





Abstract
The mammalian X chromosome has unusual evolutionary dynamics compared to autosomes. Faster-X evolution of spermatogenic protein-coding genes is known to be most pronounced for genes expressed late in spermatogenesis, but it is unclear if these patterns extend to other forms of molecular divergence. We tested for faster-X evolution in mice spanning three different forms of molecular evolution—divergence in protein sequence, gene expression, and DNA methylation—across different developmental stages of spermatogenesis. We used FACS to isolate individual cell populations and then generated cell-specific transcriptome profiles across different stages of spermatogenesis in two subspecies of house mice (Mus musculus), thereby overcoming a fundamental limitation of previous studies on whole tissues. We found faster-X protein evolution at all stages of spermatogenesis and faster-late protein evolution for both X-linked and autosomal genes. In contrast, there was less expression divergence late in spermatogenesis (slower late) on the X chromosome and for autosomal genes expressed primarily in testis (testis-biased). We argue that slower-late expression divergence reflects strong regulatory constraints imposed during this critical stage of sperm development and that these constraints are particularly acute on the tightly regulated sex chromosomes. We also found slower-X DNA methylation divergence based on genome-wide bisulfite sequencing of sperm from two species of mice (M. musculus and M. spretus), although it is unclear whether slower-X DNA methylation reflects development constraints in sperm or other X-linked phenomena. Our study clarifies key differences in patterns of regulatory and protein evolution across spermatogenesis that are likely to have important consequences for mammalian sex chromosome evolution, male fertility, and speciation.
THE X chromosome plays a disproportionately large role in adaptation and speciation (Bachtrog et al. 2011; Ellegren 2011; Charlesworth 2013), but the underlying molecular and evolutionary drivers of these patterns remain unclear. On one hand, the X chromosome often shows strong signatures of evolutionary constraint. For example, the evolution of dosage compensation via epigenetic X-chromosome inactivation (XCI) in females (Lyon 1961, 1962) imposes regulatory constraints that select for strong conservation of X-linked gene content in placental mammals (Ohno 1967; Kohn et al. 2004). On the other hand, these inherent constraints are punctuated by strong specialization in X-linked gene content (Emerson et al. 2004; Mueller et al. 2008, 2013; Potrzebowski et al. 2008; Zhang et al. 2010; Sin et al. 2012) and numerous examples of rapid X-linked evolution (Torgerson and Singh 2003, 2006; Baines and Harr 2007; Kousathanas et al. 2014; Nam et al. 2015).
The X chromosome is predicted to evolve faster than the autosomes if beneficial mutations are on average recessive because selection will act more efficiently on X-linked mutations exposed in hemizygous males (Charlesworth et al. 1987). Under this model, faster-X evolution should be most intense for male-specific genes (Rice 1984; Vicoso and Charlesworth 2009). Indeed, the strongest evidence for faster-X evolution comes from patterns of protein-coding evolution during spermatogenesis (Torgerson and Singh 2006; Baines et al. 2008; Grath and Parsch 2012; Sin et al. 2012; Kousathanas et al. 2014). Molecular evolution on the X chromosome may also differ from the autosomes due to a smaller effective population size (i.e., 3/4 autosomal Ne assuming equal sex ratios) (Vicoso and Charlesworth 2009; Mank et al. 2010) or sex-linked differences in mutation rates (μ) (Miyata et al. 1987; Begun and Whitley 2000; Ellegren 2007). Differences in Ne and μ aside, the theoretical expectations for faster-X evolution should extend to other functional aspects of DNA sequence evolution. Gene regulation is usually measured through various biochemical phenotypes (e.g., transcript abundances, methylation patterns) that may not directly reflect linked sequence evolution, yet accelerated divergence of X-linked gene expression levels has been reported in fruit flies (Kayserili et al. 2012; Meisel et al. 2012a; Coolon et al. 2015; Llopart 2015), birds (Dean et al. 2015), and mammals (Khaitovich et al. 2005a; Zhang et al. 2010; Brawand et al. 2011). The evolution of other regulatory phenotypes has not been widely considered and the extent to which different forms of molecular evolution show similar patterns of X-linked evolution remains to be seen.
A critical evaluation of molecular evolution in the male germ line depends on a few important details of spermatogenesis. Spermatogenesis is defined by progressive gene specialization, with postmeiotic genes tending to be more narrowly expressed and functionally specific (Eddy 2002; Schultz et al. 2003; Good and Nachman 2005). Expression breadth and specialization influence rates of protein-coding evolution (Liao et al. 2006; Ellegren and Parsch 2007; Meisel et al. 2012b) and genes expressed later in spermatogenesis show more rapid protein-coding evolution (Good and Nachman 2005; Sin et al. 2012). Yet there remain relatively few examples where patterns of divergence have been evaluated across different stages of spermatogenesis (Good and Nachman 2005; Kousathanas et al. 2014), and how different forms of molecular evolution change in this developmental context is largely unknown. The sex chromosomes are also silenced during male meiosis through meiotic sex chromosome inactivation (MSCI) (Turner 2007). This period of inactivation results in strong selection against X-linked genes that need to be expressed during meiosis, while genes expressed prior to MSCI are enriched on the X chromosome (Wang et al. 2001; Khil et al. 2004). Gene expression remains transcriptionally repressed in postmeiotic stages of spermatogenesis [postmeiotic sex chromosome repression (PSCR)], although several X-linked genes overcome PSCR and are highly expressed in round spermatids (Namekawa et al. 2006; Mueller et al. 2008; Sin et al. 2012). In general, X-linked genes expressed during PSCR tend to be more specialized with narrower expression profiles and show more rapid protein evolution relative to coexpressed autosomal genes (Sin et al. 2012; Kousathanas et al. 2014).
While the context and timing of expression during spermatogenesis play crucial roles in the interpretation of faster-X protein evolution, most comparative expression studies have focused on whole tissues. This experimental approach implicitly assumes that differential gene expression between species is not simply an artifact of differences in cell composition. Yet spermatogenesis is usually asynchronous and overlapping in mature testis, leading to age-dependent heterogeneity in the abundances of germ cell populations through time. Testis cellular composition (i.e., testis histology) may also evolve rapidly when selection from sperm competition results in allometric shifts toward more sperm-producing seminiferous tubules (Firman et al. 2015). Such technical issues confound patterns of gene expression measured from whole testis (Good et al. 2010), especially when combined with differences in stages of maturity, levels of fertility, or comparisons between species. For example, testis expression patterns in primates cluster more strongly with mating system than evolutionary relatedness (Brawand et al. 2011; Saglican et al. 2014), indicating that testis transcriptomes are strongly influenced by convergent shifts in cellular composition associated with different reproductive strategies. When combined with considerable variation in relative enrichment for or against X linkage across different stages of spermatogenesis (Khil et al. 2004), it is apparent that a rigorous examination of faster-X gene expression evolution requires a cell- or stage-specific approach.
Here we report two experiments designed to evaluate three different forms of molecular evolution across spermatogenesis in mice. First, we used fluorescence-activated cell sorting (FACS) (Getun et al. 2011) and RNA sequencing (RNA-seq) (Wang et al. 2009) to generate transcriptomes from mitotic, meiotic, and postmeiotic spermatogenic cells in two subspecies of house mice (Mus musculus musculus and M. m. domesticus). We quantified genome-wide patterns of protein-coding and expression divergence across key developmental stages of spermatogenesis and tested for faster-X molecular evolution. Second, we performed whole genome bisulfite sequencing (BS-seq) (Frommer et al. 1992) to quantify patterns of DNA methylation divergence between sperm from house mice (M. m. musculus) and the closely related Algerian mouse (M. spretus). This second experiment allowed us to quantify the evolution of a key regulatory phenotype on and off the X chromosome for the first time in mice. Collectively, these experiments allow us to quantify different molecular evolutionary patterns in light of specific stages of sperm development.
Materials and Methods
Experimental design and choice of mouse strains
Information on protein-coding evolution between M. m. musculus, M. m. domesticus, and M. spretus is accessible using published genomic resources (Keane et al. 2011). However, the technical demands and experimental resources necessary to generate novel cell-specific transcriptome and DNA methylation data across spermatogenesis in all three lineages are reasonably beyond the scope of a single study. Therefore, we used a nested subset of evolutionary contrasts to optimize our power to quantify gene expression and methylation divergence. Divergence in gene expression levels accumulates relatively quickly (Lemos et al. 2004) and so we focused our FACS-based partitioning of gene expression across spermatogenesis on two subspecies of house mice (M. m. domesticus and M. m. musculus). We used four wild-derived inbred strains (purchased from The Jackson Laboratory, Bar Harbor, ME) to generate interstrain F1’s for each subspecies (M. m. musculus: CZECHII/EiJ females × PWK/PhJ males; M. m. domesticus: WSB/EiJ females × LEWES/EiJ males). This crossing design reduces the impacts of inbreeding depression on basic reproductive phenotypes and follows previous studies on these mice (Good et al. 2008, 2010; Campbell et al. 2013). Less in known about the evolutionary tempo of DNA methylation divergence, but patterns of methylation can be highly conserved between species (Molaro et al. 2011). Therefore, we focused our analysis of sperm DNA methylation on contrasts between M. spretus and M. m. musculus represented by four partially inbred strains of M. spretus (SFM and STF) and M. m. musculus (MPB and MBS) acquired from François Bonhomme (University of Montpellier, Montpellier, France).
Gene expression and DNA methylation experiments were initiated independently at the University of Montana (UM) (expression) and the University of Southern California (USC) (methylation) using different strains of mice. To confirm the expected evolutionary relationships among the mouse strains used in this study, we estimated a phylogeny using published whole genome data for WSB/EiJ and SPRET/EiJ (Keane et al. 2011) and new whole exome data from all other strains. Whole exomes were enriched for Illumina sequencing using an in-solution NimbleGen SeqCap EZ Mouse (Roche) exome design targeting 54.3 Mb of exonic regions in the mouse genome (Fairfield et al. 2011). This in-solution platform performs well when used across different species of Mus with minimal biases in capture efficiency and sensitivity (B. A. J. Sarver and J. M. Good, unpublished data). Custom individually barcoded Illumina libraries (Meyer and Kircher 2010) were enriched and sequenced [100 bp, paired end (PE)] at the University of Utah Microarray and Genomic Analysis Core (HiSequation 2000), the University of Oregon Genomics and Cell Characterization Core Facility (HiSequation 2500), and the USC Epigenome Center (HiSequation 2000 and NextSequation 500). We mapped quality-filtered reads to the mouse reference genome (GRCm38) using Burrows–Wheeler Aligner v0.7.12 (Li and Durbin 2009), called variants using HaplotypeCaller in the Genome Analysis Toolkit v3.3.0 (McKenna et al. 2010), filtered variants on a minimum quality score of 30 and depth of 10 reads, and combined variants using VCFtools v0.1.12b (Danecek et al. 2011). The combined set of SNPs was filtered to include sites called across all samples. We then used a concatenated alignment of all variant genotypes to estimate a maximum likelihood phylogeny using RAxML v8.2.3 (Stamatakis 2014).
Animal use was approved by USC or UM Institutional Animal Care and Use Committees. Experimental males were weaned at ∼21 days after birth and individually caged for at least 15 days to mitigate potential reproductive influences associated with dominance interactions (Snyder 1967). All experimental males were killed between 60 and 90 days using CO2 followed by cervical dislocation (UM protocol 002-13) or only cervical dislocation (USC protocol 11394).
Transcriptome and methylome sample preparation and sequencing
We used FACS to enrich individual cell populations from across the developmental timeline of spermatogenesis following Getun et al. (2011). Testes were dissected following euthanasia, decapsulated, and seminiferous tubules were digested. Cells were washed repeatedly, stained with Hoechst 33343 (Invitrogen) and propidium iodide, filtered twice through a 40-μm cell strainer, and kept on ice prior to sorting. Cell sorting was performed on a FACSAria IIu cell sorter (BD Biosciences) at the UM Center for Environmental Health Sciences Fluorescence Cytometry Core. Cell populations were sorted based on size, granularity, and fluorescence and collected in 15 μl β-mercaptoethanol (Sigma, St. Louis, MO) per milliliter of RLT lysis buffer (Qiagen, Valencia, CA). RNA was extracted from each cell population using the Qiagen RNeasy kit and quantified using a Bioanalyzer 2000 (Agilent, Santa Clara, CA). Samples with RNA integrity (RIN) ≥8 were prepared for RNA-seq using the Illumina Truseq Sample Prep Kit v2 in a design that avoided batch effects between cell populations and genotypes. Libraries were sequenced [100 bp PE and 100 bp, single end (SE)] on Illumina machines at the QB3 Vincent J. Coates Genomics Sequencing Laboratory at University of California, Berkley (HiSequation 2000), the University of Utah Microarray and Genomic Analysis Core (HiSequation 2000), the University of Oregon Genomics and Cell Characterization Core Facility (HiSequation 2500), and the USC Epigenome Center (HiSequation 2000 and NextSequation 500).
For BS-seq, sperm were isolated from caudal epididymes by incubating diced tissue in 50 μl of equilibrated M199 for 40 min at 5% CO2 at 37°. We removed tissue debris and allowed sperm to settle for 20 min. We then collected 100 μl of the top suspension and incubated the sample in 100 μl of extraction buffer (20 mM Tris Cl pH 8.0, 20 mM EDTA, 200 mM NaCl, 80 mM DTT, 4% SDS, 250 μg/ml Proteinase K) at 55°, with occasional mixing, until the sample was completely dissolved. We extracted DNA with the QIAamp DNA Mini Kit or the QIAamp DNA Blood Mini Kit (Qiagen) with a final elution in 50 μl of distilled H2O. Bisulfite treatment, which converts unmethylated cytosines to thymine, and 100 bp PE Illumina sequencing was performed at Beijing Genomics Institute.
Illumina sequence read processing and mapping
For RNA-seq data, we removed adaptors and low-quality bases using Trimmomatic v0.32 (Bolger et al. 2014). Trimmed reads were mapped using Tophat v2.0.10 (default parameters) (Kim et al. 2013) to strain-specific (PWK/PhJ and WSB/EiJ) published pseudoreferences that incorporate all known SNPs, indels, and structural variants relative to the mouse reference genome (GRCm38) based on the classic laboratory strain, C57BL/6J (Huang et al. 2014). This approach leverages the extensive annotation developed for the mouse genome while minimizing mapping bias that favors reads matching the C57BL/6J reference, which is predominantly derived from M. m. domesticus (Yang et al. 2011). We translated reads back into the GRCm38 coordinates using Lapels v1.0.5 (Holt et al. 2013; Huang et al. 2013) and counted the number of fragments uniquely mapping to protein-coding genes (GRCm38, Ensembl release 78) using featureCounts v1.4.4 (Q 20, C, primary). In addition, we counted multiple-mapped fragments within 203 multicopy/ampliconic X-linked genes (Mueller et al. 2013) and 156 Y-linked genes.
For methylome sequence data, we trimmed adaptors and mapped PE reads using rmapbs-pe in the RMAP package (Smith et al. 2008, 2009). We mapped M. m. musculus to GRCm38 and M. spretus to a custom pseudoreference generated with GATK v1.6 that combined the coordinates of GRCm38 with the sequence variation information (Sanger release version 1303) of M. spretus strain SPRET/EiJ (Keane et al. 2011).
Gene expression across spermatogenesis
All gene expression analyses were conducted using R v3.1.1(R Development Core Team 2014) and the Bioconductor v3.0 R package edgeR v3.12 (Robinson et al. 2010) with P-values adjusted to 5% false discovery rate (FDR) (Benjamini and Hochberg 1995). We restricted our analyses to protein-coding genes with greater than one fragment per kilobase of exon per million mapped reads (FPKM) in at least 4 of our 36 samples, and we also tested a range of expression thresholds (see Results). We evaluated library normalization using the weighted trimmed mean of M-values (Robinson and Oshlack 2010) or the scaling factor method (R package DESeq v1.22, Anders and Huber 2010). For brevity, we only present results using scaling factor normalization. To visualize expression data, we normalized FPKM values so that the sum of squares equals one (R package vegan v2.3-1) (Oksanen et al. 2015) or used variance-stabilizing transformation (Anders and Huber 2010). We plotted normalized FPKM values using the R packages beanplot v1.2 (Kampstra 2008) and gplots v2.17.0 (Warnes et al. 2015).
We defined a gene as “expressed” in a particular cell type (e.g., spermatogonia, round spermatids) if it had an FPKM >1 in a minimum of four individuals for a given cell type. We defined a gene as “induced” in a particular cell type if the median expression in the focal cell was higher (>2×) than the median expression of the other cell types combined. Similarly, we defined genes as induced at a particular stage of spermatogenesis (e.g., early, late) when expression was higher (>4×) than the maximum expression of all other stages (e.g., Kousathanas et al. 2014). We used Affymetrix microarray data across 96 tissues (Mouse Genome 430 2.0 Array) from the Mouse MOE430 Gene Atlas (Su et al. 2002; Lattin et al. 2008) to identify testis-biased genes (testis expression >2× median tissue expression) and to estimate tissue specificity (τ) following the recommendations of Liao and Zhang (2006). The τ-value ranges from 0 to 1 with higher values indicative of expression restricted to one or a few tissues (Yanai et al. 2005; Liao and Zhang 2006; Liao et al. 2006).
Evolutionary analyses of protein-coding and expression divergence
We used codeml in PAML v4.7 (Yang 2007) to estimate the rate of nonsynonymous substitutions (dN), synonymous substitutions (dS), and the dN/dS ratio (ω). Annotated protein-coding sequences were extracted from published whole-genome sequences of M. m. musculus (PWK/PhJ), M. m. domesticus (WSB/EiJ), and M. spretus (SPRET/EiJ) (Keane et al. 2011). We used Biostrings v2.32.1 (Pages et al. 2008) to retrieve sequences from the longest transcript for each gene, excluding transcripts with internal stop codons. For genes that are induced early and late in spermatogenesis, we estimated pairwise and global (i.e., one rate) ω from unrooted three-taxon alignments of (1) individual transcripts and (2) a concatenated set of transcripts for each chromosome. Estimates of dN and dS from one-to-one orthologs between M. m. domesticus (C57BL/6J) and Rattus norvegicus were retrieved from Ensembl Biomart release 83 (Smedley et al. 2015). For analyses of individual genes, we discarded transcripts with dS values above the 95% quantile as a filter for poor alignments and transcripts with dS estimates near zero, which are uninformative and can artificially inflate ω.
To identify differentially expressed (DE) genes between M. m. musculus and M. m. domesticus, we fit our data with negative binomial generalized linear models with Cox–Reid tagwise dispersion estimates (McCarthy et al. 2012). Our model included species and cell type as a single factor and we constructed a design matrix that contrasted each unique combination (e.g., M. m. musculus spermatogonia vs. M. m. domesticus spermatogonia). We then tested for DE using likelihood ratio tests, dropping one coefficient from the design matrix (i.e., the “null” model) and comparing that to the full model. We also calculated the correlation (1 − Spearman’s ρ) in chromosome-wide expression divergence between subspecies using pairwise median FPKM values with C.I.’s generated by bootstrapping the data 1000 times.
Sperm methylation divergence
We quantified sperm methylome divergence between M. m. musculus and M. spretus using BS-seq (Frommer et al. 1992) on eight mice (four per species). Methylation of the cytosine in cytosine–phosphate–guanine dinucleotides (CpG sites) plays a central role in the epigenetic regulation of gene expression in mammals (Reik 2007). Mammalian genomes generally show high levels of CpG methylation, punctuated by small hypomethylated regions (HMRs) associated with promoters and enhancers (Molaro et al. 2011). Our primary goal was to study methylation divergence at orthologous CpG sites. Therefore, we excluded sites if over half of the mapped reads for a given individual suggested a mutation resulting in the loss of CpG status (neither thymine-phosphate-guanine nor CpG). To control for differences in sequencing coverage, we randomly downsampled uniquely mapped autosomal reads that contained at least one CpG so that individual autosomes had average CpG coverage similar to the X-linked CpGs. HMRs and other basic statistics were called using the HMR program in MethPipe (Song et al. 2013). Genomic windows that were called HMR in one species but not the other were flagged as potential differentially methylated regions (DMRs). We then called DMRs using the DMR program in MethPipe, requiring each DMR to contain at least five orthologous CpGs that were differentially methylated between the species based on an hypergeometric test of the four possible states (methylated vs. not methylated × M. spretus vs. M. m. musculus).
We used three different null hypotheses to test whether the proportion of X-linked DMRs differed from expectations. First, the number of X-linked base pairs divided by the total number of base pairs [minus mitochondrial DNA (mtDNA) and chromosome Y (chrY); 171,032,299/2,633,777,672 = 0.065] in the sequenced mouse genome (GRCm38) provided a crude expectation of the proportion of X-linked DMRs. Second, because hypomethylation tends to cluster near protein-coding genes, we calculated the null expectation based on the proportion of protein-coding genes that are X linked (869/19,720 = 0.044). Third, to further account for potential differences in CpG sequencing coverage between the X chromosome and the autosomes, we calculated the X-to-autosome ratio of CpGs that were covered at least 3× in one species and at least 4× in the other (the minimum number of reads necessary to detect a significantly differentially methylated CpG) among the downsampled reads (348,647/9,968,595 = 0.035).
Data availability
The data reported in this paper are available through the National Center for Biotechnology Information under accession nos. SRP065082, SRP065034, SRP075865 and SRP077631.
Results
Phylogenetic relationship of the mice used in this study
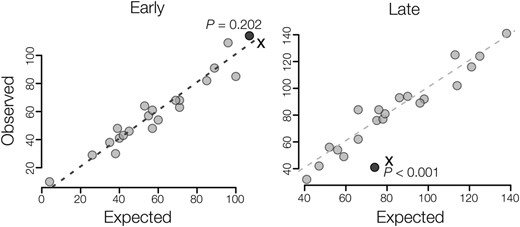
We estimated a maximum likelihood phylogeny using whole genome and targeted exome resequencing data from the mouse strains used in this study (Figure 1). The resulting tree conformed to the expected evolutionary relationships among strains of M. m. musculus, M. m. domesticus, and M. spretus. Strains within a given taxon are closely related and approximate levels of individual variation follow general expectations for these species and subspecies. Likewise, M. musculus and M. spretus were separated by about twice as much genetic divergence as M. m. musculus and M. m. domesticus. Thus, the different strains of mice used within our studies of expression, DNA methylation, and protein-coding divergence represent closely related samples from these three lineages.
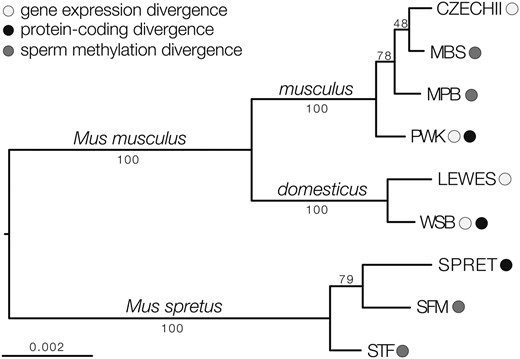
Evolutionary relationships among mouse strains and species. The phylogeny was estimated using maximum likelihood, based on genome-wide data. Bootstrap proportions are indicated for each internal branch. Shaded circles indicate which strains were used for estimates of gene expression, protein-coding, and sperm DNA methylation divergence.
Stage-specific dissection of gene expression across mouse spermatogenesis
We used FACS to generate highly enriched populations of four cell types spanning three phases of spermatogenesis (Supplemental Material, Figure S1, Figure 2A): (1) spermatogonia (mitosis), (2) leptotene/zygotene spermatocytes (meiosis, prior to MSCI), (3) diplotene spermatocytes (meiosis, after MSCI), and (4) round spermatids (postmeiosis). For each subspecies, M. m. musculus and M. m. domesticus, we mapped between 53 and 74 million unique sequencing fragments from each cell type to their respective genomes (Table S1). Each cell population had a distinct expression profile, there was strong clustering of transcription levels by cell type, and expression patterns of established cell- or stage-specific genes were specific to their cell type (Figure S2). Overall, these results indicate that our FACS experiments yielded highly enriched cell populations with low variance among individual samples.
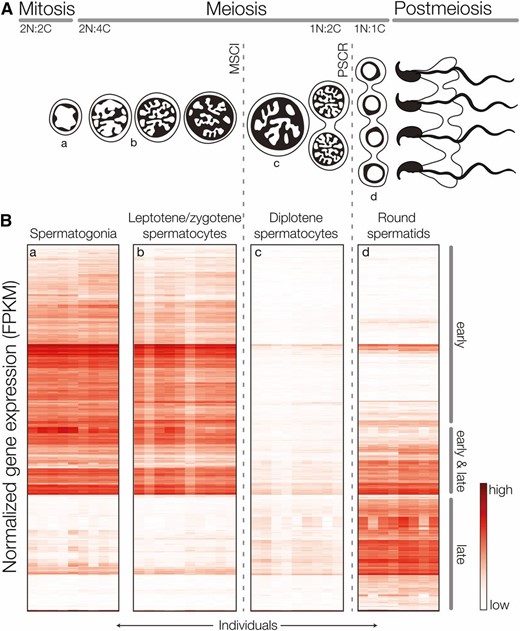
X-linked expression across spermatogenesis in M. m. musculus and M. m. domesticus. (A) Progression of dividing germ cells during spermatogenesis. In early prophase I the X chromosome is transcriptionally silenced (MSCI) for the remainder of meiosis and remains partially inactivated (PSCR) during postmeiotic development of spermatids. We used FACS to isolate cell populations spanning these three phases of spermatogenesis (a–d). (B) Patterns of X-linked gene expression in FACS isolated cell populations. Genes (rows) are grouped by the timing of induction: early (expressed prior to MSCI), early and late (inactivated at MSCI and reactivated late), and late (expressed only in postmeiotic cells).
We detected expression of 14,223 protein-coding genes across spermatogenesis. Most genes were expressed in multiple cell types (Table 1), but often with large between-cell differences in expression levels. Our developmental timeline brackets the onset of MSCI (at midpachytene), and as expected, we observed chromosome-wide repression of X-linked genes in diplotene spermatocytes and partial reactivation of the X chromosome in postmeiotic round spermatids (Figure 2). Each cell population produced distinct expression profiles (Figure S2), but early cell types (spermatogonia and leptotene/zygotene spermatocytes) showed very similar patterns of expression. Therefore, we grouped results into three expression stages relative to MSCI: (1) expressed early in spermatogonia and/or leptotene/zygotene spermatocytes and silenced at MSCI; (2) expressed early, silenced at MSCI, and reactivated late; and (3) expressed only late (Figure 3). Genes induced at each of these stages showed good overall agreement between functional annotations and developmental processes indicative of that stage of spermatogenesis (Table 1). We also observed very high gene-by-gene correspondence between our general groupings and previously described patterns of X-linked expression during spermatogenesis (Figure S3), which used different methods of cell enrichment and expression profiling (Namekawa et al. 2006). These results indicate that our FACS-based enrichment captures the dynamic changes in transcription across spermatogenesis.
The X chromosome is enriched for genes induced before and after MSCI and for testis-biased genes
| % Expresseda | % Inducedb | % Testis-biasedc | Functional annotation | ||||
|---|---|---|---|---|---|---|---|
| Auto | X | Auto | X | Auto | X | Auto and X | |
| By cell | |||||||
| Spermatogonia | 54.3 | 50.5 | 19.1 | 40.3*** | 5.6 | 11.3*** | Cell, intracellular, anatomical structure development, biosynthetic process, nucleic acid binding transcription factor activityd |
| Leptotene/zygotene spermatocytes | 52.7 | 45.6 | 15.3 | 34.0*** | 5.2 | 7.5 | Organelle, intracellular, cell, cellular nitrogen metabolic process, nucleusd |
| Diplotene spermatocytese | 48.0 | 13.2 | 16.2 | — | 48.4 | — | Cilium, reproduction, microtubule organizing center |
| Round spermatids | 50.3 | 35.6 | 22.8 | 23.0 | 42.5 | 44.9 | Cilium, reproduction |
| By stage | |||||||
| Early | NA | NA | 10.6 | 33.9*** | 3.1 | 7.5*** | Anatomical structure development, immune system process, plasma membrane, signal transduction, cell differentiationd |
| Early and late | NA | NA | 2.3 | 9.3 | 14.3 | 19.5 | Anatomical structure development, cytoskeletal organization, cytoskeletal protein binding, cell differentiation, cytoskeletond |
| Late | NA | NA | 7.9 | 19.1*** | 35.6 | 49.4*** | Reproduction, signal transducer activity, extracellular region, cell wall organization or biogenesis, neurological system process |
| % Expresseda | % Inducedb | % Testis-biasedc | Functional annotation | ||||
|---|---|---|---|---|---|---|---|
| Auto | X | Auto | X | Auto | X | Auto and X | |
| By cell | |||||||
| Spermatogonia | 54.3 | 50.5 | 19.1 | 40.3*** | 5.6 | 11.3*** | Cell, intracellular, anatomical structure development, biosynthetic process, nucleic acid binding transcription factor activityd |
| Leptotene/zygotene spermatocytes | 52.7 | 45.6 | 15.3 | 34.0*** | 5.2 | 7.5 | Organelle, intracellular, cell, cellular nitrogen metabolic process, nucleusd |
| Diplotene spermatocytese | 48.0 | 13.2 | 16.2 | — | 48.4 | — | Cilium, reproduction, microtubule organizing center |
| Round spermatids | 50.3 | 35.6 | 22.8 | 23.0 | 42.5 | 44.9 | Cilium, reproduction |
| By stage | |||||||
| Early | NA | NA | 10.6 | 33.9*** | 3.1 | 7.5*** | Anatomical structure development, immune system process, plasma membrane, signal transduction, cell differentiationd |
| Early and late | NA | NA | 2.3 | 9.3 | 14.3 | 19.5 | Anatomical structure development, cytoskeletal organization, cytoskeletal protein binding, cell differentiation, cytoskeletond |
| Late | NA | NA | 7.9 | 19.1*** | 35.6 | 49.4*** | Reproduction, signal transducer activity, extracellular region, cell wall organization or biogenesis, neurological system process |
Values represent the percentage of genes that meet a given expression threshold in each cell type and stage of spermatogenesis. Enrichment of X-linked genes are based on chromosome-wise hypergeometric tests, FDR-corrected P-values: *** P ≤ 0.001.
Expressed, genes with FPKM > 1 in a minimum of four individuals for a given cell type out of the total genes detected (14,223 genes).
Induced, by cell: genes with a median FPKM > 2× the median FPKM of all other cell types combined out of the total genes expressed. By stage: genes with a maximum FPKM > 4× the maximum FPKM of all other stages combined of the total genes expressed. (Note: a gene can be induced in more than one cell type, but can only be induced at a single stage).
Testis-biased, genes with higher expression in the testes compared 96 other tissues from the Mouse MOE430 Gene Atlas out of the total genes induced.
Only the top five gene enrichment categories are listed.
Due to MSCI there are too few genes expressed in diplotene spermatocytes to report X-linked induced genes.
| % Expresseda | % Inducedb | % Testis-biasedc | Functional annotation | ||||
|---|---|---|---|---|---|---|---|
| Auto | X | Auto | X | Auto | X | Auto and X | |
| By cell | |||||||
| Spermatogonia | 54.3 | 50.5 | 19.1 | 40.3*** | 5.6 | 11.3*** | Cell, intracellular, anatomical structure development, biosynthetic process, nucleic acid binding transcription factor activityd |
| Leptotene/zygotene spermatocytes | 52.7 | 45.6 | 15.3 | 34.0*** | 5.2 | 7.5 | Organelle, intracellular, cell, cellular nitrogen metabolic process, nucleusd |
| Diplotene spermatocytese | 48.0 | 13.2 | 16.2 | — | 48.4 | — | Cilium, reproduction, microtubule organizing center |
| Round spermatids | 50.3 | 35.6 | 22.8 | 23.0 | 42.5 | 44.9 | Cilium, reproduction |
| By stage | |||||||
| Early | NA | NA | 10.6 | 33.9*** | 3.1 | 7.5*** | Anatomical structure development, immune system process, plasma membrane, signal transduction, cell differentiationd |
| Early and late | NA | NA | 2.3 | 9.3 | 14.3 | 19.5 | Anatomical structure development, cytoskeletal organization, cytoskeletal protein binding, cell differentiation, cytoskeletond |
| Late | NA | NA | 7.9 | 19.1*** | 35.6 | 49.4*** | Reproduction, signal transducer activity, extracellular region, cell wall organization or biogenesis, neurological system process |
| % Expresseda | % Inducedb | % Testis-biasedc | Functional annotation | ||||
|---|---|---|---|---|---|---|---|
| Auto | X | Auto | X | Auto | X | Auto and X | |
| By cell | |||||||
| Spermatogonia | 54.3 | 50.5 | 19.1 | 40.3*** | 5.6 | 11.3*** | Cell, intracellular, anatomical structure development, biosynthetic process, nucleic acid binding transcription factor activityd |
| Leptotene/zygotene spermatocytes | 52.7 | 45.6 | 15.3 | 34.0*** | 5.2 | 7.5 | Organelle, intracellular, cell, cellular nitrogen metabolic process, nucleusd |
| Diplotene spermatocytese | 48.0 | 13.2 | 16.2 | — | 48.4 | — | Cilium, reproduction, microtubule organizing center |
| Round spermatids | 50.3 | 35.6 | 22.8 | 23.0 | 42.5 | 44.9 | Cilium, reproduction |
| By stage | |||||||
| Early | NA | NA | 10.6 | 33.9*** | 3.1 | 7.5*** | Anatomical structure development, immune system process, plasma membrane, signal transduction, cell differentiationd |
| Early and late | NA | NA | 2.3 | 9.3 | 14.3 | 19.5 | Anatomical structure development, cytoskeletal organization, cytoskeletal protein binding, cell differentiation, cytoskeletond |
| Late | NA | NA | 7.9 | 19.1*** | 35.6 | 49.4*** | Reproduction, signal transducer activity, extracellular region, cell wall organization or biogenesis, neurological system process |
Values represent the percentage of genes that meet a given expression threshold in each cell type and stage of spermatogenesis. Enrichment of X-linked genes are based on chromosome-wise hypergeometric tests, FDR-corrected P-values: *** P ≤ 0.001.
Expressed, genes with FPKM > 1 in a minimum of four individuals for a given cell type out of the total genes detected (14,223 genes).
Induced, by cell: genes with a median FPKM > 2× the median FPKM of all other cell types combined out of the total genes expressed. By stage: genes with a maximum FPKM > 4× the maximum FPKM of all other stages combined of the total genes expressed. (Note: a gene can be induced in more than one cell type, but can only be induced at a single stage).
Testis-biased, genes with higher expression in the testes compared 96 other tissues from the Mouse MOE430 Gene Atlas out of the total genes induced.
Only the top five gene enrichment categories are listed.
Due to MSCI there are too few genes expressed in diplotene spermatocytes to report X-linked induced genes.
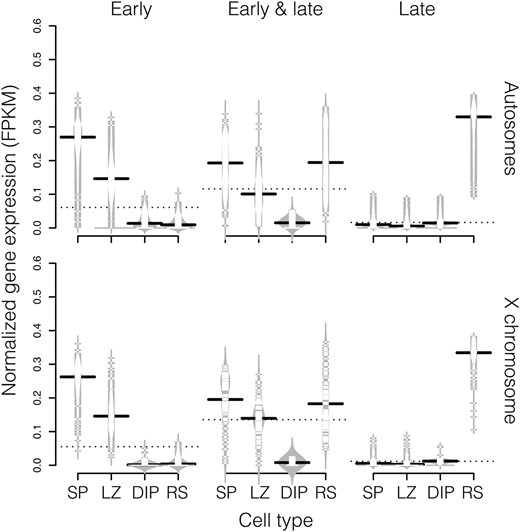
Expression patterns for genes induced at different stages of spermatogenesis in M. musculus. The normalized FPKM values for each gene is plotted as a horizontal tick mark and the distribution density for each cell type is plotted as a gray outline. Lines represent the median FPKM for each cell type (solid) and stage of spermatogenesis (dashed). Genes induced early have higher expression before the onset of MSCI (LZ, leptotene/zygotene spermatocytes and SP, spermatogonia). Genes induced both early and late are silenced at MSCI (DIP, diplotene spermatocytes) and are reactivated in postmeiotic cells (RS, round spermatids). Genes induced late are only highly expressed in postmeiotic cells.
Next, we combined our data with published multitissue expression data and found that testis-biased genes made up a larger proportion of the genes induced late in spermatogenesis (early 4.0%, late 35.4%), consistent with increasing gene specificity during postmeiotic development (Schultz et al. 2003). The tissue specificity of gene expression (τ) also increased late (median ± SE τ early, 0.831 ± 0.005; late, 0.903 ± 0.009, Wilcoxon test P ≤ 0.001). This pattern was particularly striking on the X chromosome where nearly half of late-induced genes were also testis-biased (Table 1). In addition, the X chromosome was enriched for genes induced early. These results are in agreement with previous studies indicating that X-linked gene content has been shaped by natural selection for spermatogenic genes expressed before or after MSCI (Khil et al. 2004; Mueller et al. 2008; Sin et al. 2012).
Faster-late and faster-X protein evolution
We calculated rates of protein-coding evolution (ω) between M. m. musculus, M. m. domesticus, and M. spretus for genes induced at different stages of spermatogenesis. These contrasts revealed two striking patterns. First, late-induced autosomal and X-linked genes evolved more rapidly than early-induced genes (i.e., faster-late, Figure 4A, Table 2), confirming a previous finding of a positive correlation between rates of protein evolution and timing of expression during spermatogenesis (Good and Nachman 2005). Second, X-linked genes showed significantly higher ω when compared to autosomal genes (i.e., faster-X, Figure 4A). These patterns of faster-late and faster-X protein evolution held when considering only pairwise differences within M. musculus (M. m. musculus vs. M. m. domesticus) (Table S2) or when considering much more divergent contrasts between the house mouse and the Norwegian rat (Table 2). They were also robust to different expression level thresholds (Table S3).
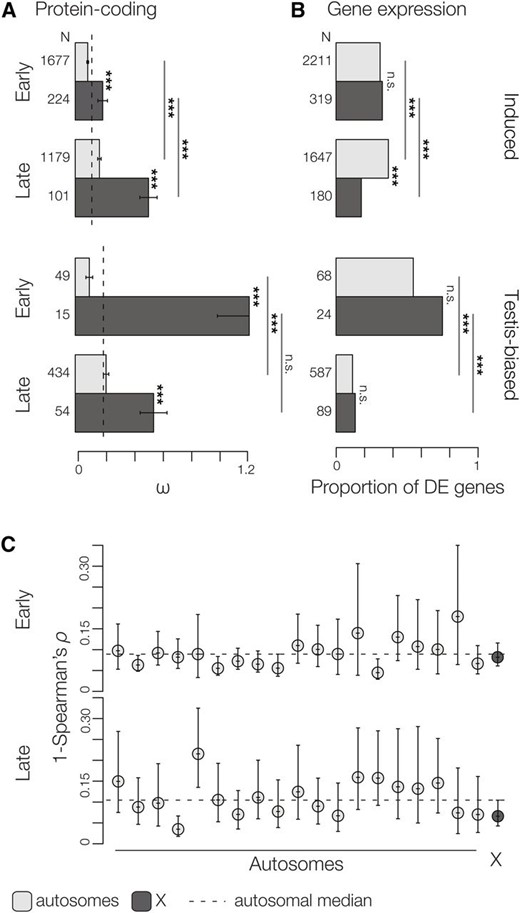
Evolutionary divergence in protein-coding sequence and expression level. (A) Median (±SE) estimates of protein-coding divergence (ω) among subspecies of M. musculus and M. spretus is higher for genes induced late and for X-linked genes. Gene expression divergence between subspecies of M. musculus estimated as (B) the proportion of induced genes that are DE and (C) the pairwise correlations of gene expression per chromosome (1 − ρ, ± 95% C.I.). The evolution of X-linked genes is either equivalent or slower than autosomal genes and there is a marked drop in the proportion of testis-biased genes induced late in spermatogenesis that are DE. N = total number of genes. Significant differences in ω (Wilcoxon test) and proportion of DE genes (χ2) are indicated for each contrast. FDR-corrected P-values: *** P ≤ 0.001.
Protein-coding divergence on the X chromosome and the autosomes
| Global estimates among Mus species | M. m. domesticus vs. R. norvegicus | |
|---|---|---|
| N | ||
| Auto | 9898 | 10,565 |
| X | 416 | 311 |
| ω | ||
| Auto | 0.114 ± 3.00 × 10−3 | 0.119 ± 1.61 × 10−3 |
| X | 0.235*** ± 2.82 × 10−2 | 0.192*** ± 1.33 × 10−2 |
| dN | ||
| Auto | 0.003 ± 5.48 × 10−5 | 0.023 ± 3.38 × 10−4 |
| X | 0.004*** ± 4.99 × 10−4 | 0.029*** ± 2.82 × 10−3 |
| dS | ||
| Auto | 0.024 ± 1.10 × 10−4 | 0.196 ± 5.73 × 10−4 |
| X | 0.018*** ± 4.90 × 10−4 | 0.156*** ± 3.52 × 10−3 |
| dSX/dSauto | 0.750 | 0.796 |
| Global estimates among Mus species | M. m. domesticus vs. R. norvegicus | |
|---|---|---|
| N | ||
| Auto | 9898 | 10,565 |
| X | 416 | 311 |
| ω | ||
| Auto | 0.114 ± 3.00 × 10−3 | 0.119 ± 1.61 × 10−3 |
| X | 0.235*** ± 2.82 × 10−2 | 0.192*** ± 1.33 × 10−2 |
| dN | ||
| Auto | 0.003 ± 5.48 × 10−5 | 0.023 ± 3.38 × 10−4 |
| X | 0.004*** ± 4.99 × 10−4 | 0.029*** ± 2.82 × 10−3 |
| dS | ||
| Auto | 0.024 ± 1.10 × 10−4 | 0.196 ± 5.73 × 10−4 |
| X | 0.018*** ± 4.90 × 10−4 | 0.156*** ± 3.52 × 10−3 |
| dSX/dSauto | 0.750 | 0.796 |
Estimates of median (±SE) omega (ω), nonsynonymous substitution rate (dN), and synonymous substitution rate (dS) among M. m. musculus, M. m. domesticus, M. spretus, and R. norvegicus. N = the number of genes in each comparison. Significant differences (Wilcoxon test) between the autosomes and X chromosome are indicated by FDR-corrected (Benjamini and Hochberg 1995) P-values: *** P < 0.001.
| Global estimates among Mus species | M. m. domesticus vs. R. norvegicus | |
|---|---|---|
| N | ||
| Auto | 9898 | 10,565 |
| X | 416 | 311 |
| ω | ||
| Auto | 0.114 ± 3.00 × 10−3 | 0.119 ± 1.61 × 10−3 |
| X | 0.235*** ± 2.82 × 10−2 | 0.192*** ± 1.33 × 10−2 |
| dN | ||
| Auto | 0.003 ± 5.48 × 10−5 | 0.023 ± 3.38 × 10−4 |
| X | 0.004*** ± 4.99 × 10−4 | 0.029*** ± 2.82 × 10−3 |
| dS | ||
| Auto | 0.024 ± 1.10 × 10−4 | 0.196 ± 5.73 × 10−4 |
| X | 0.018*** ± 4.90 × 10−4 | 0.156*** ± 3.52 × 10−3 |
| dSX/dSauto | 0.750 | 0.796 |
| Global estimates among Mus species | M. m. domesticus vs. R. norvegicus | |
|---|---|---|
| N | ||
| Auto | 9898 | 10,565 |
| X | 416 | 311 |
| ω | ||
| Auto | 0.114 ± 3.00 × 10−3 | 0.119 ± 1.61 × 10−3 |
| X | 0.235*** ± 2.82 × 10−2 | 0.192*** ± 1.33 × 10−2 |
| dN | ||
| Auto | 0.003 ± 5.48 × 10−5 | 0.023 ± 3.38 × 10−4 |
| X | 0.004*** ± 4.99 × 10−4 | 0.029*** ± 2.82 × 10−3 |
| dS | ||
| Auto | 0.024 ± 1.10 × 10−4 | 0.196 ± 5.73 × 10−4 |
| X | 0.018*** ± 4.90 × 10−4 | 0.156*** ± 3.52 × 10−3 |
| dSX/dSauto | 0.750 | 0.796 |
Estimates of median (±SE) omega (ω), nonsynonymous substitution rate (dN), and synonymous substitution rate (dS) among M. m. musculus, M. m. domesticus, M. spretus, and R. norvegicus. N = the number of genes in each comparison. Significant differences (Wilcoxon test) between the autosomes and X chromosome are indicated by FDR-corrected (Benjamini and Hochberg 1995) P-values: *** P < 0.001.
Interpreting patterns of ω requires consideration of the synonymous (dS) and nonsynonymous (dN) per site substitution rates in isolation. The mammalian X chromosome has a lower per-nucleotide mutation rate than the autosomes as a consequence of male-driven molecular evolution (Ellegren and Parsch 2007). Consistent with this, we observed lower dS for X-linked genes across all evolutionary contrasts (Table 2, Table S2) and for X-linked genes expressed early or late in spermatogenesis (Table S2). The X chromosome is also predicted to have a lower Ne relative to the autosomes and thus shallower coalescent depths on average, which could have a particularly strong impact on the comparison of X vs. autosomal divergence between closely related lineages. Consequently, we observed the greatest difference in X-linked vs. autosomal estimates of dS between M. m. musculus and M. m. domesticus (dSX/dSauto = 0.572 vs. dSX/dSauto ∼0.8 for contrasts involving M. spretus or rat). Finally, we found that the X chromosome shows higher dN compared to the autosomes (Table 2), with the greatest difference among genes induced late in spermatogenesis (Table S2). Thus, we found consistently faster-late and faster-X protein-coding evolution for mouse spermatogenic genes across different levels of evolutionary divergence and when considering both relative and absolute rates of nonsynonymous divergence.
Slower-late and slower-X expression evolution
We observed strikingly different patterns of expression divergence relative to protein evolution. Although there was a general trend toward more DE genes induced late in spermatogenesis (24% of spermatogonia genes vs. 37% of round spermatids genes were DE) this trend was restricted to autosomal genes and there were actually fewer X-linked DE genes induced late (i.e., slower-X, Figure 4, B and C). The slower-X pattern late in spermatogenesis was robust to different temporal classifications (Table S4), different expression thresholds (Table S5), different methods of accounting for X-linked ampliconic/multicopy genes (Figure S4), and was also apparent in per-chromosome comparisons (Figure 5). As discussed above, testis-biased genes tended to be expressed late in spermatogenesis (Table 1). When we restricted our analysis to testis-biased genes, we found that expression divergence was dramatically reduced late and was similarly constrained between the X chromosome and the autosomes (i.e., slower-late, Figure 4B). If we compare expression divergence on the autosomes and the X chromosome while excluding testis-biased genes, the pattern of slower-X late is even more apparent (Figure S5). Thus, slower-late expression evolution appears to be a general feature of X-linked spermatogenic genes, but was restricted to testis-biased genes on the autosomes.
Contrasting patterns of gene expression divergence on the X chromosome. Early in spermatogenesis the X chromosome has a similar proportion of DE genes compared to the autosomes, while there are fewer X-linked DE genes late in spermatogenesis. The shift in the number of observed/expected X-linked genes between early and late reflects the enrichment of X-linked genes induced early. Significance is based on chromosome-wise hypergeometric test for enrichment with FDR-corrected P-values.
These estimates of differential expression reflect divergence of a biochemical phenotype that do not account for the potential impacts of lower mutation rates and/or shallower coalescent times on the X chromosome. If we conservatively scale our expectation for X-linked differential expression by the X-to-autosome ratio of synonymous divergence (dSX/dSauto = 0.572 for the contrast between M. m. musculus and M. m. domesticus) then we find that the X chromosome becomes significantly enriched for DE genes induced early (P < 0.001) and the slower-X pattern becomes nonsignificant late (P = 0.38). Likewise, for testis-biased genes, the X chromosome is significantly enriched for DE genes early (P < 0.001) but not late (P = 0.924). Thus, the strongest pattern in our data is that expression divergence changes considerably across spermatogenesis both on and off the X chromosome, but at each stage the rate of evolution of the X chromosome relative to the autosomes depends on the extent to which mutation and effective population size influence evolution of this phenotype.
Overall patterns of protein divergence and expression divergence were qualitatively different across this timeline. Protein divergence increased late in spermatogenesis and was elevated on the X chromosome, while the opposite was true for expression divergence. Interpretation of X vs. autosomal expression divergence is clearly dependent on the assumptions one makes with respect to null expectations. Given this, it is informative to focus on the relationships between gene expression and protein evolution across spermatogenesis. We calculated correlations and partial correlations between protein evolution (ω), expression level (normalized FPKM), log2 fold change between subspecies (logFC), and tissue specificity (τ) (Figure S6). There was a strong positive relationship between expression level and logFC on and off the X chromosome for genes induced early and late in spermatogenesis. LogFC and τ were positively correlated on the autosomes early, indicating that tissue-specific genes tended to also show larger changes in expression levels between M. m. musculus and M. m. domesticus. In contrast, logFC was negatively correlated with both τ and ω on the autosomes late in spermatogenesis, but only when testis-biased genes were included (Figure S6). These results support our general finding of elevated protein-coding divergence coupled with more constrained gene expression for testis-biased genes late in spermatogenesis.
Slower-X sperm methylome evolution
For BS-seq, we obtained an average genomic coverage of 12.7× in M. m. musculus and 12.3× in M. spretus (Table S6, Table S7). Methylation levels across individual CpG sites were most strongly correlated among individuals within strains, followed by strains within species, followed by between-species comparisons (Table S8), as expected based on patterns of DNA sequence divergence (Figure 1). Approximately 6.0% of the genome lies underneath X-linked HMRs, which is reasonably close to expectations based on the relative length of the X chromosome (6.4%). We defined methylome divergence by identifying DMRs as genomic regions with adequate coverage in both species but where an HMR was called in only one of the species. Only 2.3% of the 9580 DMRs between M. m. musculus and M. spretus were X linked, a strongly significant reduction given the proportion of X-linked CpGs (χ2 = 23.2, P < 10−5; Figure 6, Table S9). We observed considerable variation in DMR enrichment among chromosomes (Figure 6), likely because of the sensitivity of enrichment tests when sample sizes are large. Nine of 20 linkage groups showed significant skews in observed vs. expected DMRs but the X chromosome always showed the strongest de-enrichment of DMRs across a range of test configurations. We emphasize here that we downsampled autosomal reads to match X-linked coverage; therefore, our results cannot be explained by differential coverage. Our results remained qualitatively identical across five different downsampling schemes. Thus, in mouse sperm, the X chromosome shows less absolute divergence in sperm DNA methylation status relative to the autosomes.

Evolution of the mouse sperm methylome. Chromosomal distributions of DMRs between M. m. musculus and M. spretus. The observed and expected numbers of DMRs are plotted to the left. Expectations are based on the proportion of CpG sites on each chromosome multiplied by the total number of DMRs. Darkly shaded chromosomes have significantly more or less DMRs than expected based on chromosome-wise hypergeometric tests. The X chromosome has significantly fewer DMRs compared to all of the autosomes (P < 0.001).
Finally, we evaluated the potential influence of lower X chromosome substitution rates on sperm methylome evolution. Similar to our expression analyses above, we corrected our expectation for the number of X-linked DMRs by the X-to-autosome ratio of synonymous substitutions (dSX/dSauto = 0.77 for the contrast between M. m. musculus and M. m. spretus). Using this correction, the reduction in the frequency of DMRs on the X chromosome becomes nonsignificant (χ2, P = 0.088).
Discussion
Forty years ago, King and Wilson argued that protein sequence and gene expression represent distinct levels of evolution (King and Wilson 1975). This influential paper popularized the ideas that evolution proceeds through different molecular mechanisms along the transition from genotype to phenotype and that gene expression may play a predominant role in organismal evolution. In our study we found striking contrasts in patterns of divergence between different forms of molecular evolution dependent on chromosome origin and developmental stage of spermatogenesis. Our cell-specific data yield new insights into the evolution of spermatogenesis and the critical role that developmental context plays in molecular evolution on and off the sex chromosomes. Below we discuss the implications of our results for the evolution of spermatogenesis, the X chromosome, and speciation.
Protein evolution and spermatogenesis
Faster-X protein evolution has now been found across a broad range of taxa, including mammals (Hvilsom et al. 2012; Veeramah et al. 2014), birds (Mank et al. 2007), flies (Begun et al. 2007; Langley et al. 2012; Garrigan et al. 2014), and other insects (Jaquiery et al. 2012; Sackton et al. 2014). Both higher rates of adaptive substitutions (Kousathanas et al. 2014) and relaxed constraint (Wright et al. 2015) likely contribute to these patterns. X-linked sequence evolution is not always unusual (reviewed in Vicoso and Charlesworth 2006; Ellegren and Parsch 2007; Meisel and Connallon 2013), but there is strong support for faster-X protein evolution for genes expressed in male reproductive tissues (Torgerson and Singh 2003, 2006; Baines et al. 2008; Grath and Parsch 2012; Sin et al. 2012; Kousathanas et al. 2014). From these data, gene expression has emerged as a defining factor in faster-X protein evolution, with breadth of expression, tissue specificity, and degree of sex bias all strongly influencing evolutionary rates (Meisel 2011; Meisel et al. 2012b).
Given these general findings, the details of spermatogenesis should strongly dictate patterns of molecular evolution on and off the sex chromosomes, yet most evolutionary studies have not utilized a strong developmental framework (but see Kousathanas et al. 2014). Our FACS data allowed us to partition genes across spermatogenesis, yielding strong support for faster-X protein evolution (Figure 4A) based on relative (ω) and absolute (dN) estimates of amino acid divergence. Faster-X protein evolution was most striking when considering ω estimates that used dS to approximate neutral divergence. This assumption is violated in many species due to selection for biased codon usage. For example, the X chromosome shows stronger codon usage bias in Drosophila (Singh et al. 2005), which in turn may inflate X-linked estimates of ω (Campos et al. 2013). There is also weak codon usage bias in mice, but unlike Drosophila, it is significantly weaker on the X chromosome (Kessler and Dean 2014) and therefore conservative with respect to the observation of faster-X protein evolution. Further, both lower mutation rates and shallower coalescent times likely contribute to less nucleotide divergence on the mouse X chromosome (Table S2), making the observation of higher X-linked dN conservative.
Some aspects of our faster-X protein-coding results are seemingly contrary to the basic dominance predictions of faster-X theory. Postmeiotic cells are haploid, leading to the prediction that faster-X evolution in the germ line should be restricted to genes expressed prior to meiosis (Kousathanas et al. 2014). However, spermatids form a multicellular syncytium connected through intercellular bridges that enable functional equivalence through cytoplasmic exchange (Braun et al. 1989; Caldwell and Handel 1991). Exchange of gene products between spermatids is likely a functional necessity given that many sex-linked genes are essential to the postmeiotic stages of spermatogenesis. Assuming exchange of autosomal gene products maintains functional diploidy, then our finding of faster-X protein evolution across the diploid and haploid stages of spermatogenesis is generally consistent with the dominance predictions of faster-X theory (Charlesworth et al. 1987; Vicoso and Charlesworth 2009).
Regulatory evolution and spermatogenesis
In contrast to protein evolution, X-linked postmeiotic genes (i.e., induced late) showed less differential expression than comparable autosomal genes (Figure 4). As with expression divergence, sperm methylome evolution also appeared slower on the X chromosome (Figure 5). Given the strong signature of faster-X protein evolution, why do we find evidence for equivalent or slower-X gene expression and DNA methylation evolution across the same developmental timeline? One simple explanation is that one or more of the assumptions of the faster-X model sequence evolution do not hold for these complex biochemical traits. First, for the predictions of faster-X theory to hold, these regulatory phenotypes must reflect linked-sequence evolution. For faster-X expression divergence, this would require that differences in transcript abundances are due to divergence in cis-regulatory elements and/or X-linked trans-regulatory elements (Kayserili et al. 2012; Meisel et al. 2012a; Meisel and Connallon 2013). Several studies suggest that divergence in transcript abundances is largely determined by evolution in cis (Wittkopp et al. 2004; Landry et al. 2005; Wittkopp et al. 2008; Graze et al. 2009; Goncalves et al. 2012; Shi et al. 2012; Shen et al. 2014; Mack et al. 2016), while other research indicates that trans-regulatory divergence is more common (Emerson et al. 2010; McManus et al. 2010; Schaefke et al. 2013; Coolon et al. 2014; Meiklejohn et al. 2014; Combes et al. 2015) or that the inferred mode of regulatory divergence is dependent on taxon and experimental design (Coolon and Wittkopp 2013; Guerrero et al. 2016). The mechanisms underlying the evolution of DNA methylation are even more poorly understood, but levels of DNA methylation within a given genomic region appear strongly dependent on underlying (i.e., cis) genetic sequences (Hernando-Herraez et al. 2015).
Faster-X sequence evolution also assumes that beneficial mutations are on average at least partially recessive (Charlesworth et al. 1987), while theory (Gibson and Weir 2005) and several empirical studies (Lemos et al. 2008; McManus et al. 2010; Schaefke et al. 2013) suggest that cis-regulatory elements should on average act additively. Nonetheless, these studies also find a substantial proportion of cis-regulatory elements that act nonadditively or, in some cases, a greater overall proportion of nonadditive cis-regulatory elements (Coolon et al. 2014). Unfortunately, these issues remain largely unresolved on the X chromosome because both dominance relationships and cis- vs. trans-regulatory evolution are usually assessed through allele-specific expression in F1 genotypes, which cannot be evaluated for X-linked genes during spermatogenesis.
It is also important to consider what fraction of X-linked substitutions that influence gene expression or DNA methylation reflect new mutations that have been targeted by positive directional selection. One formal possibility is that divergence of regulatory traits simply reflects neutral sequence evolution. Both patterns of slower-X gene expression and methylation divergence become nonsignificant when expectations are corrected by lower synonymous substitution rates on the X chromosome. This simple correction is likely highly conservative, but does suggest that differences in mutation rates and/or the effective population sizes may partially account for patterns of divergence on the X chromosome. We still lack a strong theoretical framework for differentiating between adaptive and neutral divergence of expression phenotypes (Khaitovich et al. 2005b; Meisel and Connallon 2013). Recent genomic analyses of patterns of sequence polymorphism and divergence in mice indicate widespread adaptive evolution of both coding (Halligan et al. 2010) and noncoding regions (Halligan et al. 2011, 2013). Most putatively adaptive autosomal substitutions in mouse genomes occur within candidate regulatory regions (i.e., untranslated exons and conserved noncoding elements), although the individual fitness effects of amino acid substitutions appear to be much larger (Halligan et al. 2013). These studies did not consider X-linked patterns directly but do suggest that a substantial fraction of substitutions within regulatory regions may be targeted by positive selection. However, if selection on gene expression acts on standing genetic variation, as opposed to new mutations, then substitution rates are predicted to be lower on the X chromosome (Meisel and Connallon 2013), given lower levels of X-linked genetic diversity in mice (Geraldes et al. 2008). The relative contribution of new mutations vs. standing genetic variation to adaptive regulatory divergence has not yet been explicitly addressed (Coolon and Wittkopp 2013).
In sum, it is clear that extending the faster-X model sequence evolution to regulatory phenotypes depends on several assumptions that are both restrictive and remain largely unresolved. However, if regulatory divergence between subspecies of mice were simply a consequence of the genetic architecture of these traits, mutational processes, or the origin of adaptive variation, then we might reasonably expect patterns on and off the X chromosome to be consistent across different stages of spermatogenesis. Instead we found that X-linked expression divergence, both overall and relative to the autosomes, changed dynamically over the timeline of spermatogenesis with a strong signature of less divergence late (Figure 4B). Given this finding, we propose that slower-late regulatory evolution may be best explained by strong developmental constraints on gene expression phenotypes during the later stages of spermatogenesis that are particularly acute on the sex chromosomes.
Evolution of the mammalian X chromosome likely reflects a balance between the inherent constraints of dosage compensation (Ohno 1967; Kohn et al. 2004) and spermatogenesis (Schultz et al. 2003; Shima et al. 2004; Chalmel et al. 2007) with the conflicting forces of sexual and antagonistic selection that favor X-linked male-biased genes (Rice 1984). During spermatogenesis, the sex chromosomes are tightly regulated through the generally repressive epigenetic environments of MSCI and PSCR (Turner 2007; Hu and Namekawa 2015). For example, PSCR in mice is partially maintained by the multicopy Y-linked Sly gene (Cocquet et al. 2009, 2010). Sly represses postmeiotic sex chromosome expression and favors Y transmission in spermatids. The multicopy X-linked genes Slx/Slxl1 seem to counter this by increasing sex chromosome expression and X transmission. This direct antagonism appears to have driven a copy-number arms race (Scavetta and Tautz 2010; Ellis et al. 2011) where each added copy leads to an incremental increase in relative expression and subsequent counterselection for upregulation of the other gene group (Cocquet et al. 2012). So while the underlying genomic architecture evolves rapidly via gene duplication (Ellis et al. 2011), expression level phenotypes appear to be under strong stabilizing selection for a dosage equilibrium between sex-linked postmeiotic genes that if disrupted leads to male sterility. This is evidence for strong constraint on the regulatory phenotypes of X-linked genes that overcome PSCR to be expressed during this critical period of sperm development. Our data suggest that such constraints likely extend more broadly across the X chromosome and to testis-biased autosomal genes.
We also found that patterns of DNA methylation diverged more slowly on X chromosomes compared to the autosomes (Figure 5). Dynamic methylation plays a key role in mammalian genome regulation (Li et al. 1992; Okano et al. 1999; Reik et al. 2001), yet very little is known about either the tempo or mechanisms of DNA methylome evolution. Sperm are the first mature cells following a major “erasure”; nearly the entire genome is demethylated in spermatogonia stem cells and then remethylated late in spermatogenesis (Reik et al. 2001). A study involving primates found the methylomes of mature sperm to differ from those of somatic cells in several important ways. Most notably, the HMRs are more numerous and extend for longer genomic intervals in sperm compared to somatic cells (Molaro et al. 2011). We propose that the observation of less X-linked sperm methylome divergence may be an extension of the same regulatory constraints that lead to slower gene expression divergence late in spermatogenesis, as well as other X-linked phenomena such as mutational environment. Comparable methylome data from other tissues and other stages in spermatogenesis will be helpful in resolving these outstanding questions.
Cis-regulatory evolution is thought to proceed with fewer pleiotropic constraints than protein-coding divergence (e.g., Carroll 2008). However, our results indicate that the stages of spermatogenesis that show the fastest rates of protein evolution show less expression divergence for both X-linked and testis-biased genes. Protein evolution is strongly influenced by underlying patterns of gene expression; there are generally higher rates of protein-coding divergence for genes that have lower expression (Nguyen et al. 2015) and for narrowly expressed genes (Liao et al. 2006; Meisel 2011). Expression specificity increases late in spermatogenesis and the X chromosome is enriched for testis-biased postmeiotic genes. Therefore expression specificity may be the primary factor underlying faster-late and faster-X protein evolution. While divergence in gene expression (logFC) and tissue specificity (τ) were also positively correlated early in spermatogenesis, they were negatively correlated for autosomal testis-biased genes expressed late (Figure S6). Thus, specificity may actually constrain expression divergence late in spermatogenesis, especially for postmeiotic testis-biased genes that presumably play critical roles in sperm development and maturation. There is typically a positive association between differential expression and tissue specificity (Meisel et al. 2012a), suggesting that our results reflect a unique feature of sperm development rather than a general molecular evolutionary trend.
Our observation of less X-linked expression divergence late in spermatogenesis differs some from studies in mammals reporting elevated (uncorrected) X-linked expression divergence primarily in testis (Khaitovich et al. 2005a; Zhang et al. 2010; Brawand et al. 2011). These previous studies have focused on various whole tissue contrasts spanning different taxa and phylogenetic depths. However, Zhang et al. (2010) did report greater relative X-linked expression divergence in spermatids between mouse and rat, the exact cell population that we found to be the most conserved between subspecies of mice (Figure 4). While this could reflect changes in X-linked expression evolution over deeper evolutionary timescales, this previous result was based on a metaanalysis of microarray data collected independently in mouse (Chalmel et al. 2007) and rat (Johnston et al. 2008) using different cell-enrichment procedures. A full assessment of testis gene expression evolution between mouse and rat awaits the comparison of cell-specific transcriptomes that control for potential experimental artifacts.
How broadly the slower-late X and testis-biased autosomal patterns of regulatory evolution apply to other taxa remains to be seen. Expression divergence has not been systematically evaluated in nonmammalian systems using cell- or stage-specific data, making the relative contributions of cellular composition and developmental constraint difficult to determine. This technical limitation aside, the influence of developmental timing on spermatogenic expression evolution will also be dependent on the existence or extent of MSCI/PSCR-like phenomena in other taxa. For example, faster-X expression divergence has also been reported in Drosophila (Kayserili et al. 2012; Meisel et al. 2012a; Coolon et al. 2015; Llopart 2015) where the X chromosome appears to be transcriptionally repressed during spermatogenesis (Meiklejohn et al. 2011). Whether this repression reflects a process akin to MSCI has been contentious, but it does suggest that regulatory constraints likely play an important role in sex chromosome evolution (Vibranovski 2014).
Implications for speciation
Faster-X theory was originally proposed in part to explain two observations that invoke a large role for sex chromosomes in speciation (Charlesworth et al. 1987). First, when hybrids between recently diverged lineages show sex-specific sterility or inviability, it is usually the heterogametic sex (Haldane 1922). Second, heterogametic sterility is disproportionately linked to the X or Z chromosomes, a pattern known as the large-X effect (Coyne 1992). Faster-X evolution remains one of the predominant mechanisms invoked to explain the evolution of hybrid sterility (Kousathanas et al. 2014), and the link between rapid evolution and speciation is intuitive. A long-standing alternative hypothesis is that spermatogenesis is an inherently sensitive process that may be easily disrupted in hybrids (Lifschytz and Lindsley 1972; Jablonka and Lamb 1991; Wu and Davis 1993). Disruptions of MSCI/PSCR are strong a priori candidate regulatory mechanisms for the sensitivity of spermatogenesis (Lifschytz and Lindsley 1972). Studies in mice (Good et al. 2010; Bhattacharyya et al. 2013; Campbell et al. 2013; Turner et al. 2014) and other mammals (Davis et al. 2015) have recently found a strong link between regulatory disruption of the X chromosome during spermatogenesis and the evolution of hybrid male sterility. Our results reveal that the same developmental stages that have disrupted gene expression in mouse hybrids (Good et al. 2010; Bhattacharyya et al. 2013; Campbell et al. 2013) show the strongest evolutionary constraints in gene expression levels (Figure 4). While these data do not discount an important role for faster-X protein-coding evolution in speciation, they do suggest that inherent developmental constraints on the regulation of gene expression late in spermatogenesis may play a central and underappreciated role in the evolution of hybrid male sterility.
Acknowledgments
We thank Pamela K. Shaw, Irina Getun, Bivian Torres, Colin Callahan, and Brent Young for assistance with FACS and other sample preparations; Francois Bonhomme for mice; and the staff of the University of Montana (UM) Laboratory Animal Research facility. Members of the J.M.G. and M.D.D. labs gave us helpful feedback on experimental results, and comments from Bret Payseur and multiple anonymous reviewers improved previous versions of this manuscript. We thank the UM Fluorescence Cytometry Core, supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) (P30-GM103338) and the UM Genomics Core, supported by a grant from the M. J. Murdock Charitable Trust and the Vincent J. Coates Genomics Sequencing Laboratory at the University of California, Berkeley, supported by NIH S10 instrumentation grants S10-RR029668 and S10-RR027303. This research was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01-HD073439; to J.M.G.), the National Institute of General Medical Sciences (R01-GM098536; to M.D.D.), and the National Science Foundation (1146525; to M.D.D.).
Footnotes
Communicating editor: B. A. Payseur
Supplemental material is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.116.186825/-/DC1.
Literature Cited
Oksanen J., F. G. Blanchet, R. Kindt, P. Legendre, P. R. Minchin et al., 2015 vegan: community ecology package. R package version 2.3–1.
Pages H., R. Gentleman, P. Aboyoun, and S. DebRoy, 2008 Biostrings: string objects representing biological sequences, and matching algorithms R package version 2.38.2.
Warnes G. R., B. Bolker, L. Bonebakker, R. Gentleman, W. Huber et al., 2015 gplots: various R programming tools for plotting data. R package version 2.17.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}