





Abstract
As the only catalytic member of the Sir-protein gene-silencing complex, Sir2’s catalytic activity is necessary for silencing. The only known role for Sir2’s catalytic activity in Saccharomyces cerevisiae silencing is to deacetylate N-terminal tails of histones H3 and H4, creating high-affinity binding sites for the Sir-protein complex, resulting in association of Sir proteins across the silenced domain. This histone deacetylation model makes the simple prediction that preemptively removing Sir2’s H3 and H4 acetyl substrates, by mutating these lysines to unacetylatable arginines, or removing the acetyl transferase responsible for their acetylation, should restore silencing in the Sir2 catalytic mutant. However, this was not the case. We conducted a genetic screen to explore what aspect of Sir2’s catalytic activity has not been accounted for in silencing. Mutation of a nonsirtuin histone deacetylase, Rpd3, restored Sir-protein-based silencing in the absence of Sir2’s catalytic activity. Moreover, this antagonism could be mediated by either the large or the small Rpd3-containing complex. Interestingly, this restoration of silencing appeared independent of any known histone H3 or H4 substrates of Rpd3. Investigation of Sir-protein association in the Rpd3 mutant revealed that the restoration of silencing was correlated with an increased association of Sir proteins at the silencers, suggesting that Rpd3 was an antagonist of Sir2’s function in nucleation of Sir proteins to the silencer. Additionally, restoration of silencing by Rpd3 was dependent on another sirtuin family member, Hst3, indicating multiple antagonistic roles for deacetylases in S. cerevisiae silencing.
SIR2 is the founding member of the sirtuin enzyme family, which deacetylates acetylated lysines on a variety of proteins. Unlike other deacetylase enzymes that catalyze the hydrolysis of acetyl-lysine, sirtuins couple lysine deacetylation to NAD+ hydrolysis (Imai et al. 2000; Tanner et al. 2000; Sauve et al. 2006). Utilizing this high-energy cofactor for deacetylation allows sirtuins to be potentially regulated by the energy balance and/or redox balance within the cell. Sirtuins regulate diverse processes across all biological kingdoms—ranging from life-span regulation and fat metabolism to chromatin modifications (Sauve et al. 2006). Saccharomyces cerevisiae has five sirtuins: Sir2 and its four paralogs named Homologs of Sir2, Hst 1–4, which are involved in cell-cycle regulation, chromatin modification, and perhaps other functions as well (Brachmann et al. 1995; Celic et al. 2006; Sauve et al. 2006; Downey et al. 2013).
Of the five yeast sirtuins, Sir2, the catalytic member of the Sir-protein silencing complex, is the most thoroughly studied. Sir2’s catalytic activity is necessary for silencing HML, HMR, and some genes near telomeres. A structurally intact, but catalytically inactive Sir2 protein, encoded by the sir2N345A allele, cannot support gene silencing (Imai et al. 2000) but can assemble into a Sir-protein complex along with Sir3 and Sir4 and bind to silencers (Rusché et al. 2002; Thurtle and Rine 2014). The catalytic activity of Sir2 is necessary for the deacetylation of histone H4 N-terminal lysine residue 16 (H4K16) in nucleosomes at silenced loci. Point mutations changing H4K16 to glutamine, which mimics the acetylated state, disrupt silencing at HML (Park and Szostak 1990). This H4K16-to-glutamine silencing defect can be suppressed by a point mutation in Sir3, indicating that acetylation of H4K16 likely disrupts the Sir3–nucleosome association (Johnson et al. 1990), as confirmed by structural studies of the Sir3 BAH domain cocrystallized with the nucleosome core (Armache et al. 2011; Wang et al. 2013). Additionally, although Sir2 can deacetylate H3K9-acetyl and H3K14-acetyl, it shows preference for H4K16-acetyl over other acetylated positions on histones in vitro (Imai et al. 2000), and deacetylation of H4K16 at HML and HMR in vivo is Sir2 dependent (Kimura et al. 2002; Suka et al. 2002). Sas2, a member of the MYST family of histone acetyltransferases, functions in a complex with Sas4 and Sas5 to acetylate H4K16, thus functioning in opposition to Sir2 (Meijsing and Ehrenhofer-Murray 2001; Osada et al. 2001; Kimura et al. 2002; Suka et al. 2002).
This evidence for Sir2 deacetylation of H4K16-acetyl combined with other biochemical and genetic studies led to the nucleation and spreading model for yeast gene silencing: Sir2, Sir3, and Sir4 are recruited to the silent mating loci through interactions with the ORC, Abf1, and Rap1 proteins, which bind the silencers in addition to their other individual roles in the cell. In the standard model for silencing, once nucleated at the silencers, Sir2 deacetylates the H3 and H4 tails on the neighboring nucleosome, allowing the Sir-protein complex to bind to the hypoacetylated nucleosome. Iterative rounds of deacetylation lead to the spreading of Sir protein complexes across the silenced loci, resulting in silencing at HML and HMR (Hoppe et al. 2002; Rusché et al. 2002) largely by occluding transcription factors (Loo and Rine 1994; Steakley and Rine 2015; Wang et al. 2015). The nucleation and spreading model makes the simple prediction that, although Sir2’s catalytic activity is necessary, it should be bypassed by the absence of H4K16 acetylation. However, genetically removing H4K16 acetylation either by deleting SAS2, the gene encoding the H4K16 acetylase, or by making the H4K16R mutant, mimicking the hypoacetylated state, does not restore the silencing defect of the Sir2 catalytic mutant, sir2N345A (Yang and Kirchmaier 2006). Thus, the standard model is incomplete and missing a key role(s) for Sir2’s catalytic activity in silencing.
There are several possible unaccounted roles that Sir2’s catalytic activity might play in silencing. For example, Sir2 may be critical for the deacetylation of other histone residues that are not targets of Sas2. In addition to H3 and H4 N-terminal tail lysine residues, a core lysine acetylation, H3K56-acetyl, has been implicated in silencing based upon the ability of H3K56 point mutations to disrupt telomeric silencing (Xu et al. 2007; Yang et al. 2008). H3K56-acetyl is more abundant at silenced loci in cells lacking Sir2 function, suggesting a possible role for Sir2 in deacetylating this core position (Xu et al. 2007). However, there is contradictory evidence as to whether Sir2 can deacetylate H3K56 in vitro (Xu et al. 2007; Yang et al. 2008; Oppikofer et al. 2011). In vivo, there is strong evidence that H3K56-acetyl is deacetylated after deposition into the chromatin by either of two other sirtuins, Hst3 and Hst4 (Celic et al. 2006; Yang et al. 2008). Alternatively, Sir2 may deacetylate an undiscovered nonhistone protein with a role in silencing. Sir2-dependent deacetylation of the nonhistone proteins Ifh1 and Pck1 certainly expands the possibilities (Lin et al. 2009; Downey et al. 2013).
The sirtuin catalytic reaction itself, rather than a specific deacetylation per se, could conceivably be necessary for silencing. Sirtuins couple lysine deacetylation to the hydrolysis of NAD+, producing the metabolites, 2-O-acetyl-ADP-ribose (OAAR) and nicotinamide (NAM) (Tanner et al. 2000). In mammalian cells, the histone variant macroH2A of the inactive X binds OAAR, generated from the sirtuin SIRT1, with a Kd of the same order of magnitude as the concentration of OAAR in a cell (Kustatscher et al. 2005; Lee et al. 2008). One study with yeast proteins reported that in vitro OAAR can promote structural changes in the Sir protein complex, as assayed by electron microscopy (Liou et al. 2005). Additionally, OAAR increases the affinity of the Sir protein complex for reconstituted chromatin (Martino et al. 2009), suggesting that OAAR could enhance silencing by promoting a critical level of chromatin compaction. However, an elegant genetic study has shown that a sirtuin reaction is not necessary for gene silencing in Saccharomyces (Chou et al. 2008). Thus there is no evidence for an in vivo role of OAAR in silencing or anything else in yeast, but the possibility remains open.
To shed light on what additional proteins or processes require Sir2’s catalytic function for silencing, we performed a genetic screen for mutants that could restore silencing of HML and HMR in cells with a catalytically inactive form of Sir2, encoded by sir2N345A as the only source of Sir2. In addition, in the parental stain, H4K16 acetylation was preemptively blocked by deletion of SAS2, which encodes the relevant H4K16 acetylase. A priori, there was one known gene, SUM1, in which a particular mutation (SUM1-1) could, in principle, be recoverable from this screen. The SUM1-1 mutation can restore silencing in cells lacking any of the four SIR genes by causing the assembly of a different type of silenced chromatin (Klar et al. 1985; Laurenson and Rine 1991; Chi and Shore 1996; Rusché and Rine 2001). Although the mechanism of SUM1-1’s function was not germane to our goal, the existence of this very rare mutation provided a positive control for the effectiveness of the screen. The mutants recovered from the screen revealed unappreciated roles for multiple histone deacetylases with antagonistic functions at HMR and HML and Sir2’s catalytic function in the nucleation of the Sir proteins at the silencers.
Materials and Methods
Yeast strains, plasmids, and culture
All yeast strains were derived from the W303 background and genotypes are indicated in Supplemental Material, Table S1. Deletions were constructed through one-step integration of knockout cassettes (Longtine et al. 1998). The hmra1∆::URA3 reporter was previously described (Zill et al. 2010). All plasmids used are indicated in Table S2. Plasmids bearing the histone point mutations were introduced into strains containing a pRS316 plasmid bearing wild-type H3 and H4 (pJR2657). Plasmid swap using 5-FOA counterselection against the pRS316 plasmid was used to recover yeast with only the mutant histone genes. The rpd3P264L point mutant (JRY9568) was determined through cloning by complementation with a previously described yeast plasmid library containing both TRP1 and G418-resistance selectable markers (Jauert et al. 2005). To determine silencing phenotype, fivefold serial dilutions of overnight cultures grown in rich medium were spotted onto appropriate media at an initial approximate density of 4 × 107/ml as in Zill et al. (2010). To test for mating ability, cells of the strain of interest were manually mixed on a YPD plate with an a-mating tester (JRY2728) or the α-mating tester (JRY2726), grown overnight at 30°, and then replica plated onto minimal medium plates (YM), to select for successful mating events.
EMS mutagenesis
EMS mutagenesis was conducted as described (Amberg et al. 2005). Briefly, 10 OD units (Absorbance = 600 nm) were pelleted, washed with distilled water, and resuspended in 0.1 M sodium phosphate buffer (pH 7). A total of 30 µl of EMS was added to the sample and incubated at 30° for 1 hr. Cells were washed with 5% sodium thiosulfate and resuspended in PBS and plated on YPD. Plates were grown overnight on YPD, then replica plated to 5-FOA. After mutagenesis, yeast was grown at room temperature to allow for the possibility of conditional alleles of essential genes.
Whole-genome-sequencing library preparation and data analysis
DNA for whole-genome sequencing analysis was isolated following the yeast DNA extraction protocol in (Hoffman and Winston 1987). For the JRY9567 (sir2N345A, sas2∆, hmra1Δ::URA3, sum1-1) libraries, three double-backcrossed segregants exhibiting the mutant phenotype were grown to saturation in a liquid culture and DNA extracted as described. The three segregants, JRY9614, JRY9615, and JRY9616, were barcoded separately and sequenced with the unmutagenized parent, JRY9097, and the original mutant, JRY9567. For the JRY9569 (sir2N345A, sas2∆, hmra1Δ::URA3, rpd3P397H) and JRY9570 (sir2N345A, sas2∆, hmra1Δ::URA3, rpd3G290N, orc3A333V) libraries, bulk segregant analysis as in Birkeland et al. (2010) was performed to determine causative mutations. Ten segregants with a silencing restoration phenotype and 10 segregants with a nonsilencing phenotype from the JRY9617 and JRY9618 diploids were pooled equally according to the OD600 of each culture. DNA was then isolated from these pooled cultures. A total of 4 µg of DNA was sheared with the S220 Focused-ultrasonicator (Covaris, Woburn, MA) and the libraries were constructed using the Illumina Tru-Seq library preparation kit. Samples were indexed and sequenced in two lanes on the Illumina HiSequation 2000 as 100 bp single end reads. Reads were mapped using Burrows-Wheeler Alignment tool (BWA) (Li and Durbin 2009) to a modified SacCer 2 genome. SNPs were detected using VCFtools (Danecek et al. 2011) and visualized on the Integrative Genomics Viewer (Thorvaldsdóttir et al. 2013). Number of reads mapped for each sample is recorded in Table S3. Reads have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) at http://www.ncbi.nlm.nih.gov/sra under accession no. SRP077398.
RNA isolation and analysis
RNA was prepared as in Ozaydin and Rine (2010). Complementary DNA (cDNA) was synthesized using Superscript III First-Strand Synthesis (Invitrogen, Carlsbad, CA), utilizing oligo-dT primers. Amplification values for all RNA primer sets were normalized to ACT1 cDNA amplification values for that sample. RNA analysis was performed on four independent RNA preparations and error bars represent standard error.
Chromatin isolation and immunoprecipitation
A total of 70 OD units (Absorbance = 600 nm) of logarithmically growing cells were cross-linked with 1% formaldehyde for 30 min at room temperature. Cells were lysed with 0.5-mm zirconia beads in 1 ml FA lysis buffer using the MP Fastprep-24. Chromatin was prepared as previously described (Aparicio et al. 2005). Chromatin was immunoprecipitated with 25 μl of anti-c-Myc agarose (Sigma, St. Louis, MO; cat. no. 7470) overnight at 4°.
Washes, elutions, and isolation of DNA were performed as previously described (Aparicio et al. 2005). After the reversal of cross-links with Proteinase K, DNA was isolated with QIAGEN PCR purification kit (Valencia, CA).
Quantitative PCR
Quantitative PCR (qRT-PCR) analysis was conducted on the MX3000P machine from Stratagene (La Jolla, CA) using DyNAmo HS SYBR Green qPCR kit (Thermo Fisher Scientific, Waltham, MA). All primer sequences for RNA and chromatin immunoprecipitation (ChIP) analysis are listed in Table S4.
Data availability
Strains and plasmids are available upon request. Table S1 contains detailed genotypes of all strains used in this study.
Results
Mutations in a few genes could restore silencing independently of Sir2’s catalytic activity
To investigate what function of Sir2’s catalytic activity is not accounted for in the gene silencing model, we screened for mutations able to suppress the silencing defect of a sas2∆ sir2N345A strain. As a silencing reporter, the a1 open reading frame at HMR was replaced with URA3 (Figure 1A). URA3 expression causes sensitivity to the drug 5-FOA, allowing for selection of cells that have silenced URA3 at HMR by their ability to grow on 5-FOA (Boeke et al. 1984). The sir2N345A sas2∆ MATa strain (JRY9097) was mutagenized with EMS and mutants were initially identified by growth on 5-FOA medium. To distinguish mutants able to grow on 5-FOA because of silencing restoration from mutants with lesions in the URA3 reporter gene, a secondary screen was conducted for putative mutant’s restoration of mating ability, which required silencing at HMLα as well as at HMRa1∆::URA3 (Figure 1A).
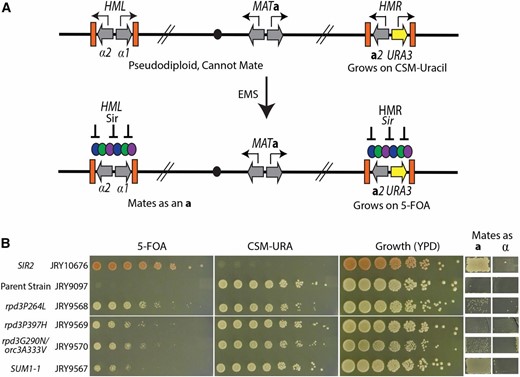
A genetic screen identified suppressors of Sir2’s catalytic activity. (A) Schematic of the screen to identify mutations that restored silencing in the absence of Sir2 catalytic activity. A sir2N345A sas2Δ strain had the URA3 reporter integrated in place of the a1 ORF at HMR. Expression of the URA3 gene resulted in cells that grow on complete synthetic media lacking uracil but are sensitive to 5-FOA. Expression of the α-genes from HML and a-genes from MAT result in a pseudodiploid cell identity that cannot mate. This strain was mutagenized with EMS and mutants were picked that reestablished silencing as assayed by resistance to 5-FOA and the ability to mate. (B) The phenotypes of a SIR2 wild-type strain (JRY10676) exhibiting complete silencing, the parent strain (JRY9097), and the four mutants that were isolated from the suppressor screen. Fivefold serial dilutions assayed for growth phenotypes on the indicated media. Growth in the patches on the right indicates successful matings as either an a- or α-cell identity.
From 1500 5-FOA resistant colonies, four mutants were recovered that also exhibited some restoration of mating ability (Figure 1B). All four mutants were able to grow both on solid medium lacking uracil in addition to medium containing 5-FOA, indicating that silencing at HMRa1∆::URA3 was only partially restored. Molecular quantitation of the extent of restored silencing is discussed below. Silencing was also not restored to wild-type levels at HML as evidenced by the inefficient mating of the mutants (see also Figure 2B). Mutants were mated to an isogenic strain of the opposite mating type, and the corresponding diploids were sensitive to 5-FOA. Thus the silencing phenotype of the mutants was recessive (data not shown).

Mutation of RPD3 and SIN3 restored silencing. (A) Fivefold serial dilutions of yeast were spotted on plates to determine their growth phenotype. Growth on 5-FOA indicates silencing of the hmra1Δ::URA3 reporter. All strains are in the sir2N345A sas2Δ background. The first row is the unmutagenized, RPD3 parent (JRY9097). The next two rows are replicates of the rpd3P264L point mutant (JRY9568). The fourth row is isogenic to the unmutagenized parent except for the opposite mating type, MATα, and ade2-1 (JRY9572). In the fifth row, RPD3 is deleted in the parent strain (JRY9573). In the last row, RPD3 is deleted in the MATα ade2-1 strain isogenic to the parent (JRY9574). (B) The parental strain (JRY9097) and the recovered rpd3P264L mutant (JRY9568), and the rpd3Δ (JRY9573), were crossed with the MATα mating tester (JRY2728) to test for silencing at HML. Growth on the media indicates mating and thus silencing. (C) The Rpd3 subunit gene indicated was deleted in JRY9097 to determine if deletion of that subunit could also restore silencing. Green labels indicate Rpd3L subunits and red labels indicate Rpd3S subunits. (D) The sin3Δ strain that exhibited silencing restoration was transformed with plasmids expressing full-length SIN3 or one of the four SIN3 PAH domain deletions (M1635–M1639). Silencing was assayed on media lacking histidine to select for the plasmid.
Several different strategies were used to identify the causative mutation. For one of the mutants, the causative mutation was determined by individually sequencing multiple backcrossed segregants. The causative mutation was determined as the SNP that all of the segregants with restored silencing shared in common, but was absent in the unmutagenized parent strain. This mutant had exactly the same point mutation as SUM1-1 (Klar et al. 1985; Chi and Shore 1996). As noted before, this mutation appears dominant by some assays and recessive by others (Laurenson and Rine 1991).
The other three mutants had different point mutations in the gene encoding the nonsirtuin histone deacetylase, RPD3. One of these mutants (JRY9568) was amenable to cloning by complementation and a plasmid with a genomic fragment containing RPD3 complemented the silencing phenotype of this mutant. Sanger sequencing of the RPD3 gene in this mutant identified a transition mutation causing a proline-to-leucine change at amino acid 264 (hereafter referred to as rpd3P264L). The causative mutation of the two additional mutants (JRY9569 and JRY9570) was determined through pooled linkage analysis (Birkeland et al. 2010). Both mutants analyzed by pooled linkage analysis revealed a mutation in RPD3 specific to the mutant pool (Figure S1, A and B). Thus, three of four mutants recovered from this screen identified nonsynonymous mutations in RPD3 (Figure 1B).
We focused most of the following analyses on understanding how rpd3 mutations could partially bypass the need for Sir2’s catalytic activity. However, interestingly, one of the two mutants (JRY9570) analyzed by pooled linkage analysis, in addition to the rpd3G290N mutation, also contained an alanine-to-valine substitution at amino acid 333 in ORC3, which encodes a subunit of ORC, that segregated at 94% frequency with the mutant pool (46 of 49 reads) (Figure S2). All other mutations showed a heterozygous distribution in both the mutant and wild-type pools between 35 and 65% frequency and hence were discarded from consideration. ORC3 and RPD3 are present on different chromosomes so the association of the ORC3 mutation was not due to physical linkage to RPD3. The tetrad analysis used to isolate single segregants for pooling showed that the restoration-of-silencing phenotype showed a 2:2 segregation, indicating a single Mendelian locus for the causative mutation. However, the segregants that were pooled were those with the most vigorous growth on 5-FOA. Thus the cosegregation of the rpd3 and orc3 mutations implied that the orc3A333V mutation enhanced the rpd3 mutant’s ability to restore silencing.
RPD3 as an antagonist of Sir-protein-based silencing
Rpd3 has been implicated in silencing in several ways. Rpd3 restricts the spread of Sir proteins at the boundary of silent chromatin domains (Sun and Hampsey 1999; Zhou et al. 2009). An rpd3 mutation suppresses the silencing defect of an HMR-E silencer with a defective Rap1 binding site (Vannier et al. 1996). Additionally, mutations in the Drosophila Rpd3 ortholog, as well as deletion of yeast RPD3, result in increased position-effect variegation and telomere position effect, respectively (De Rubertis et al. 1996). To determine whether the point mutations isolated here had some novel property leading to the silencing phenotype, RPD3 was deleted in the original sir2N345A sas2∆ parent strain and a strain of similar genotype, but opposite mating type. As with the point mutant, the deletion also suppressed the silencing defect of the sir2N345A mutation (Figure 2, A and B). Hence the point mutation likely caused loss of Rpd3 function. Interestingly, as judged by resistance to 5-FOA, silencing at HMR was slightly better in the rpd3P264L mutant than the rpd3∆. However, this point mutant was derived from the original EMS mutagenesis and may have had background mutations improving silencing slightly beyond the silencing achieved by the complete loss of Rpd3 function.
Rpd3 is a histone deacetylase found in two distinct complexes: the Rpd3 large complex (Rpd3L) and Rpd3 small complex (Rpd3S). Rpd3S deacetylates histone H4 in nucleosomes in the coding regions of actively transcribed genes to prevent intragenic transcription (Carrozza et al. 2005). The large complex is involved in general transcriptional repression of many genes (Kasten et al. 1997; Lechner et al. 2000). To determine if Rpd3L or Rpd3S was specifically involved in the restoration of silencing, we deleted components specific to the small complex, EAF3 and RCO1, or the large Complex, PHO23, SAP30, ASH1, and SDS3, in the sir2N345A sas2∆ strain to determine if, as in rpd3∆, these deletions could also restore silencing. Neither deletions specifically affecting the small complex nor those specifically affecting the large complex restored silencing in the sir2N345A sas2∆ strain (Figure 2C). However, deletion of SIN3, the scaffolding platform for both the Rpd3S and Rpd3L complexes, phenocopied the rpd3∆. Thus, neither the Rpd3L nor Rpd3S specifically blocked silencing in the sir2N345A sas2∆ strain.
Sin3 contains four paired amphipathic helix (PAH) domains that facilitate protein–protein interactions. To test which of these domains were required for the restoration of silencing when SIN3 is deleted, we transformed plasmids expressing SIN3 with each of the PAH domains deleted (Wang and Stillman 1993). Only the pah3 mutant was still able to silence HMR (Figure 2D), indicating that the deletion of PAH3 was required for the restoration of silencing by sin3∆ and rpd3∆.
Both Rpd3 complexes are involved in transcriptional repression, with mutation of RPD3 resulting in increased expression of genes (Kurdistani et al. 2004; Carrozza et al. 2005; Chen et al. 2012). Therefore it was odd that silencing should be partially restored in the absence of Rpd3. To test directly whether the effect of the rpd3 mutation on silencing was transcriptional, and if so, specific to HML and HMR, or a more general effect on MAT gene expression, we measured RNA expression from the original unmutagenized parent strain (RPD3), the rpd3P264L point mutant, and the rpd3∆ and compared the expression from the MAT locus to the expression from HML and HMR. If the restoration of silencing were due to general down regulation of the a1 promoter, then both the a1 gene at MAT and the URA3 gene at HMR would show decreased expression in the mutant. In contrast, if the apparent suppression of sir2N345A were due to silencing restoration, then the decrease in expression would be specific to HMR and not apparent at the MAT locus. Consistent with a silencing-specific role for rpd3 mutants rather than a transcriptional down-regulation, decreased expression was observed only at HML and HMR and not the MAT locus for both the recovered rpd3P264L point mutant and the rpd3∆ (Figure 3A). Interestingly, as was observed by growth on 5-FOA, the rpd3P264L point mutant showed more complete silencing of URA3 at HMR than the rpd3∆, showing concordance between the assays. Therefore, the phenotypes described here of the rpd3 mutations were due to a restoration of silencing.
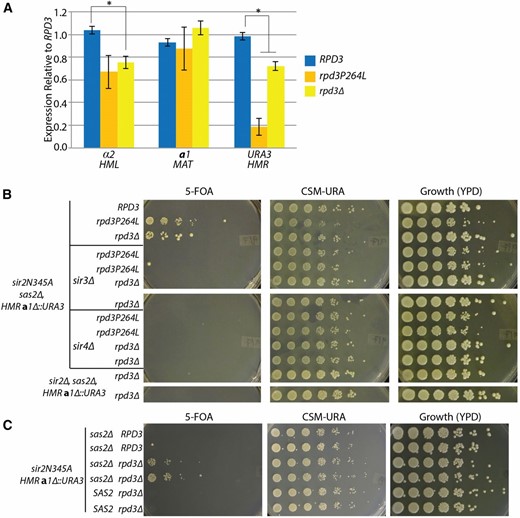
The rpd3 mutant restored Sir-based silencing. (A) α2, a1, and URA3 transcripts were measured from the RPD3 parent strain (JRY9097), the rpd3P264L point mutant (JRY9568), and the rpd3Δ (JRY9573) to determine expression levels at HML, MAT, and HMR, respectively. RNA was normalized to ACT1 for each sample and reported as relative to the RPD3 sample. Error bars are the standard error of four biological replicates. The asterisks indicate a P-value of <0.05 (Student’s t-test). (B) Fivefold serial dilutions of the indicated genotypes were spotted to determine the growth phenotype. All strains are sir2Δ, sas2Δ, hmra1Δ::URA3, and all but the bottom two rows also express sir2N345A (JRY9588 and JRY9589). SIR3 and SIR4 were deleted as indicated (JRY9580–JRY9587). The replicates represent individual transformants for the knockouts or individual segregants from the same tetrad dissection. (C) Restoration of silencing at HMR by rpd3Δ was tested in a SAS2 wild-type strain. Fivefold serial dilutions were plated on the indicated media to determine growth phenotype. All strains express the catalytically incactive SIR2 allele, sir2N345A, and have the hmra1Δ::URA3 reporter. Six individual segregants derived from a single tetrad dissection were tested for their ability to restore silencing depending on their SAS2 genotype (from top to bottom: JRY9598, JRY9599, JRY9600, JRY9601, JRY9602, and JRY9603). Strains with the same indicated genotype represent two different segregants from the tetrad dissection to test for reproducibility of the silencing phenotype.
As described above, the SUM1-1 mutation restores silencing independently of Sir proteins. Since the SUM1-1 mutation was recovered in this very screen, it was possible that the rpd3 mutants also restored silencing through a Sir-protein-independent mechanism. Since we were interested in understanding unaccounted roles for Sir2’s catalytic activity in silencing, we tested whether the rpd3 mutants’ restoration of silencing was Sir-protein dependent, or whether they functioned by some independent means, like SUM1-1. Silencing restoration by the rpd3 mutants was dependent on both Sir3 and Sir4, indicating that Sir protein-dependent silencing had been restored (Figure 3B). To determine if restoration of Sir silencing by the rpd3∆ was due to misregulation of Sir3 and Sir4, we also measured messenger RNAs (mRNAs) from SIR3 and SIR4 in sir2N345A sas2∆ strains, with and without the rpdP264L point mutant or rpd3∆. SIR3 showed a modest 1.5-fold increase in expression in both the rpd3∆ and rpd3P264L mutants (Figure S3); no increase was noted for SIR4. Previous overexpression of SIR3 to ∼16-fold wild-type levels had only marginal effects on silencing (Yang and Kirchmaier 2006), thus this modest increase in SIR3 expression in the rpd3 mutant was unlikely to be responsible for the silencing restoration. Additionally, sir2N345A itself was also required to restore silencing by rpd3∆. A sir2∆ rpd3∆ strain that was not expressing the catalytically dead sir2N345A allele remained unable to silence (Figure 3B). Thus the entire Sir-protein complex, but not Sir2’s catalytic activity, was required for rpd3∆ to restore silencing.
Sir proteins have been implicated in silencing of synthetic telomeric constructs (Gottschling et al. 1990; Renauld et al. 1993; Fourel et al. 1999). However, more recent studies have shown that Sir-protein-based silencing at native telomeres is less pervasive than originally thought (Loney et al. 2009; Ellahi et al. 2015). To test if the restoration of silencing by the rpd3 mutant also occurred at native telomeres, we measured RNA expression of a native telomeric gene, COS8, shown to have Sir2-dependent silencing. We observed no significant change in expression of COS8 in the rpd3 point mutant or rpd3∆ (Figure S4), indicating that this mutant does not broadly affect telomeric silencing.
Additionally, the restoration of silencing by the rpd3∆ mutant was observed only in the absence of wild-type SAS2 (Figure 3C). Thus rpd3∆ was unable to compensate for the loss of Sir2 catalytic activity when H4K16 was acetylated. These results illustrate that the phenotype revealed by rpd3∆ implied the existence of an unaccounted function(s) for Sir2’s catalytic activity in silencing.
Restoration of silencing by rpd3∆ was independent of Rpd3 histone targets
Rpd3 deacetylates histone H4K5-acetyl, H4K8-acetyl, and H4K12-acetyl (Suka et al. 2001). Because these are the known substrates for Rpd3 in yeast nucleosomes, we tested if the restoration of silencing by the rpd3∆ was dependent on H4K5, H4K8, and H4K12 acetylation. Silencing was assayed at HMR by the ability of cells of various genotypes to mate with a MATα strain to form prototrophic diploids. To test if the restoration of silencing by rpd3∆ resulted from the retention of acetylated H4K5, K8, or K12, we expressed plasmids with the nonacetylatable mimic H4K5,8,12R and tested if rpd3∆ restored silencing in this background (Figure 4). In these strains the endogenous copies of histones H3 and H4 genes were deleted and either wild-type or mutant H3 and H4 were expressed from a single-copy plasmid. It was evident that altered dosage of histones from the single source of H3 and H4, per se, did not disrupt the rpd3 mutant’s ability to restore silencing. Likewise, the rpd3∆ still restored silencing in cells with H4K5,8,12R. Thus, the acetylation status of these positions on H4 were not part of the mechanism by which rpd3∆ restored silencing (Figure 4).

Silencing by rpd3Δ was independent of known Rpd3 histone H4 targets. Endogenous copies of H3 and H4 were deleted and wild-type or mutant H3 and H4 were expressed from a CEN-ARS plasmid. Complete genotypes for all strains tested are in Table S1 (JRY9605, JRY9607–JRY9613, and JRY10644–JRY10655). Silencing was tested by mating to a MATa tester strain (JRY2726) and replica plated to minimal media. Growth indicates successful mating, which reflected extent of silencing.
Rpd3 also deacetylates histone H3 tail residues (Rundlett et al. 1996) that have been shown to affect silencing in combination with H4 tail mutations (Kelly et al. 2000). To test whether multiple histone acetylations may be critical for the mechanism of silencing restoration by the rpd3∆, we mutated H3 tail residues K9, K14, and K27 each in turn to R or Q in the H4K5,8,12R background and tested if silencing was affected in the original strain or whether the rpd3∆ restoration of silencing was affected. None of the H3 tail mutations in combination with the H4 tail mutations affected silencing restoration by rpd3∆. Therefore H3 and H4 lysine tail acetylations were likely not important for silencing restoration by rpd3∆.
Increased nucleation of Sir proteins in the rpd3 mutant
High-resolution analysis of Sir3- and Sir4-protein distribution in a cell with the catalytically inactive sir2N345A mutant protein revealed that association of Sir3 and Sir4 is reduced at the silencers relative to cells with wild-type Sir2, though the reason for this decreased association is unknown (Thurtle and Rine 2014). The origin recognition complex (ORC) binds to all four silencers and interacts with Sir1 to nucleate the Sir-protein complex (Bell et al. 1993; Zhang et al. 2002; Hou et al. 2005). Since a mutation in ORC3, orc3A333V (Figure 1B and Figure S2) also cosegregated with the strongest restoration-of-silencing phenotype in a cross with one of the rpd3 point mutants, we considered the possibility that Rpd3 somehow antagonized Sir protein nucleation at silencers. If so, in the absence of Rpd3 function, Sir protein association at the silencers would be expected to increase. To test this hypothesis, we ChIPed myc-tagged Sir4 in chromatin from SIR2 sas2∆ (JRY10635), sir2N345A sas2∆ (JRY10632), and the sir2N345A rpd3∆ sas2∆ (JRY10633) strains to test the association of Sir proteins with silencers. As previously reported, Sir4 association was greatly reduced at the silencers in sir2N345A as compared to the enrichment in SIR2 cells (Figure 5, comparing purple and blue bars) (Rusché et al. 2002; Thurtle and Rine 2014). rpd3∆ restored Sir4 association at the HML-E and HMR-I silencers to near wild-type levels, whereas there was a significant, but more limited increase of Sir4 association within HMR and HML. Association of Sir4 at HMR-E was not restored to wild-type levels, which is consistent with the incomplete restoration of silencing observed in the rpd3∆ sir2N345A strain. Thus rpd3∆ restored silencing in the sir2N345A strain by increasing Sir-protein association at the silencers and to a lesser degree within the silenced cassette.
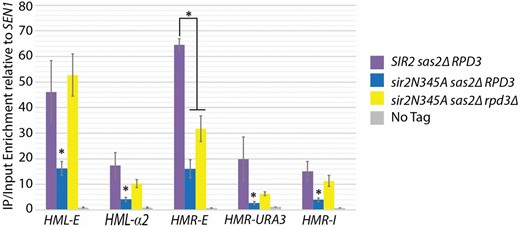
Deletion of RPD3 increased Sir4 association with the silencers. Sir4-myc ChIP was performed in the MATa sas2Δ hmra1Δ::URA3 with the following SIR2 and RPD3 genotypes: SIR2 RPD3 (JRY10635), sir2N345A RPD3 (JRY10632), and sir2N345A rpd3Δ (JRY10633). No enrichment was observed in a strain not expressing the Sir4-myc (JRY10634). Enrichment is shown as the Immunoprecipitation (IP)/input values relative to the SEN1 IP/input values as a negative control. Error bars represent standard error of four biological replicates. The asterisks indicate a P-value of <0.05 (Student’s t-test).
Restoration of silencing was dependent on Hst3, but independent of H3K56
Silencing is lost when yeast is grown in the presence of NAM, a competitive inhibitor of sirtuins (Landry et al. 2000; Bitterman et al. 2002). This silencing loss has been attributed to inhibition of Sir2. However, NAM inhibits the four yeast Hst proteins in addition to Sir2. Hst3 and Hst4 have been implicated in silencing (Yang et al. 2008), suggesting the possibility that NAM’s effect on silencing could be due to more than just inhibition of Sir2. Silencing in the rpd3 mutant provided a unique opportunity to determine if other yeast sirtuins contributed to silencing in this mutant, which was independent of Sir2’s catalytic activity. Surprisingly, silencing restoration by rpd3P264L was sensitive to NAM, thus implicating another sirtuin in silencing (data not shown). Silencing restoration by the rpd3 mutant depended specifically on HST3 (Figure 6A). The hst3∆ mutant showed a strong phenotype, whereas the hst4∆ mutant had no discernible phenotype, even though both deacetylate the same substrate, H3K56-acetyl, although HST4 is the minor partner in this role (Celic et al. 2006). To test whether the lack of growth on 5-FOA in the rpd3P264L hst3∆ was due to loss of silencing, or an unrelated sensitivity to the 5-FOA drug, as has been previously reported to interfere with reporter assays (Rossmann et al. 2011; Takahashi et al. 2011), we measured HML and HMR RNA levels in the rpd3P264L hst3∆ sirN345A strain. As expected if Hst3 were required for the restoration of silencing, expression at HML and HMR in this mutant was significantly elevated as compared to the rpd3P264L sir2N345A strain (Figure 6B). Additionally, loss of silencing in hst3∆ was not due to misregulation of SIR3 or SIR4 as their mRNA levels were similar to their levels in the rpd3P264L and rpd3∆ mutants (Figure S3). These results identified a specific role for Hst3 in HMR silencing that was independent of Hst1, Hst2, and Hst4.
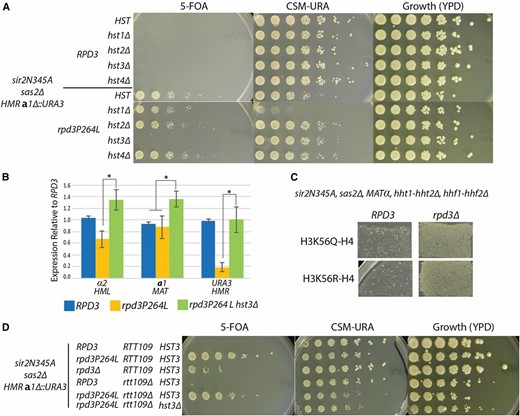
Silencing by the rpd3 mutant was dependent on Hst3. (A) Fivefold serial dilutions were spotted on the indicated media to determine growth phenotype. All strains are sir2Δ, sir2N345A, sas2Δ, and have the hmra1Δ::URA3 reporter to measure silencing as growth on 5-FOA. RPD3 and HST genotypes are as indicated (JRY9590–JRY9597). (B) α2, a1, and URA3 transcripts were measured to determine expression levels at HML, MAT, and HMR, respectively in RPD3 (JRY9097), rpd3P264L (JRY9568), and rpd3P264L hst3Δ (JRY9596). RNA was normalized to ACT1 for each sample and reported as relative to the RPD3 sample. Error bars are the standard error of four biological replicates. The asterisks indicate a P-value of <0.05 (Student’s t-test). (C) Endogenous copies of H3 and H4 were deleted and mutant H3 was expressed from a CEN-ARS plasmid (JRY9604 and JRY9606). Silencing was tested by mating to a MATa tester strain (JRY2726) and replica plated to minimal media. Growth indicates successful mating and silencing. (D) Fivefold serial dilutions of the indicated RPD3, HST3, and RTT109 genotypes were spotted to determine the growth phenotype. The strains assayed are (from top to bottom) JRY9097, JRY9567, JRY9573, JRY10636, JRY10637, and JRY10638. All strains are sir2Δ, sas2Δ, hmra1Δ::URA3, and sir2N345A.
Hst3 deacetylates histone H3K56 (Celic et al. 2006). To determine if the dependence of silencing by the rpd3 mutant on Hst3 was due to a requirement for deacetylation of H3K56-acetyl, we tested silencing in appropriately marked strains expressing either the hyperacetylated mimic (Q) or hypoacetylated mimic (R) of H3K56. As reported previously, expression of the H3K56R could not rescue the silencing defect in sir2N345A (Yang et al. 2008) (Figure 6C). Additionally, neither H3K56 acetylation-state mimic interfered with the silencing restoration in the sir2N345A sas2∆ strain provided by rpd3∆ (Figure 6C). In some contexts, the arginine and glutamine mutations may not be adequate mimics of the histone acetylation states. Thus, to provide an independent test of whether H3K56-acetyl blocked the ability of the rpd3 mutant to restore silencing when HST3 was deleted, we tested whether deletion of RTT109, which encodes the histone acetyltransferase that acetylates H3K56, could restore silencing in the rpd3P264L hst3∆ strain. The rtt109∆ mutation did not restore silencing in cells lacking HST3 (Figure 6D). Thus, there appeared to be another substrate of Hst3 whose hypoacetylation promoted silencing, at least in the absence of SIR2 and RPD3 catalytic function.
Discussion
There were two ways to restore silencing independent of Sir2 catalytic activity
The genetic screen identified four mutants that restored silencing independent of Sir2’s catalytic activity. One caused the very same amino acid substitution of SUM1 previously reported, and the other three were all in RPD3. Thus few genes can be mutated to restore silencing independent of Sir2’s catalytic activity. The silencing restoration observed by SUM1-1 in the sir2N345A sas2∆ double mutant was dependent on Hst1 (data not shown), indicating that it restored silencing through the previously described mechanism (Rusché and Rine 2001). Hst1 was not required for the silencing restored in rpd3 mutants, described here. The Rpd3 protein is part of two different complexes, suggesting that mutations in genes for other members of the two complexes could potentially have been recovered. However, of the genes tested, only those encoding subunits of both complexes, RPD3 and SIN3, when deleted, restored silencing. Mutations in the genes encoding subunits specific to either Rpd3L or Rpd3S, had no impact on silencing. Hence, it appeared that both complexes prevented silencing in the sir2N345A sas2 double mutant.
We note that rpd3∆ is reported to be synthetically lethal in combination with sas2∆ in two previous reports (Collins et al. 2007; Ehrentraut et al. 2010). This synthetic lethality appears to result from the silencing of some essential gene(s), presumably near telomeres, given that the lethality is suppressed by mutations in SIR genes. Conceivably, such synthetic lethality would be a problem in our strains in which silencing was restored in the sas2∆ sir2N345A rpd3 mutant, yet we did not see any growth defect associated with the triple mutants. Presumably the level of silencing restored in these strains was not sufficient to interfere with the function of a telomere proximal essential gene(s). Additionally, we did not observe restoration of telomeric silencing in rpd3 mutants.
Rpd3 antagonized Sir-protein-based silencing
Rpd3 is part of two complexes in S. cerevisiae: the large complex (Rpd3L) and the small complex (Rpd3S), each with distinct activities. Previous studies in yeast indicate that Rpd3 antagonizes silencing by restricting Sir-protein spreading beyond the boundary of silent loci, into adjacent euchromatic domains flanking HML and HMR and adjacent to telomeres (Zhou et al. 2009; Ehrentraut et al. 2010). However, this function of Rpd3 seems to be mediated exclusively by the Rpd3L complex (Sun and Hampsey 1999; Loewith et al. 2001; Zhou et al. 2009). In contrast, the block to silencing in the sas2∆ sir2N345A mutant showed no dependence on any of the Rpd3L-specific complex members tested. Although we tested those members previously shown to have the largest silencing effects in our assay, there is the possibility that other Rpd3L complex members are necessary for the silencing restoration. Additionally, there is the possibility that multiple large-complex-specific members must be deleted in combination to restore silencing in the sir2N345A sas2∆ strain. The Rpd3S complex functions to deacetylate nucleosomes within coding regions of actively transcribed genes to prevent aberrant transcription initiation (Carrozza et al. 2005), which is dependent on the small complex-specific subunit, Eaf3. Our data clearly showed that eaf3∆ provided no silencing restoration (Figure 2). We tested all members specific to the RpdS complex and none were able to restore silencing in the sir2N345A sas2∆ strain; thus it is unlikely that the Rpd3S complex is involved in the restoration of silencing. Additionally, we also tested which of the four PAH domains on Sin3 were necessary for the silencing restoration by the sin3∆. Only PAH3 was required to block silencing in the sir2N345A sas2∆ mutant (Figure 2D). Unlike PAH1, PAH2, and PAH4, which are necessary for interactions with other Sin3 complex members (Wang and Stillman 1993; Kasten et al. 1996; Washburn and Esposito 2001; Mallory et al. 2010), PAH3 has been shown in other organisms to specifically mediate interactions with Rpd3 (Laherty et al. 1997). Since none of the large or small complex-specific members had an effect on the silencing restoration, either by deletion of the subunit or deletion of the specific interacting surface on Sin3, the role of RPD3 in antagonizing silencing at HMR and HML appeared mechanistically distinct from the roles of these complexes restricting the spread of heterochromatic silencing and suppression of aberrant transcription initiation.
Rpd3 has also been shown to control origin firing in budding yeast. Deletion of RPD3 or SIN3 causes early activation of late origins and, similar to our observations, this phenotype is independent of any of the other complex members tested (Aparicio et al. 2004). Additionally, Rpd3’s repression of late origins is in opposition to Sir2, which promotes early origin firing (Yoshida et al. 2014). The silencers at HMR are late firing origins and the HML silencers can act as origins of replication on plasmids, but typically do not do so in the chromosome (Rusché et al. 2003). Additionally, establishment of Sir-based silencing is dependent on passage through S phase; however, this S-phase requirement is independent of replication (Miller and Nasmyth 1984; Kirchmaier and Rine 2001; Li et al. 2001). Nonetheless, our observations that Sir proteins show increased association at the silencers in the rpd3∆ (Figure 5) and that a mutation in ORC3 cosegregated with one of the Rpd3 mutations recovered (Figure S2) may indicate that Rpd3’s effect on origins is antagonistic to Sir-based silencing.
Sir2 catalytic activity promoted Sir-protein nucleation at silencers
Previously, it had been assumed that Sir2’s catalytic activity was necessary for the spreading of Sir proteins across the silenced cassette and that nucleation of Sir proteins was independent of Sir2’s catalytic activity. This view emerged from nonquantitative ChIP studies that showed Sir-protein association at the silencers, but not across HMR, in the absence of Sir2’s catalytic activity (Rusché et al. 2002). However, a more recent, quantitative study showed that Sir-protein association is reduced at the silencers of HMR and HML in sir2N345A mutants (Thurtle and Rine 2014). The results reported here confirmed that Sir-protein nucleation at silencers is defective in sir2N345A mutants and increased in rpd3 mutants commensurate with the partial restoration of silencing.
What is the protein target of Rpd3 whose acetylation promotes Sir-protein binding at the silencers? We investigated all known histone H3 and H4 targets of Rpd3, individually and in combinations, which revealed no involvement in the restoration of silencing observed here (Figure 4). Additionally, the silencers are nucleosome-free regions (Weiss and Simpson 1998; Ravindra et al. 1999). These data suggest an alternative acetylation as the target of Sir2 and Rpd3. Although historically named histone deacetylases, Sir2 and Rpd3 deacetylate proteins other than histones (Lin et al. 2009; Henriksen et al. 2012; Kaluarachchi Duffy et al. 2012; Downey et al. 2013). For example, Rpd3 deacetylates Snf2, a subunit of the SWI/SNF chromatin remodeling complex (Kim et al. 2010). Another possible candidate of a nonhistone Rpd3 target is the ORC. ORC binds to each of the silencers and the HMR silencers can act as origins of replication. A mutation in ORC3 cosegregated with the stronger restoration-of-silencing phenotypes of one of the RPD3 mutations. The Orc2 subunit is acetylated in human cells (Iizuka et al. 2006), the Hat1 acetyl transferase associates with Saccharomyces ORC, and mutations in HAT1 exacerbate the defect of temperature-sensitive orc mutations (Suter et al. 2007). Moreover, N-terminal acetylation of Orc1 in Saccharomyces contributes to its roles in replication and silencing (Geissenhöner et al. 2004). Perhaps the catalytically inactive Sir2 can still recognize and bind to acetylated Orc subunits, especially if their level is elevated in the rpd3 mutant, and contribute to the recruitment of the Sir-protein complex to the silencer.
Hst3 activity independent of H3K56 deacetylation was critical for silencing
Previous studies have implicated two other sirtuins, Hst3 and Hst4, as important for silencing at telomeres through their role in deacetylation of H3K56-acetyl (Yang et al. 2008). Additionally, H3K56-acetyl and Hst3 have a role in the stability of silencing at HML (Dodson and Rine 2015). The work reported here clearly highlighted a role for Hst3 in silencing at HML and HMR that was independent of H3K56-acetyl and perhaps of Hst4. The lack of an effect of an hst4 mutation may simply reflect a lower activity level of Hst4 relative to Hst3, or possibly a nonshared substrate. Nevertheless, it seems inescapable that Hst3’s role in silencing in the rpd3 mutant reflects a heretofore-overlooked substrate.
How could two sirtuins (Sir2 and Hst3), the broadly acting Rpd3 deacetylase, and the H4K16 acetyltransferase (Sas2) collaborate to produce the phenotypes described in this study? If we restrict speculation to models involving direct effects at HML and HMR, the models converge on a paradox. Mutations in Rpd3 result in elevated acetylation of most acetylated positions in core histones, with the exception of H4K16 (Robyr et al. 2002). Nevertheless, all such positions are hypoacetylated in heterochromatin. So how can the hyperacetylation resulting from the rpd3 mutation, at all positions except H4K16 (due to the sas2∆ mutation), restore silencing in a way that depends upon the sirtuin Hst3? The function of sirtuins is NAD+ dependent and results in the production of two cleavage products of NAD+: NAM and 2-OAAR (Jackson and Denu 2002). The 2-OAAR has been proposed to be a small molecule effector of silencing. To date there is only in vitro evidence that this molecule has the potential for affecting chromatin structure (Liou et al. 2005; Martino et al. 2009; Ehrentraut et al. 2010), and one in vivo study shows that silencing can be established without 2-OAAR in the presence of cleverly designed fusion proteins (Chou et al. 2008). Nevertheless there is still room to consider the possibility that 2-OAAR might be a critical ligand to promote silencing. In this view, the rpd3 mutation would result in elevated levels of acetylation of a substrate for Hst3, which would then produce 2-OAAR. To test this hypothesis, we mutated combinations of H3 and H4 histone N terminus residues individually and in combination (Figure 4) to test if their acetylation was necessary for the restoration of silencing by rpd3 mutants, and none were. However our study did not evaluate the entire acetylome, so an as yet unidentified Hst3 substrate could be deacetylated by the Hst3 sirtuin to generate 2-OAAR locally and promote silencing.
Additionally, there is the possibility that global, rather than local, increases in 2-OAAR production in the cell are necessary for silencing. In the absence of Rpd3, many of its targets are now acetylated and may become substrates for Hst3, generating 2-OAAR, which may promote silencing through some allosteric mechanism. As a partial test of this hypothesis, we deleted the Nudix hydrolase YSA1 in the sir2N345A sas2∆ strain. This deletion has been shown to increase intracellular 2-OAAR (Tong et al. 2009), but did not restore silencing (data not shown). However, Ysa1 localizes to the mitochondria, thus its activity may not alter nuclear 2-OAAR levels. Hence, for the time being, the 2-OAAR hypothesis remains a possible explanation for some or all of the phenotypes described here.
Acknowledgments
We thank Erin Osborne Nishimura for initial conception of the screen; Gavin Schlissel and Kelsey Van Dalfsen for strain contributions; David Stillman for the plasmids containing the paired amphipathic helix domain mutations in SIN3; members of the Rine lab for critical discussions, advice, and support; and Minyong Chung and the Vincent J. Coates Genomics Sequencing Laboratory at the University of California, Berkeley, which is supported by National Institutes of Health (NIH) S10 Instrumentation grants S10-RR029668 and S10-RR027303. This work has been supported by National Science Foundation predoctoral fellowships (to D.M.T.-S. and A.E.D.); a University of California, Berkeley’s training grant from the NIH (T32 GM 007232 34); and by a grant from the NIH National Institute of General Medical Sciences (NIGMS-31105 to J.R.).
Footnotes
Supplemental material is available online at http://www.genetics.org/lookup/suppl/doi:10.1534/genetics.116.190835/-/DC1.
Communicating editor: M. Hampsey
Literature Cited
Aparicio, O., J. V. Geisberg, E. Sekinger, A. Yang, Z. Moqtaderi et al., 2005 Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. Curr. Protoc. Mol. Biol. Chapter 21: Unit 21.3.
Burke, D. J., D. C. Amberg, and J. N. Strathern (Editors), 2005 Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Laboratory Press, Cold Spring Harbor, NY.
Geissenhöner A., C. Weise, and A. E. Ehrenhofer-Murray, 2004 Dependence of ORC Silencing Function on NatA-Mediated N α Acetylation in Saccharomyces cerevisiae. Mol. Cell Biol.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}