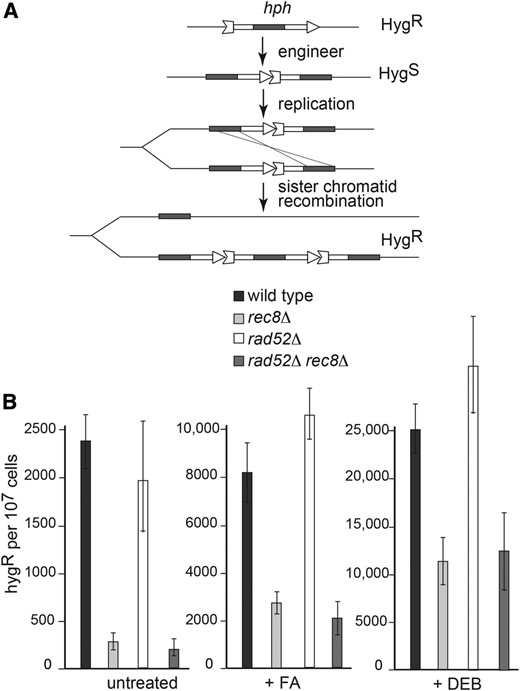
Sister chromatid recombination assay. (A) Schematic illustrates the construction of the SCR reporter from the hygromycin phosphotransferase gene. Hygromycin resistant recombinants arise from an unequal SCR event. (B) SCR frequency determinations were performed on the indicated different strains grown in the absence or presence of FA or DEB as described in Materials and Methods. Median values of triplicates from three to five independent experiments were used to determine SD (error bars).
These findings imply that Rec8 is expressed to some degree in mitotic cells of U. maydis, and suggest that it plays a contributing role, most likely in the context of cohesin in promoting intersister chromatid dynamics. It could be imagined, for instance, that, in U. maydis, the pool of mitotic cohesin is comprised of a population of molecules with both Rad21 and Rec8 as kleisin components. In this event, perhaps the Rec8-containing subset provides some subtle architectural contribution to mitotic chromosome structure that would not be evident under most circumstances. This contribution would not be required for establishing cohesion after DNA DSBs as in budding yeast (Ström et al. 2004; Heidinger-Pauli et al. 2008), but perhaps could be called into play under certain specific conditions of stress. In budding yeast, mutants defective in Scc1/Mnd1/Rad21, the mitotic kleisin, accumulate reduced levels of X-shaped sister chromatid junctions compared with cohesin-competent strains (Fumasoni et al. 2015). These structures are believed to form as intermediates during HR-mediated damage bypass by template switching as a consequence of replication stress (Giannattasio et al. 2014). Given that the role of the cohesin complex in template switching resulting from fork stalling is likely in maintaining a certain structural conformation and/or configuration of newly replicated sister chromatids, we entertained the question of whether depleting Rec8 might be consequential in DPC or ICL repair.
The requirements for HR mediators and FR components in DPC repair change in the absence of Rec8
Intrigued by the notion that, in the absence of Rec8, the architectural configuration of newly replicated sister chromatids might be altered due to a disturbance in the makeup of the cohesin pool, we re-examined the contribution of HR and associated FR factors to FA and DEB in a rec8∆ mutant background (Figure 4). In this situation, the requirement for HR and FR factors shifted. Most striking was the finding that while resistance to FA was dependent on Rad51 and Rad52, it was no longer dependent on the primary mediator Brh2, or the Rad51 paralog Rec2, or the fork remodeler Rad5. Yet resistance to DEB still required Rad51, Brh2, Rec2, and Rad5, but not Rad52. These observations suggest that, in the absence of Rec8, a pathway involving template switching with branches that enable recombinational bypass with or without fork regression bypass of DPCs is blocked, leaving a Rad52-dependent channel as an alternative mode for utilizing Rad51 in maintaining resistance to FA. Without Rec8, in other words, Rad52 takes over in providing the mediator function for Rad51 in repair or tolerance of DPCs. It also remains possible that the strand annealing and strand pairing activities of Rad52 (Mortensen et al. 1996; Kagawa et al. 2001) might also be important in this Rec8 independent pathway, but in ways that are by no means clear. In this light, it is appropriate to consider a modification to the original scheme of pathways proposed above in which an additional alternative mode of Rad51 action is put into play.
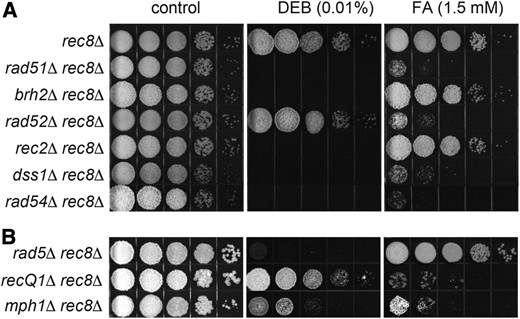
Eliminating Rec8 alters HR component requirements in cross-link repair. Mutant strains defective in HR core components (A) or FR components (B) were tested for sensitivity to chronic exposure to FA and DEB as described in Materials and Methods. Representative results of at least three independent determinations for each strain are shown.
Notable also in the rec8∆ mutant background was the continued requirement for Dss1 in the absence of Brh2 in promoting FA resistance. This marks a separation in function between Brh2 and Dss1 (Kojic et al. 2003), and is probably indicative of the wider role of Dss1 beyond HR, possibly into the realm of mRNA processing (Kragelund et al. 2016). These observations support the idea that the constellation of HR and FR factors dedicated to rescuing and restoring the integrity of the genome corrupted by cross-links is dynamic, and responds not only to the nature of the cross-link but also to changes in sister chromatid architecture governed by cohesin. In particular, the preference for Rad52 as mediator for Rad51 over Brh2 revealed in the absence of Rec8 raises the possibility that Rad51 contributes to DPC repair or tolerance through a mode other than through its canonical DNA strand invasion function.
A noncanonical role for Rad51 in DPC repair
Besides its catalytic role in DNA repair in promoting strand invasion, Rad51 has been demonstrated to serve in a protective capacity to prevent destruction of replication forks stalled by hydroxyurea poisoning (Hashimoto et al. 2010; Petermann et al. 2010; Schlacher et al. 2011; Spies et al. 2016; Bhat and Cortez 2018; Feng and Jasin 2018). For this noncanonical mode of action, BRCA2 has been shown to serve as mediator in promoting assembly of Rad51 filaments on stalled forks to shield them against Mre11-dependent degradation (Schlacher et al. 2011; Kolinjivadi et al. 2017; Feng and Jasin 2018). In light of this alternative Rad51 function, it seems that our original notion of DPC repair through pathways involving recombination and fork regression is too simplistic, and that another branch involving Rad51 in its noncanonical role of template stabilization is warranted. A scenario can be envisioned as follows. While the Brh2-dependent recombination branch and the Rad5-dependent fork regression branch appear to require Rec8, the additional branch is revealed when access to those two branches is blocked by elimination of Rec8. This third branch is dependent on Rad52, presumably through action as mediator for Rad51 in a fork protection role.
We approached the question of whether Rad52 works with Rad51 in a noncanonical fork protection mode rather than with Rad51 in its canonical DNA strand exchange role along two avenues. First, we asked if Rad52 was involved in SCR either spontaneous or induced by exposure to FA or DEB. In line with previous findings on Rad52 and interchromosomal recombination (Kojic et al. 2008) we found no role for Rad52 in SCR (Figure 3B). This result, while negative, is consistent with the notion that repair or tolerance of DPCs involving Rad52-mediated delivery of Rad51 to DNA is unlikely to proceed through a mechanism involved in DNA strand switching dynamics.
In the second approach, we asked whether certain Rad51 variants inactivated in specific functions by different mutations could illuminate a possible fork protection branch. It could be imagined, for instance, that fork protection would require Rad51 to bind DNA but not necessarily to exchange strands. We chose three different mutant variants of Rad51 to examine (Figure 5A). The first contained the K133R mutation in the Walker A box. This K133R variant is able to bind DNA, but is defective in ATP hydrolysis and is unable to dissociate from DNA once bound (Chi et al. 2006). While still capable of catalyzing DNA strand exchange in vitro (Chi et al. 2006), it is deficient in supporting DNA repair and recombination in vivo (Morgan et al. 2002), and confers a dominant negative phenotype when expressed in cells containing wild-type Rad51 by interfering with nucleoprotein filament dynamics (Stark et al. 2002; Kim et al. 2012). The second variant contained the F232A mutation in the L1 loop, which is crucial in DNA binding. Mutation of the corresponding conserved residue in the human variant Y232A was shown to cause reduced binding to both single-stranded and double-stranded DNA (Matsuo et al. 2006). The third variant contained three mutations R130A, R303A, and R313A corresponding to the three conserved residues in the budding yeast Rad51 II3A variant shown able to bind single-stranded DNA and support ATP hydrolysis, but defective in DNA strand exchange (Cloud et al. 2012). In studies on replication fork blockage with an inducible fork barrier in Schizosacchromyces pombe, the equivalent mutant was found to be proficient in protecting nascent strands from degradation (Ait Saada et al. 2017).
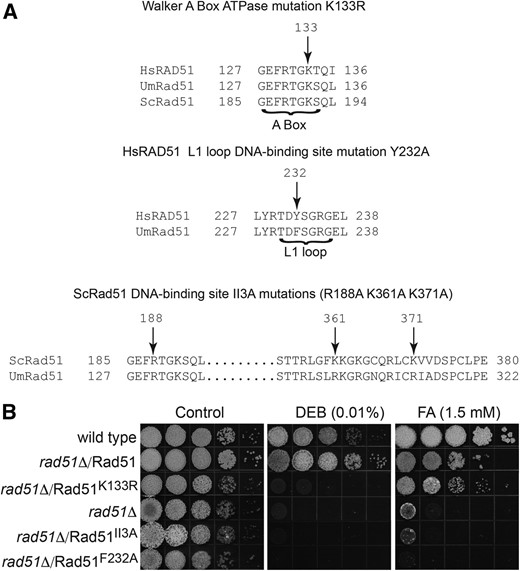
Rad51 mutant variants in cross-link repair. (A) Individual residues altered in Rad51 variants are shown in the context of specific protein sequences with alignments to yeast (Sc) or human (Hs) to demonstrate sequence conservation. Residue numbers of UmRad51 are the same as HsRad51, but differ from ScRad51 due to the presence of an extended leader sequence in the latter. The UmRad51II3 variant is Rad51R130A R303A R313A. (B) rad51∆ strains expressing different Rad51 variants as indicated were tested for ability to complement sensitivity to DEB or FA.
The three Rad51 variants were tested for activity in supporting DNA repair by expression in rad51∆ strains (Figure 5B). None was capable of supporting resistance to DEB indicating loss of functional activity in ICL repair. However, Rad51K133R was effective in promoting a substantial level of resistance to FA, although the other two variants were not. The implication is that, while Rad51K133R is defective in canonical DNA repair activity, it retains the capacity to function in a noncanonical protective role. The failure of the Rad51F232A and Rad51II3A variants to support FA resistance is consistent with the notion that both the primary and secondary DNA-binding sites of Rad51 are required to potentiate full fork protection under the conditions tested. These findings suggest that a Rad51 filament that is locked on DNA and unable to support HR, can serve effectively in maintaining resistance to FA, while Rad51 mutants defective in DNA binding remain ineffective. That Rad52 serves as primary mediator of Rad51 when Rec8 is absent supports the notion of a specialized Rad52-dependent fork protection pathway that is called into play when cells are challenged with FA.
A caveat to the interpretation above is the assumption that the U. maydis Rad51 variants under testing here behave functionally in the same manner as those variants from human and budding yeast, which were analyzed in detail biochemically and used first as models in the studies described. It remains a possibility that the U. maydis variants differ in ways not yet known, although this seems unlikely to us given the stringent conservation in amino acid sequence among these orthologs. It should also be noted that, while the engineered gene construct expressing Rad51 was able to complement fully the DEB sensitivity of rad51∆, it was only partially effective in complementing the FA sensitivity. We do not have an explanation for this, but duly note that the Rad51 construct introduced into the rad51∆ mutant is expressed ectopically and not from the natural promoter. It is possible that the Rad51 level in these cells is sufficient for DEB resistance but insufficient to mount full resistance to FA.
Toward genetic analysis of Rad52 function
The requirement for Rad52 in resistance to FA toxicity in U. maydis affords a singular opportunity to genetically dissect Rad52 and to learn of its function in a BRCA2-family-containing organism. By reintroducing Rad52 mutant variants into the rad52Δ mutant and monitoring complementation of the FA sensitivity, it should be possible to assess the importance of individual protein domains and residues of Rad52 in DPC repair.
As an expeditious illustration of the potential offered by this approach, we tested the ability of the cloned Rad52 gene and two different deletion variants to restore FA resistance to the rad52Δ mutant (Figure 6A). Structural arrangement of the Rad52 protein family members features a highly conserved protomer–protomer self-association and DNA-binding domain of ∼200 residues in the N-terminal region (Iyer et al. 2002; Kagawa et al. 2002), but less well defined Rad51 and RPA interaction domains in the divergent C-terminal region (Park et al. 1996; Seong et al. 2008). Introduction of a construct encoding the full-length Rad52 protein into the rad52Δ mutant restored FA resistance (Figure 6B). Constructs encoding the N-terminal and C-terminal fragments showed marginal and partial ability, respectively, to complement the FA sensitivity, suggesting that functional activity requires both DNA-binding and Rad51/RPA-interaction abilities. With more sophisticated mutational targeting, it should be possible to define more precisely the functionally important regions of Rad52.
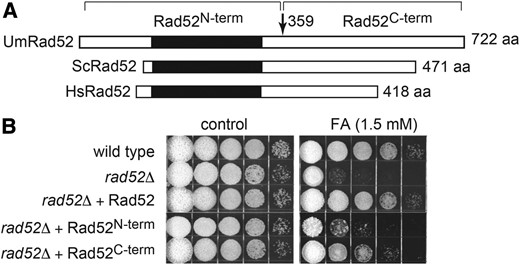
rad52Δ mutant complementation. (A) Schematic shows the region of the conserved DNA-binding/protomer-protomer interaction domain (black rectangle) in Rad52 from U. maydis (Um), S. cerevisiae (Sc), and Homo sapiens (HS) and the N-terminal and C-terminal fragments of UmRad52 that were expressed separately. (B) rad52∆ strains expressing the different Rad52 variants as indicated were tested for ability to complement sensitivity FA.
Discussion
We made three primary findings in this investigation. First, resistance to FA toxicity in U. maydis depends on a set of HR and replication FR factors that includes Rad52 in addition to the primary Rad51 mediator Brh2. Second, in the absence of Rec8, resistance to FA no longer depends on Brh2, Rec2, or Rad5, but remains dependent upon Rad52 and Mph1. Third, a Rad51 variant with a Walker A box mutation that is reported able to bind but not dissociate from DNA in vitro, and that is completely defective in ICL repair in vivo, can support resistance to FA.
Our findings are in accord with the emerging paradigm of DPC repair and tolerance. Namely that systems in place for coping with DPCs include those for enabling DNA synthesis to bypass damaged or modified sites by switching templates through recombination or FR modes. The salient finding from the survey of HR factors was the requirement for Rad52 in resistance to FA, but not DEB, unlike the other bona fide core HR components that were necessary for resistance to FA and DEB as well as other DNA clastogens. This observation raises the possibility that Rad52 does not serve in some minor capacity merely as a backup for Brh2 in mediating DNA repair processes, but rather that it functions in a specialized role in repair or management of DPCs. Support for a specialized role of Rad52 in DPC repair comes from the fortuitous finding that elimination of Rec8 abolishes the need for Brh2, Rec2, and Rad5, but not Rad52, in maintaining resistance to FA–Brh2, and Rec2 being the primary Rad51 mediators in DNA DSB and ICL repair in U. maydis, and Rad5 being a core component of the postreplicational repair system and a principal fork remodeler in template switching-dependent damage bypass. The clear implication is that, during DPC repair, Rec8 enables a pathway of molecular transactions involving repartnering DNA strands. In the absence of Rec8 with access to this pathway denied, due presumably to some change in architecture around the replication fork junction, there is still a need for Rad51, but for its role in fork protection rather than in DNA strand invasion. Channeling of Rad51 into this alternative, noncanonical mode involving DNA binding but not strand pairing is facilitated by Rad52. The mechanism directing this switch of Rad51 from strand pairing to protection mode is unknown, but likely results from some structural perturbation in sister chromatid configuration due to a subtle alteration in cohesion.
We imagine that replication forks stalled by a DPC can be shunted into two primary pathways (Figure 7). One that is Rec8 dependent uses the homologous pairing activity of Rad51 to bypass DPC lesions (Figure 7 two upward branches). This might occur through pairing of nascent strands to form an X-shaped intermediate (Figure 7b) or through strand invasion to form a D-loop recombination intermediate after Rad5-promoted fork regression (Figure 7e). Both of these branches or subpathways would be expected to rely on HR components involved in promoting strand transfer reactions as in DNA DSB repair, namely Brh2, Rec2, and Rad54. The second primary pathway, which is Rec8 independent (Figure 7 two downward branches), relies on protective activity afforded by the noncanonical role of Rad51 polymerizing on DNA to secure forks from degradation and enable restart, but with no DNA strand transfer activity involvement. This might occur through Rad51 filament formation directly on the fork structure (Figure 7g) or alternatively on a regressed fork (Figure 7, h and i) reversed by the action of Mph1. RecQ1, would then restore the fork structure to enable restart.
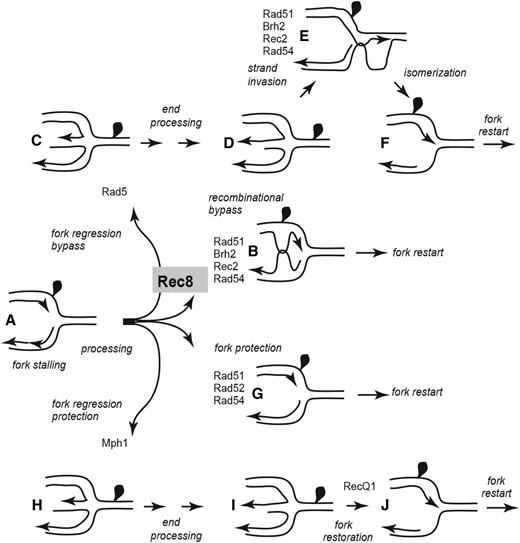
Rec8 controls access to branches in DPC repair enabling Rad51-promoted template switching. Rec8 is imagined to function as a gatekeeper that allows channeling of (a) stalled fork pictured with a DPC on the leading strand template into tolerance branches (curved arrows pointing upwards) that depend on Rad51 strand transfer activity, (b) template switching of nascent strands to bypass the lesion, (c) FR that leads to bypass of the adduct by strand invasion (d and e) of the duplicated sequence as in Figure 2 to (f) reset the replication fork. In the absence of Rec8 (curved arrows pointing downward), stalled forks are channeled into branches that (g) utilize Rad51 in its noncanonical protective function, or (h) promote regression, (i) fill-in synthesis, and (j) reversal to restore the replication fork. Individual HR and FR component requirements are tentatively placed at the relevant steps.
In the absence of any existing methodology in U. maydis that could help us visualize the structure of the FA-mediated stalled forks, we can only speculate about the mechanism of protection afforded by Rad51. Nevertheless, the analysis with the Rad51 variants offers a slight glimmer of light, albeit indirect, on the nature of the protective activity. We found that Rad51K133R was effective in promoting resistance to FA while Rad51F232A and Rad51II3A were not. Previous biochemical analysis of Rad51K133R from human revealed it to be proficient in nucleoprotein filament formation, deficient in ATP hydrolysis, and slow to dissociate from DNA, but more avid than the wild-type protein in binding both single-stranded and double-stranded DNA and in protecting DNA from restriction endonuclease cleavage (Chi et al. 2006). These findings suggest that there is minimal ATP hydrolysis-mediated turnover of Rad51K133R filaments so that the pool of free protomers available for dynamic HR reactions is limited. Rad51K133R could thus be well suited for a role in stalled/regressed fork protection by safeguarding both single-stranded and double-stranded stretches around the fork structure that could be vulnerable to nuclease attack. Rad51K133R has been demonstrated to be effective in protecting replication forks against nucleolytic degradation after hydroxyurea poisoning in human cells (Schlacher et al. 2011). That the Rad51F232A variant was ineffective in conferring FA resistance is consistent with decreased activity in binding to both single-stranded and double-stranded DNA of the corresponding Rad51Y232A mutant in human (Matsuo et al. 2006). On the other hand, it was perplexing to observe that the Rad51II3A variant was not active in promoting FA resistance given that the corresponding mutant in S. pombe was shown to be proficient in protecting nascent DNA strands from degradation after replication fork stalling caused by a protein bound at a specific site (Ait Saada et al. 2017). But it should be noted that while the Rad51II3A variant is capable of forming nucleoprotein filaments, it is slightly disturbed in DNA-binding ability (Cloud et al. 2012). Perhaps this partial defect is enough to preclude a role for the Rad51II3A variant in FA resistance. However, it could be a false comparison to equate this replication block in S. pombe formed by induction of Rtf1 protein, which mediates site-specific replication termination at an inverted duplication of the polar RTS1 barrier, to FA-induced DPCs. Rtf1 forms a complex multimeric structure likely involving different protein domains binding to multiple sites within the RTS1 locus in combination with intermolecular interactions between Rtf1 molecules and augmented by DNA looping (Eydmann et al. 2008). It seems likely that the molecular mechanisms involved in countering toxic DPCs would be different from those involved in regulating Rtf1-RTS1, which is a natural system that appears dedicated to optimizing the replication-coupled recombination event that underlies mating-type switching in S. pombe (Dalgaard and Klar 2001).
Our findings raise the question of how far conclusions of the role of Rad52 as a specialized Rad51 mediator and Rec8 as a regulator of DPC repair pathway choice can be generalized? That, of course, is a matter that only further experimentation can settle. However, we note that Rad52 has been reported to contribute to FA resistance in vertebrates (chicken DT40 cells) (Ridpath et al. 2007). That another group disagreed and reported no contribution (Rosado et al. 2011) could result from differences in the assay systems for measuring survival and possibly in the varying conditions of chronic vs. acute exposure used in the two studies. With regard to Rec8, experimentation for the most part has focused on its role in meiotic chromosome segregation. Rec8 has been noted, nevertheless, to be expressed at low levels in mitotic cells of fission yeast (Folco et al. 2017), and in human somatic cells its regulation has been found to be upregulated or downregulated in different cancer types, e.g., (Erenpreisa et al. 2009; Yu et al. 2017). The clear contribution of Rec8 to SCR in U. maydis argues strongly for a role in sister chromatid interplay in mitotic cells. Whether the findings with Rec8 and HR dependence in FA resistance can be extended beyond the U. maydis realm remain to be determined.
It was found in human cells that Rad52 inactivation is synthetically lethal with loss of BRCA2 function, and that DSB-induced HR and radiation-induced Rad51 focus formation are reduced when Rad52 was depleted in BRCA2-deficient cells (Feng et al. 2011). These findings were interpreted to mean that Rad52 operates in a BRCA2-independent pathway that mediates Rad51 in its capability for strand transfer for DNA repair. Similar findings were reported in an earlier study from our laboratory in which a synthetic loss of proficiency in DNA repair in the absence of Rad52 and Brh2 was also noted (Kojic et al. 2008) so it would appear that Rad52 can provide some alternative mediator action in the absence of Brh2. However, our suggestion that Rad52 might serve as mediator for Rad51 in the noncanonical role of fork protection, while Brh2 would serve when Rad51 is required for strand transfer, would appear to conflict with several recent findings in metazoan systems. It has been reported that BRCA2 is required for fork protection and restart after hydroxyurea induced replication stalling in human cells (Schlacher et al. 2011). This finding was confirmed and extended with use of Xenopus extracts to demonstrate that Rad51 filament formation licensed by Brca2 protected forks stalled by acute aphidicolin poisoning from degradation (Kolinjivadi et al. 2017). In this case, it was notable also that fork protection was enabled by Smarcal1, a DNA translocase motor protein related to Rad5 (Poole and Cortez 2017), most likely by enabling remodeling of forks to a reversed form that is defended from nuclease cleavage by Rad51. Perhaps, the solution to reconciling these different modes of mediator choice in Rad51 fork protection lies in the circumstance of the fork stalling and in the signaling emanating from the event. It can be imagined that depleting dNTP pools or inhibiting elongation by acute poisoning of Polα DNA polymerase creates states of DNA metabolic and structural disorder different from that of a fully operational replisome impeded in progression by encountering a physical obstacle. In this regard, the attributes of the physical blockage might also have different consequences on stalled fork structure. For instance, the effect on strand unwinding by the replicative Cdc45/Mcm2-7/Gins complex helicase (CMG) upon encountering protein adducts on the lagging strand template could differ from that by adducts on the leading strand template (Langston and O’Donnell 2017), and this in turn could differ from the impact of CMG encountering interstrand cross-links. It could well be that the nature of the stall dictates the choice of mediator, which in turn might direct or confine Rad51 action toward a particular channel for repair or tolerance.
Acknowledgments
We thank Lorraine Symington (Columbia University) and Lance Langston (Rockefeller University) for comments on the manuscript. This work was supported in part by grant 18-A8-104 from the STARR Cancer Consortium.
Footnotes
Supplemental material available at Figshare: https://doi.org/10.25386/genetics.6887282.
Communicating editor: S. Sharan

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}