





Abstract
Pre-RNA splicing is an essential step in generating mature mRNA. RNA trans-splicing combines two separate pre-mRNA molecules to form a chimeric non-co-linear RNA, which may exert a function distinct from its original molecules. Trans-spliced RNAs may encode novel proteins or serve as noncoding or regulatory RNAs. These novel RNAs not only increase the complexity of the proteome but also provide new regulatory mechanisms for gene expression. An increasing amount of evidence indicates that trans-splicing occurs frequently in both physiological and pathological processes. In addition, mRNA reprogramming based on trans-splicing has been successfully applied in RNA-based therapies for human genetic diseases. Nevertheless, clarifying the extent and evolution of trans-splicing in vertebrates and developing detection methods for trans-splicing remain challenging. In this review, we summarize previous research, highlight recent advances in trans-splicing, and discuss possible splicing mechanisms and functions from an evolutionary viewpoint.
Introduction
To create fully functional mRNA, pre-mRNA is processed into mature mRNA through three main modifications: 5′-capping, 3′-polyadenylation and RNA splicing. The last modification includes cis- and trans-splicing. Cis-splicing occurs within same pre-mRNA molecule, whereas trans-splicing uses two separate pre-mRNA molecules to form a chimeric non-co-linear RNA, which may encode novel proteins or serve as noncoding or regulatory RNAs. These novel RNAs not only increase the complexity of the proteome but also provide new regulatory mechanisms for gene activities, all of which extend the coding capacity of a genome and shape speciation.
Trans-splicing was first observed in the RNA processes in trypanosomes in which a short leader sequence is transferred to the 5′-end of the pre-mRNA for variant surface glycoprotein (Boothroyd et al. 1982; Van der Ploeg et al. 1982). A 22-nt spliced leader sequence (SL) was also found at the 5′-end of actin mRNA in Caenorhabditis elegans (Krause et al. 1987). In SL trans-splicing, a short noncoding exon is spliced to the 5′-end of mRNAs for distinct structural genes, producing mRNAs with a common leading sequence (Van Doren et al. 1988). SL trans-splicing is mediated by the spliceosome, including snRNAs (small nuclear RNAs) U2, U4, U5, and U6, but not U1 (Hannon et al. 1991). In lower eukaryotes, SL trans-splicing plays a pivotal role in mature mRNA processing (Hastings 2005), especially of polycistronic transcription units (Nilsen 1993). In addition, trans-splicing is involved in growth recovery in C. elegans (Zaslaver et al. 2011) and in nutrient-dependent translational control in the marine chordate Oikopleura dioica. (Danks et al. 2015). Through these functions, SL trans-splicing provides evolutionary advantages to prokaryotes.
Trans-splicing can occur in both prokaryotes and eukaryotes. In some archaea and bacteria, trans-splicing events probably split tRNA genes, implying an evolutionary trace of continuous tRNA genes among different species. In Drosophila, the biological significance of trans-splicing in mod (mdg4) and lola genes has been examined (Dorn et al. 2001; Horiuchi et al. 2003; McManus et al. 2010). Recently, many trans-splicing events have also been detected in vertebrates through high-throughput RNA analysis techniques (Herai et al. 2010; Frenkel-Morgenstern et al. 2013). In addition, in mammals, trans-splicing has been observed in many physiological and pathological processes including cancer (Yu et al. 1999; Li, Wang, et al. 2009). However, the occurrence, extent and implications of trans-splicing could be quite different in vertebrates compared with invertebrates. There are at least two concerns. First, the extent and scope of trans-splicing in vertebrates may be lower compared with invertebrates. Another intriguing question concerns the detection methodologies for trans-splicing, with the key challenge determining whether reverse transcription from RNA to cDNA using reverse transcriptase (RT) may result in artificial chimeras.
Trans-splicing plays important roles in many physiological and pathological processes, although it occurs at a low frequency in humans. The principle has successfully been applied in RNA-based therapy in human genetic diseases (Wally et al. 2012). However, the functions, evolution, and underlying mechanisms of trans-splicing remain unknown. In this review, we systematically discuss trans-splicing with a focus on its extent, functions, and mechanisms in vertebrates from an evolutionary viewpoint.
Pre-RNA Splicing Types: cis- and trans-Splicing
After transcription, the majority of pre-RNAs are processed through splicing to become a mature RNA. There are two types of splicing: cis- and trans-splicing. Trans-splicing involves two pre-RNA molecules, whereas cis-splicing occurs within a single pre-mRNA (fig. 1A). There are two types of trans-splicing based on the pre-RNA source: intragenic trans-splicing and intergenic trans-splicing (fig. 1B and C). In the intragenic subgroup, pre-RNAs are transcribed from the same genome locus, but chimeric RNA is spliced from different strands or exon order. Intragenic trans-splicing may occur through exon repetition, sense-antisense fusion, and exon scrambling. For example, chimeric RNAs of the mod (mdg4) and lola genes in Drosophila originate from intragenic trans-splicing (Dorn et al. 2001; Horiuchi et al. 2003; McManus et al. 2010). In intergenic trans-splicing, exons from diverse genes, even those on different chromosomes, are used to generate a chimeric RNA. For example, in humans, transcripts from the JAZF1 gene on chromosome 7p15 and the JJAZ1 gene on chromosome 17q11 can generate a chimeric JAZF1-JJAZ1 RNA (Li et al. 2008). SL splicing is a special type of trans-splicing that frequently occurs in lower eukaryotes such as nematodes (Nilsen 1993) (fig. 1D).
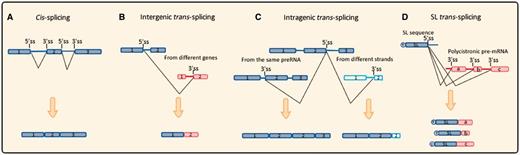
Schematic diagram of different types of pre-RNA splicing events. (A) Cis-splicing. After excision of introns, exons of the same pre-mRNA are joined together to form a linear molecule. (B) Intergenic trans-splicing. Transcripts from different genes or even different chromosomes could be spliced and generate a non-linear chimeric molecule. (C) Intragenic trans-splicing. Boxes with vertical line represent exons transcribed from the other strand. In the same gene, splicing reaction occurs between two identical transcripts, alternatively, transcripts from different strands leading to exon-duplication and sense–antisense fusion. (D) SL trans-splicing. Red boxes represent structural genes, while T represents for the TMG cap on Spliced-leader (SL) mini-exon. SL exon produced from tandem repeated SL gene cluster, splicing reaction occurs between SL exon and distinct structural genes of a ploycistronic pre-mRNA to generate an array of mature “capped” transcripts.
Evolutionary Trends of trans-Splicing
Trans-splicing frequently occurs in lower organisms, such as dinoflagellates (e.g., Karlodinium micrum), euglenozoa (e.g., Trypanosoma brucei), and some species of nematodes (e.g., C. elegans), with more than 70% of genes participating in the process. Trans-splicing even occurs in viruses such as bacteriophage T4, demonstrating an early origin (Galloway Salvo et al. 1990). In archaea, tRNA generation could occur through trans-splicing, for example, in Thermosphaera aggregans (Chan et al. 2011) and Nanoarchaeum equitans (Randau et al. 2005). Split tRNAs were found in some archaea species (Randau et al. 2005; Fujishima et al. 2009; Chan et al. 2011), and tRNA half homologs were detected in the genomes of archaea, bacteria, and eukaryotes (Zuo et al. 2013). Recent studies indicate that small guide RNA could be involved in tRNA splicing (Randau 2015). Thus, some split tRNAs are proposed to be transcribed from different loci and trans-spliced to generate mature tRNAs. Intriguingly, SL splicing has a much higher frequency, up to approximately 100% compared with other types, including inter/intragenic splicing events, which are observed mainly in dinoflagellates (e.g., 100% in Amphidinium carterae and K. micrum), euglenozoa (e.g., 100% in T. brucei), nematodes (e.g., 90% in Ascaris sp. and 70% in C. elegans), and rotifers (e.g., 60% in Adineta ricciae). In addition, in a recent mega-data study, a total of 1,627 trans-splicing events involving 2,199 genes were identified in insects, which accounts for 1.58% of the total genes (Kong et al. 2015). This finding, together with many other studies, provides new evidence against the hypothesis that trans-splicing events are merely ‘splicing noise.’
Trans-splicing frequency peaks in protozoa, radiate, and protostomia and then decreases, with a dramatic decline in vertebrates (fig. 2). The high percentage of trans-splicing events observed in invertebrates represents SL-type splicing, which can occur in 100% of genes in A. carterae, K. micrum, and T. brucei. The valleys in the percentage of trans-splicing events indicate non-SL species, mainly vertebrates. The analyses imply that along the evolutionary process, trans-splicing has experienced dynamic changes.
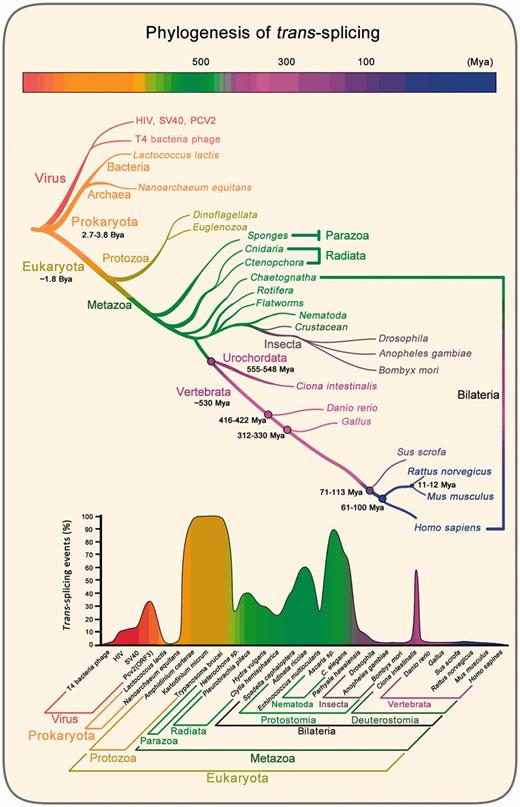
Phylogenetic analysis of trans-splicing events. Evolutionary tree and time scale refer to Benton et al. ( 2007). Ba, billion years ago; Ma, million years ago. In low panel, the percentage of trans-splicing events and trans-spliced gene numbers are relative to the total amount of gene numbers for a species. Trans-splicing data are from published literatures: Parhyale hawaiensis (Douris et al. 2010), Clytia hemisphaerica (Derelle et al. 2010), Echinococcus multilocularis (Brehm et al. 2000), Heterochone sp. (Douris et al. 2010), Hydra vulgaris (Stover et al. 2001), Pleurobrachia pileus (Derelle et al. 2010), Spadella cephaloptera (Marletaz et al. 2008; Marletaz and Le Parco 2008), Ciona intestinalis (Vandenberghe et al 2001; Satou et al. 2006; Satou et al. 2008; Matsumoto et al. 2010), Adineta ricciae (Pouchkina-Stantcheva et al. 2005), C. elegans (Krause et al. 1987; Huang et al. 1989; Zorio et al. 1994), Ascaris sp. (Nilsen et al. 1989; Maroney et al. 1995), Trypanosoma brucei (Murphy et al. 1986; Sutton et al. 1986; Perry et al. 1987; Liang et al. 2003), Amphidinium carterae (Bachvaroff et al. 2008; Zhang et al. 2009) and Karlodinium micrum (Zhang et al. 2007). In the other species, the percentage of trans-splicing events are calculated by counting known trans-splicing molecules including T4 bacteria phage (Galloway Salvo et al. 1990), HIV (Caudevilla, Da Silva-Azevedo, et al. 2001), SV40 (Caudevilla, Da Silva-Azevedo, et al. 2001), Pv2 (ORF3) (Gao et al. 2013), Lactococcus lactis (Belhocine et al. 2007), Nanoarchaeum equitans (Randau et al. 2005), Drosophila (Dorn et al. 2001,Horiuch et al. 2003), Anopheles gambiae (Robertson et al. 2007), Bombyx mori (Shao et al. 2012; Duan et al. 2013), Danio rerio (Cadieux et al. 2005), Gallus (Vellard et al. 1991), Sus scrofa (Ma et al. 2012), Rattus norvegicus (Sullivan et al. 1991; Caudevilla et al. 1998; Akopian et al. 1999; Takahara et al. 2002; Zhang et al. 2003; Fitzgerald et al. 2006; Ni et al. 2011), Mus musculus (Hirano et al. 2004; Zhang et al. 2010) and Homo sapiens (Vellard et al. 1991; Breen et al. 1997; Yu et al. 1999; Chatterjee et al. 2000; Takahara et al. 2000; Finta et al. 2002; Flouriot et al. 2002; Jehan et al. 2007; Guerra et al. 2008; Li et al. 2008; Brooks et al. 2009; Kannan et al. 2011; Kowarz et al. 2011, 2012; Fang et al. 2012; Hu et al. 2013; Kawakami et al. 2013; Yuan, Qin, et al. 2013; Li et al. 2014; Wu et al. 2014). Percentages in insects are consistent with recent mega-data study (Kong et al. 2015).
Conservation of Splicing Machinery
We believe that trans-splicing shares most characteristics with cis-splicing. Several lines of evidence have shown that trans-splicing utilizes a similar set of splicing machinery to alternative splicing. Trans-splicing has the same splicing signals and factors as alternative splicing. For example, the spliceosome, which contains U1, U2, U4, U5, and U6 snRNAs, catalyzes pre-mRNA in cis-splicing. A recent study demonstrated that U1 snRNP binding may promote mod trans-splicing in Drosophila (Gao et al. 2015). For SL splicing, the key elements of SL snRNP are very similar to the spliceosomal snRNPs (Bruzik et al. 1988; Van Doren et al. 1988), indicating that SL RNA may originate from a splicing U snRNA in lower organisms with an ancestral cis-splicing mechanism. Additional support comes from the fact that SL trans-splicing exists in some metazoans, including cnidarians, ctenophores, rotifers, flatworms, nematodes, crustaceans, sponges, and chaetognaths. In contrast, plants, fungi, insects, most protists, and vertebrates do not exhibit SL trans-splicing (Douris et al. 2010). However, simultaneous trans-splicing events could take place between SL RNA and inherent transcripts in HeLa cells both in vivo and in vitro (Bruzik et al. 1992). In addition, SL trans-splicing is favored in adenosine-rich 5′ -UTRs in hydrozoans (Derelle et al. 2010). In vertebrates, the 5′ -UTR can be involved in the generation of some trans-spliced mRNA chimeras (Li et al. 1999), which is similar to SL trans-splicing in invertebrates.
Alternative splicing has contributed much more to proteome diversity than trans-splicing. It is noteworthy that multiple protein factors and substantial energy are required in alternative splicing, which is not used by lower organisms, for example, prokaryotes. A recent study indicated that SL trans-splicing provides an evolutionary advantage for species that depend on translational control to regulate early embryogenesis, growth, and oocyte production in response to nutrient levels (Danks et al. 2015). Overall, we suggest that the biological significance of trans-splicing especially SL trans-splicing may be more vital for lower eukaryotes than vertebrates. In higher evolutionary phyla, the more complicated genome structure needs an adaptable regulatory mechanism using preexisting machineries to promote the evolutionary shift from trans-splicing to alternative splicing. In vertebrates, these trans-splicing events focus mainly on some key physiological processes, including gene expression regulation for cell viability and growth. Moreover, dysregulation of trans-splicing could induce pathological events such as cancer (Li et al. 2014).
The spliceosome consists of proteins and U snRNAs complexes that participate in the splicing process. Serine/arginine-rich proteins (SR proteins) and heterogeneous nuclear ribonucleoproteins (hnRNPs) are two of the involved protein families that join in complex A to participate in cross-exon assembly by regulating U1 and U2 snRNP binding to the prespliceosome (Wahl et al. 2015). Both SR proteins and hnRNPs contain RNA-binding domains that can bind to exonic/intronic splicing enhancer (ESE/ISE) sequences and ESS/ISS sequences on pre-mRNAs, respectively (Zhu et al. 2001). Moreover, the SR protein is a key factor for alternative splicing (Gupta et al. 2014); it is intriguing that it can also enhance the efficiency of trans-splicing (Bruzik et al. 1995; Shao et al. 2012). Given these data, we analyzed the evolutionary conservation of spliceosome-associated proteins hnRNPA1, hnRNPI (PTBP1), SRSF1, and SRSF2 from T. brucei, C. elegans, insects, fish, chicken, and mammals (fig. 3). Although the SL type of trans-splicing is obviously separated from the non-SL type, both cis- and trans-splicing can utilize the same set of splicing factors. Thus, the splicing mechanisms of cis- and trans-splicing seem to be evolutionarily conserved.
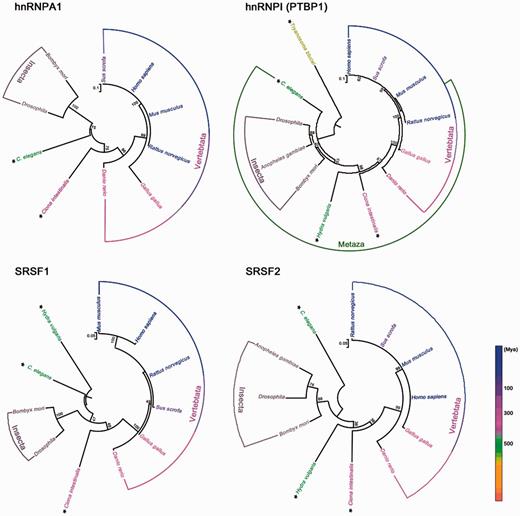
Phylogenetic analysis of spliceosome-associated proteins hnRNPA1, hnRNPI (PTBP1), SRSF1, and SRSF2. Ma, million years ago. Species with SL trans-splicing are marked with asterisks. Phylogenetic analysis was performed with MEGA 6 using maximum likelihood method. Numbers on the branches represent the bootstrap values from 1,000 replicates obtained. The scale bar corresponds to the estimated evolutionary distance units. GenBank accession numbers are as follows: Homo sapiens, NP_002127.1 (hnRNPA1), NP_002810.1 (PTBP1), NP_001071634.1 (SRSF1), NP_001182356.1 (SRSF2); Mus musculus, NP_001034218.1 (hnRNPA1), NP_001070831.1 (PTBP1), NP_001071635.1 (SRSF1); NP_035488.1 (SRSF2); Rattus norvegicus, NP_058944.1 (hnRNPA1), NP_001257986.1 (PTBP1), NP_001103022.1, (SRSF1), NP_001009720.1 (SRSF2); Sus scrofa, NP_001070686.1 (hnRNPA1), NP_999396.1 (PTBP1), NP_001033096.1 (SRSF1), NP_001070697.1 (SRSF2); Gallus gallus, XP_004950342.1 (hnRNPA1), NP_001026106.1 (PTBP1), NP_001107213.1 (SRSF1), NP_001001305.1 (SRSF2); Danio rerio, NP_956398.1 (hnRNPA1), NP_001116126.1 (PTBP1), NP_956887.2 (SRSF1), NP_998547.1 (SRSF2); Drosophila, NP_001262538.1 (hnRNPA1), NP_001097994.1 (PTBP1), NP_001247139.1 (SRSF1), NP_001188794.1 (SRSF2); Bombyx mori, NP_001093319.1 (hnRNPA1), XP_012546585.1 (PTBP1), XP_012548197.1 (SRSF1), NP_001040152.1 (SRSF2); C. elegans, NP_500326.2 (hnRNPA1), NP_741041.1 (PTBP1), NP_499649.2 (SRSF1), NP_495013.1 (SRSF2); Ciona intestinalis, XP_002128542.1 (hnRNPA1), XP_002127727.3 (PTBP1), XP_002124933.3 (SRSF1), XP_004227013.1 (SRSF2); Hydra vulgaris, XP_002156158.1 (PTBP1), XP_002159641.1 (SRSF1), XP_002161458.1 (SRSF2); Anopheles gambiae, XP_318405.4 (PTBP1), XP_318826.3 (SRSF2); Trypanosoma brucei, XP_827198.1 (PTBP1).
From SL1, SL2 to Non-SL trans-Splicing
Although vertebrates and invertebrates exhibit similarities in trans-splicing, there are some distinct features, indicating that trans-splicing is evolutionarily dynamic. In most unicellular organisms and nematodes, SL trans-splicing is an exclusive splicing mode, while no SL trans-splicing occurs in vertebrates. Trans-splicing in vertebrates shows more complexities in magnitude and regulation compared with invertebrates. One example is the donor-acceptor sequence diversity in vertebrates. In mice, the donor-acceptor sequences of the first intron in the Msh4 β and ϵ pre-mRNAs are “TG-GT” and “TC-CA,” respectively, which do not match the consensus sequence of U2-type (GT-AG) or U12-type (AT-AC) (Hirano et al. 2004). In addition, SL trans-splicing does not increase the complexity of proteomes, whereas trans-splicing in vertebrates does, suggesting that this process may generate proteins with functions that differ from those of the parental genes in vertebrates. These observations imply that trans-splicing may have evolved, probably from a broad SL splicing to a precise regulation of non-SL trans-splicing, which matches the complexity of gene regulation in vertebrates.
SL trans-splicing has evolved continually in nematodes. Two types of SL trans-splicing have been identified, namely SL1 and SL2 (Harrison et al. 2010). SL2 RNAs appeared only in the Rhabditina clade of nematodes, including C. elegans, which indicates that the SL2 RNAs evolved relatively late in nematode evolution (Guiliano et al. 2006). SL trans-splicing is associated with the evolution of operons (Blumenthal 2005). Operons evolved before SL2-like spliced leaders; nematodes can use trans-splicing to resolve their operonic transcripts into single-gene mRNAs (Guiliano et al. 2006). SL2 in C. elegans evolved from SL1; notably, in processing operon pre-mRNAs, SL2 is much more efficient (Blumenthal 2005). Nevertheless, vertebrates lack SL trans-splicing and instead form non-SL type trans-splicing. SL trans-splicing occurs in the organisms that utilize operons, and there are rarely operons in vertebrates. In fact, SL trans-splicing arose after operons. Although the evolutionary mechanisms of trans-splicing remain unknown, we speculate that along with loss of operons and formation of genome complexity, trans-splicing may have shifted to cis-splicing. Thus, trans-splicing is reserved and limited to some essential processes in vertebrates but at a much lower frequency.
Trans-Splicing in Vertebrates
In mammals, the first characterized case of trans-splicing was a novel small T antigen transcript in HeLa cells (Eul et al. 1995). To date, trans-splicing has been observed in various species, including Danio rerio, Gallus, Rattus norvegicus, Mus musculus, Sus scrofa, and Homo sapiens (Vellard et al. 1991; Caudevilla et al. 1998 ; Hirano et al. 2004; Cadieux et al. 2005; Li et al. 2008; Ma et al. 2012). Trans-splicing occurs in genes involved in some physiological processes (table 1), which expands our understanding of the repertoire of genes and their regulation.
Typical trans-Splicing Chimeras
| Organisms | Involved Genes or Chimeras | Function Description | References | Experiments Verified |
|---|---|---|---|---|
| C. elegans | eri-6/7 | Superfamily I helicase | (Fischer et al. 2008) | RT-PCR; Sequencing |
| Drosophila (Fruit fly) | lola | Transcription factor | (Horiuchi et al. 2003) | RT-PCR; Sequencing |
| mod (mdg4) | Transcription factor | (Dorn et al. 2001) | RT-PCR; Sequencing | |
| Anopheles gambiae (Mosquito) | Bursicon | Coding bursicon | (Robertson et al. 2007) | Bioinformatics analysis |
| Bombyx mori (Silk worm) | mod (mdg4) | Transcription factor | (Shao et al. 2012) | RT-PCR; Sequencing |
| Dsx-dsr2 | Sexual development | (Duan et al. 2013) | RT-PCR; Sequencing | |
| Danio rerio (Zebrafish) | Grn1-Grn2 | Hybrid granulin | (Cadieux et al. 2005) | RT-PCR; Northern blot |
| Gallus (Chicken) | C-myb | Proto-oncogene | (Vellard et al. 1991) | (?) |
| Rattus norvegicus (Rat) | 1038 mRNA | Unknown | (Fitzgerald et al. 2006) | RT-PCR; Northern blot |
| ABP-HDC | Unknown | (Sullivan et al. 1991) | RT-PCR; Northern blot | |
| COT | Gene expression regulation | (Caudevilla et al. 1998) | RT-PCR; Northern blot; in vitro trans-splicing | |
| HongrE2 | Gene expression regulation | (Ni et al. 2011) | RT-PCR; Northern blot (?) | |
| LAR tyrosine phosphatase receptor | Gene expression regulation | (Zhang et al. 2003) | RT-PCR; RNase protection assay; Northern blot | |
| SNS-A | Unknownn | (Akopian et al. 1999) | PCR; RNase protection assay; Northern blot | |
| Sp1 | Transcription factor | (Takahara et al. 2002) | RT-PCR; RNase protection assay; Northern, Southern blot | |
| Mus musculus (Mouse) | Dmrt1-Dmr | Gene expression regulation | (Zhang et al. 2010) | RT-PCR; Northern blot; Southern blot |
| Msh4-Hspa5 Msh4-Pcbp3 | Cell death | (Hirano et al. 2004) | RT-PCR; Northern blot | |
| Sus scrofa (Pig) | AK238425, AK351564 and other 667 putative chimeras | Unknownn | (Ma et al. 2012) | Systematic analysis; RT-PCR; RNA-Seq |
| Homo sapiens (Human) | ATAC-1- Exon Xa/Xb | Gene expression regulation | (Yu et al. 1999) | RT-PCR; RNase protection assay; Northern blot |
| ATAC-1-Ampr | Gain antibiotics resistance | (Hu et al. 2013) | RT-PCR; in vitro trans-splicing | |
| CAMK2G-SRP72 | Unknownn | (Breen et al. 1997) | PCR; Genetic mapping; Western blot; (?) | |
| CDC2L2 | Transcriptional regulation | (Jehan et al. 2007) | FISH; RT-PCR | |
| C-myb | Proto-oncogene | (Vellard et al. 1991) | RT-PCR; Sequencing | |
| CoAA-RBM4 | Regulate stem/progenitor cell differentiation | (Brooks et al. 2009) | RT-PCR; in vitro trans-splicing | |
| CYCLIND1-TROP2 | Cell growth | (Guerra et al. 2008) | RT-PCR; Northern blot; RNase protection assay (?) | |
| CYP3A4, 5, 7, 43 | Catalytic activity | (Finta et al. 2002) | RT-PCR; Northern blot; RNase protection assay | |
| hER alpha | Gene expression regulation | (Flouriot et al. 2002) | RT-PCR; Southern blot | |
| JAZF1-JJAZ1 | Anti-apoptotic protein | (Li et al. 2008) | RT-PCR; Southern blot; in vitro trans-splicing | |
| PJA2-FER | Cancer biomarker | (Kawakami et al. 2013) | RT-PCR | |
| PAX3-FOXO1 | Cancer biomarker | (Yuan, Qin, et al. 2013) | RT-PCR; FISH | |
| RGS12 | G protein signaling | (Chatterjee et al. 2000) | RT-PCR | |
| Sp1 | Transcription factor | (Takahara et al. 2000) | RT-PCR; Southern blot; RNase protection assay | |
| ZC3HAV1L-CHMP1A | Genome rearrangement | (Fang et al. 2012) | RT-PCR (?) | |
| AF4, AF9, ELL, ENL, MLL, ETV6, NUP98, RUNX1, EWSR1 | DNA repair and chromosomal translocation | (Kowarz et al. 2012; Kowarz et al. 2011) | RT-PCR (?) | |
| TMEM79-SMG5 | Cancer biomarker | (Kannan et al. 2011) | PCR; Sequencing (?) | |
| tsRMST | Pluripotency maintenance of hESCs | (Wu et al. 2014) | RT-PCR; RNase protection assay | |
| TSNAX-DISC1 | G1/S transition and endometrial carcinoma (EC) development | (Li et al. 2014) | PCR; RNA-Seq (?) |
| Organisms | Involved Genes or Chimeras | Function Description | References | Experiments Verified |
|---|---|---|---|---|
| C. elegans | eri-6/7 | Superfamily I helicase | (Fischer et al. 2008) | RT-PCR; Sequencing |
| Drosophila (Fruit fly) | lola | Transcription factor | (Horiuchi et al. 2003) | RT-PCR; Sequencing |
| mod (mdg4) | Transcription factor | (Dorn et al. 2001) | RT-PCR; Sequencing | |
| Anopheles gambiae (Mosquito) | Bursicon | Coding bursicon | (Robertson et al. 2007) | Bioinformatics analysis |
| Bombyx mori (Silk worm) | mod (mdg4) | Transcription factor | (Shao et al. 2012) | RT-PCR; Sequencing |
| Dsx-dsr2 | Sexual development | (Duan et al. 2013) | RT-PCR; Sequencing | |
| Danio rerio (Zebrafish) | Grn1-Grn2 | Hybrid granulin | (Cadieux et al. 2005) | RT-PCR; Northern blot |
| Gallus (Chicken) | C-myb | Proto-oncogene | (Vellard et al. 1991) | (?) |
| Rattus norvegicus (Rat) | 1038 mRNA | Unknown | (Fitzgerald et al. 2006) | RT-PCR; Northern blot |
| ABP-HDC | Unknown | (Sullivan et al. 1991) | RT-PCR; Northern blot | |
| COT | Gene expression regulation | (Caudevilla et al. 1998) | RT-PCR; Northern blot; in vitro trans-splicing | |
| HongrE2 | Gene expression regulation | (Ni et al. 2011) | RT-PCR; Northern blot (?) | |
| LAR tyrosine phosphatase receptor | Gene expression regulation | (Zhang et al. 2003) | RT-PCR; RNase protection assay; Northern blot | |
| SNS-A | Unknownn | (Akopian et al. 1999) | PCR; RNase protection assay; Northern blot | |
| Sp1 | Transcription factor | (Takahara et al. 2002) | RT-PCR; RNase protection assay; Northern, Southern blot | |
| Mus musculus (Mouse) | Dmrt1-Dmr | Gene expression regulation | (Zhang et al. 2010) | RT-PCR; Northern blot; Southern blot |
| Msh4-Hspa5 Msh4-Pcbp3 | Cell death | (Hirano et al. 2004) | RT-PCR; Northern blot | |
| Sus scrofa (Pig) | AK238425, AK351564 and other 667 putative chimeras | Unknownn | (Ma et al. 2012) | Systematic analysis; RT-PCR; RNA-Seq |
| Homo sapiens (Human) | ATAC-1- Exon Xa/Xb | Gene expression regulation | (Yu et al. 1999) | RT-PCR; RNase protection assay; Northern blot |
| ATAC-1-Ampr | Gain antibiotics resistance | (Hu et al. 2013) | RT-PCR; in vitro trans-splicing | |
| CAMK2G-SRP72 | Unknownn | (Breen et al. 1997) | PCR; Genetic mapping; Western blot; (?) | |
| CDC2L2 | Transcriptional regulation | (Jehan et al. 2007) | FISH; RT-PCR | |
| C-myb | Proto-oncogene | (Vellard et al. 1991) | RT-PCR; Sequencing | |
| CoAA-RBM4 | Regulate stem/progenitor cell differentiation | (Brooks et al. 2009) | RT-PCR; in vitro trans-splicing | |
| CYCLIND1-TROP2 | Cell growth | (Guerra et al. 2008) | RT-PCR; Northern blot; RNase protection assay (?) | |
| CYP3A4, 5, 7, 43 | Catalytic activity | (Finta et al. 2002) | RT-PCR; Northern blot; RNase protection assay | |
| hER alpha | Gene expression regulation | (Flouriot et al. 2002) | RT-PCR; Southern blot | |
| JAZF1-JJAZ1 | Anti-apoptotic protein | (Li et al. 2008) | RT-PCR; Southern blot; in vitro trans-splicing | |
| PJA2-FER | Cancer biomarker | (Kawakami et al. 2013) | RT-PCR | |
| PAX3-FOXO1 | Cancer biomarker | (Yuan, Qin, et al. 2013) | RT-PCR; FISH | |
| RGS12 | G protein signaling | (Chatterjee et al. 2000) | RT-PCR | |
| Sp1 | Transcription factor | (Takahara et al. 2000) | RT-PCR; Southern blot; RNase protection assay | |
| ZC3HAV1L-CHMP1A | Genome rearrangement | (Fang et al. 2012) | RT-PCR (?) | |
| AF4, AF9, ELL, ENL, MLL, ETV6, NUP98, RUNX1, EWSR1 | DNA repair and chromosomal translocation | (Kowarz et al. 2012; Kowarz et al. 2011) | RT-PCR (?) | |
| TMEM79-SMG5 | Cancer biomarker | (Kannan et al. 2011) | PCR; Sequencing (?) | |
| tsRMST | Pluripotency maintenance of hESCs | (Wu et al. 2014) | RT-PCR; RNase protection assay | |
| TSNAX-DISC1 | G1/S transition and endometrial carcinoma (EC) development | (Li et al. 2014) | PCR; RNA-Seq (?) |
Note.—“?”, Probably trans-splicing.
Typical trans-Splicing Chimeras
| Organisms | Involved Genes or Chimeras | Function Description | References | Experiments Verified |
|---|---|---|---|---|
| C. elegans | eri-6/7 | Superfamily I helicase | (Fischer et al. 2008) | RT-PCR; Sequencing |
| Drosophila (Fruit fly) | lola | Transcription factor | (Horiuchi et al. 2003) | RT-PCR; Sequencing |
| mod (mdg4) | Transcription factor | (Dorn et al. 2001) | RT-PCR; Sequencing | |
| Anopheles gambiae (Mosquito) | Bursicon | Coding bursicon | (Robertson et al. 2007) | Bioinformatics analysis |
| Bombyx mori (Silk worm) | mod (mdg4) | Transcription factor | (Shao et al. 2012) | RT-PCR; Sequencing |
| Dsx-dsr2 | Sexual development | (Duan et al. 2013) | RT-PCR; Sequencing | |
| Danio rerio (Zebrafish) | Grn1-Grn2 | Hybrid granulin | (Cadieux et al. 2005) | RT-PCR; Northern blot |
| Gallus (Chicken) | C-myb | Proto-oncogene | (Vellard et al. 1991) | (?) |
| Rattus norvegicus (Rat) | 1038 mRNA | Unknown | (Fitzgerald et al. 2006) | RT-PCR; Northern blot |
| ABP-HDC | Unknown | (Sullivan et al. 1991) | RT-PCR; Northern blot | |
| COT | Gene expression regulation | (Caudevilla et al. 1998) | RT-PCR; Northern blot; in vitro trans-splicing | |
| HongrE2 | Gene expression regulation | (Ni et al. 2011) | RT-PCR; Northern blot (?) | |
| LAR tyrosine phosphatase receptor | Gene expression regulation | (Zhang et al. 2003) | RT-PCR; RNase protection assay; Northern blot | |
| SNS-A | Unknownn | (Akopian et al. 1999) | PCR; RNase protection assay; Northern blot | |
| Sp1 | Transcription factor | (Takahara et al. 2002) | RT-PCR; RNase protection assay; Northern, Southern blot | |
| Mus musculus (Mouse) | Dmrt1-Dmr | Gene expression regulation | (Zhang et al. 2010) | RT-PCR; Northern blot; Southern blot |
| Msh4-Hspa5 Msh4-Pcbp3 | Cell death | (Hirano et al. 2004) | RT-PCR; Northern blot | |
| Sus scrofa (Pig) | AK238425, AK351564 and other 667 putative chimeras | Unknownn | (Ma et al. 2012) | Systematic analysis; RT-PCR; RNA-Seq |
| Homo sapiens (Human) | ATAC-1- Exon Xa/Xb | Gene expression regulation | (Yu et al. 1999) | RT-PCR; RNase protection assay; Northern blot |
| ATAC-1-Ampr | Gain antibiotics resistance | (Hu et al. 2013) | RT-PCR; in vitro trans-splicing | |
| CAMK2G-SRP72 | Unknownn | (Breen et al. 1997) | PCR; Genetic mapping; Western blot; (?) | |
| CDC2L2 | Transcriptional regulation | (Jehan et al. 2007) | FISH; RT-PCR | |
| C-myb | Proto-oncogene | (Vellard et al. 1991) | RT-PCR; Sequencing | |
| CoAA-RBM4 | Regulate stem/progenitor cell differentiation | (Brooks et al. 2009) | RT-PCR; in vitro trans-splicing | |
| CYCLIND1-TROP2 | Cell growth | (Guerra et al. 2008) | RT-PCR; Northern blot; RNase protection assay (?) | |
| CYP3A4, 5, 7, 43 | Catalytic activity | (Finta et al. 2002) | RT-PCR; Northern blot; RNase protection assay | |
| hER alpha | Gene expression regulation | (Flouriot et al. 2002) | RT-PCR; Southern blot | |
| JAZF1-JJAZ1 | Anti-apoptotic protein | (Li et al. 2008) | RT-PCR; Southern blot; in vitro trans-splicing | |
| PJA2-FER | Cancer biomarker | (Kawakami et al. 2013) | RT-PCR | |
| PAX3-FOXO1 | Cancer biomarker | (Yuan, Qin, et al. 2013) | RT-PCR; FISH | |
| RGS12 | G protein signaling | (Chatterjee et al. 2000) | RT-PCR | |
| Sp1 | Transcription factor | (Takahara et al. 2000) | RT-PCR; Southern blot; RNase protection assay | |
| ZC3HAV1L-CHMP1A | Genome rearrangement | (Fang et al. 2012) | RT-PCR (?) | |
| AF4, AF9, ELL, ENL, MLL, ETV6, NUP98, RUNX1, EWSR1 | DNA repair and chromosomal translocation | (Kowarz et al. 2012; Kowarz et al. 2011) | RT-PCR (?) | |
| TMEM79-SMG5 | Cancer biomarker | (Kannan et al. 2011) | PCR; Sequencing (?) | |
| tsRMST | Pluripotency maintenance of hESCs | (Wu et al. 2014) | RT-PCR; RNase protection assay | |
| TSNAX-DISC1 | G1/S transition and endometrial carcinoma (EC) development | (Li et al. 2014) | PCR; RNA-Seq (?) |
| Organisms | Involved Genes or Chimeras | Function Description | References | Experiments Verified |
|---|---|---|---|---|
| C. elegans | eri-6/7 | Superfamily I helicase | (Fischer et al. 2008) | RT-PCR; Sequencing |
| Drosophila (Fruit fly) | lola | Transcription factor | (Horiuchi et al. 2003) | RT-PCR; Sequencing |
| mod (mdg4) | Transcription factor | (Dorn et al. 2001) | RT-PCR; Sequencing | |
| Anopheles gambiae (Mosquito) | Bursicon | Coding bursicon | (Robertson et al. 2007) | Bioinformatics analysis |
| Bombyx mori (Silk worm) | mod (mdg4) | Transcription factor | (Shao et al. 2012) | RT-PCR; Sequencing |
| Dsx-dsr2 | Sexual development | (Duan et al. 2013) | RT-PCR; Sequencing | |
| Danio rerio (Zebrafish) | Grn1-Grn2 | Hybrid granulin | (Cadieux et al. 2005) | RT-PCR; Northern blot |
| Gallus (Chicken) | C-myb | Proto-oncogene | (Vellard et al. 1991) | (?) |
| Rattus norvegicus (Rat) | 1038 mRNA | Unknown | (Fitzgerald et al. 2006) | RT-PCR; Northern blot |
| ABP-HDC | Unknown | (Sullivan et al. 1991) | RT-PCR; Northern blot | |
| COT | Gene expression regulation | (Caudevilla et al. 1998) | RT-PCR; Northern blot; in vitro trans-splicing | |
| HongrE2 | Gene expression regulation | (Ni et al. 2011) | RT-PCR; Northern blot (?) | |
| LAR tyrosine phosphatase receptor | Gene expression regulation | (Zhang et al. 2003) | RT-PCR; RNase protection assay; Northern blot | |
| SNS-A | Unknownn | (Akopian et al. 1999) | PCR; RNase protection assay; Northern blot | |
| Sp1 | Transcription factor | (Takahara et al. 2002) | RT-PCR; RNase protection assay; Northern, Southern blot | |
| Mus musculus (Mouse) | Dmrt1-Dmr | Gene expression regulation | (Zhang et al. 2010) | RT-PCR; Northern blot; Southern blot |
| Msh4-Hspa5 Msh4-Pcbp3 | Cell death | (Hirano et al. 2004) | RT-PCR; Northern blot | |
| Sus scrofa (Pig) | AK238425, AK351564 and other 667 putative chimeras | Unknownn | (Ma et al. 2012) | Systematic analysis; RT-PCR; RNA-Seq |
| Homo sapiens (Human) | ATAC-1- Exon Xa/Xb | Gene expression regulation | (Yu et al. 1999) | RT-PCR; RNase protection assay; Northern blot |
| ATAC-1-Ampr | Gain antibiotics resistance | (Hu et al. 2013) | RT-PCR; in vitro trans-splicing | |
| CAMK2G-SRP72 | Unknownn | (Breen et al. 1997) | PCR; Genetic mapping; Western blot; (?) | |
| CDC2L2 | Transcriptional regulation | (Jehan et al. 2007) | FISH; RT-PCR | |
| C-myb | Proto-oncogene | (Vellard et al. 1991) | RT-PCR; Sequencing | |
| CoAA-RBM4 | Regulate stem/progenitor cell differentiation | (Brooks et al. 2009) | RT-PCR; in vitro trans-splicing | |
| CYCLIND1-TROP2 | Cell growth | (Guerra et al. 2008) | RT-PCR; Northern blot; RNase protection assay (?) | |
| CYP3A4, 5, 7, 43 | Catalytic activity | (Finta et al. 2002) | RT-PCR; Northern blot; RNase protection assay | |
| hER alpha | Gene expression regulation | (Flouriot et al. 2002) | RT-PCR; Southern blot | |
| JAZF1-JJAZ1 | Anti-apoptotic protein | (Li et al. 2008) | RT-PCR; Southern blot; in vitro trans-splicing | |
| PJA2-FER | Cancer biomarker | (Kawakami et al. 2013) | RT-PCR | |
| PAX3-FOXO1 | Cancer biomarker | (Yuan, Qin, et al. 2013) | RT-PCR; FISH | |
| RGS12 | G protein signaling | (Chatterjee et al. 2000) | RT-PCR | |
| Sp1 | Transcription factor | (Takahara et al. 2000) | RT-PCR; Southern blot; RNase protection assay | |
| ZC3HAV1L-CHMP1A | Genome rearrangement | (Fang et al. 2012) | RT-PCR (?) | |
| AF4, AF9, ELL, ENL, MLL, ETV6, NUP98, RUNX1, EWSR1 | DNA repair and chromosomal translocation | (Kowarz et al. 2012; Kowarz et al. 2011) | RT-PCR (?) | |
| TMEM79-SMG5 | Cancer biomarker | (Kannan et al. 2011) | PCR; Sequencing (?) | |
| tsRMST | Pluripotency maintenance of hESCs | (Wu et al. 2014) | RT-PCR; RNase protection assay | |
| TSNAX-DISC1 | G1/S transition and endometrial carcinoma (EC) development | (Li et al. 2014) | PCR; RNA-Seq (?) |
Note.—“?”, Probably trans-splicing.
Functions of Chimeric Transcripts in Vertebrates
Chimeric RNA is abundant in both normal and cancer tissues (Romani et al. 2003; Frenkel-Morgenstern et al. 2013). Chimeric RNA may be produced not only by trans-splicing but also by cis-splicing of adjacent genes (Zhang et al. 2012), chromosomal translocation (Mori et al. 2002), and cotranscription across neighboring loci (Magrangeas et al. 1998; Communi et al. 2001). Trans-spliced chimeric RNA was previously ignored as the byproduct of aberrant transcription or “splicing noise” due to its rarity (Maniatis et al. 2002; Tasic et al. 2002), but now it seems to be a “hidden” component of the genome. Evidence suggests that trans-splicing generates an additional layer of genome complexity (Gingeras 2009; Kowarz et al. 2012). These chimeric RNAs are engaged in a versatile range of physiological processes as either protein-coding or noncoding RNAs. Here, we summarize the functions of trans-spliced chimeric RNAs in vertebrates.
Trans-Splicing and Cancer
Although some trans-spliced chimeric RNAs are associated with cancers (Guerra et al. 2008; Kowarz et al. 2011), the causal relationship between trans-splicing and cancer remains unclear. JAZF1-JJAZ1 in cancer cells is derived from chromosomal translocation. However, it was also detected in normal endometrial stromal cells, indicating that the chimeric RNA is trans-spliced in normal cells (Li et al. 2008; Li,Wang, et al. 2009). The same situation is also found in the chimeric RNA PAX3-FOXO1 (Yuan, Qin, et al. 2013). In addition, intermolecular recombination events are involved in the tissue-specific expression of the C-myb proto-oncogene (Vellard et al. 1991). In human prostate cancer, most partner genes involved in chimeric RNAs have a low expression level (Kannan et al. 2011).
It has been postulated that a trans-spliced RNA molecule may serve as a scaffold to facilitate genomic interactions, which could lead to chromosomal translocations (Zaphiropoulos 2011). Kowarz et al. observed premature transcriptional termination as a common feature of genome rearrangements, and early terminated RNAs have an “unsaturated” splice donor site that gives rise to trans-splicing events (Kowarz et al. 2011, 2012). In this hypothesis, in case of DNA damage, these chimeric RNAs may direct broken chromosomes to align to the corresponding gene loci and guide chromosomal translocation. It seems that trans-spliced chimeric RNA is a precondition for chromosomal exchange; this may be a good explanation of why some patients have recurrent genetic rearrangements between AF4 (exon 4) and MLL (exon 9) (Kowarz et al. 2011). Because chromosomal translocation is a common event in neoplastic cells (Kowarz et al. 2011), some trans-spliced chimeras may be indicative of tumorigenesis (Guerra et al. 2008; Yuan, Qin, et al. 2013). Indeed, chimeric RNA molecules have been proposed as potential biomarkers for tumor diagnosis (Zhou et al. 2012). For example, the chimeric TMEM79-SMG5 molecule occurs in approximately 90% of prostate cancer samples, which may enable it to serve as a diagnostic biomarker for that type of cancer (Kannan et al. 2011).
Gene Expression Regulation
As indicated in table 1, trans-spliced chimeric RNAs are involved in the regulation of gene expression. For example, a 4.3-kb mRNA of human Acyl-CoA cholesterol acyltransferase 1 (ACAT-1) is derived from both chromosomes 1 and 7 (Li et al. 1999). The trans-spliced additional exons Xa and Xb, serve as the 5′ -UTR upstream the exon 1 and may account for its unconventional translation initiation. This chimeric RNA encodes a 56-kDa isoform protein with reduced activity (∼30%) compared with the common form (Chen et al. 2008). Another case is the epididymis-specific HongrES2, composed of exons from different chromosomes, which was found to share a common 3′ -end with the CES7 gene (Ni et al. 2011). Meanwhile, HongrES2 can give rise to miRNA-like small RNA (mil-HongrES2) that downregulates CES7 gene expression.
Signal Transduction
Trans-spliced chimeric RNAs are associated with signal transduction. For example, the SNS-A transcript comprises a repeat sequence of exons 12, 13, and 14, which encodes four trans-membrane regions of domain II (Akopian et al. 1999). Nerve growth factor can induce SNS-A transcript expression. The regulation is probably associated with nervous signal transduction. Similarly, a truncated isoform (γSRP) of CaM kinase II acquires six amino acids (RNNYKL) from the SRP72 gene (Breen et al. 1997). Although it has most of the catalytic properties of the holoenzyme, this isoform lacks an association domain, which may change its targeting ability. In addition, the RGS (regulators of G-protein signaling) protein family has several distinct chimeric transcripts of RGS12 in COS-7 cells, suggesting that trans-splicing may be a novel mechanism in the regulation of G-protein signaling pathways (Chatterjee et al. 2000).
Cell Viability and Growth
As mentioned above, the antiapoptotic JAZF1-JJAZ1 protein is associated with aberrant proliferation of neoplastic cells. The chimeric Msh4 δ variant is generated by trans-splicing between the Hspa5 and Msh4 pre-mRNAs, which could induce programed cell death during spermatogenesis (Hirano et al. 2004). In addition, some trans-spliced RNAs play a role in cell growth. For example, a low level of expression of the CYCLIN D1-TROP2 chimera was shown to be sufficient to induce cell proliferation and to extend the life span of primary culture cells, while high expression of the chimera can induce cell transformation, indicating its role in the regulation of cell growth and cancer (Guerra et al. 2008). A recent study reported a chimeric TSNAX-DISC1 in human endometrial carcinoma cells, which is regulated by a long intergenic noncoding RNA lincRNA-NR_034037 (Li et al. 2014). Notably, the regulation of TSNAX-DISC1 expression is involved in cell transition from G1 to S phase and in tumor growth.
Other Functions
In addition to previously discussed functions, trans-splicing is associated with other biological processes. Some chimeric RNAs in human are tissue-specific and can encode proteins. These proteins may compete with their parental proteins, disturbing protein interaction networks (Frenkel-Morgenstern and Valencia 2012; Frenkel-Morgenstern et al. 2012). In addition, a novel type of trans-splicing has been found in the ACAT1 transcript, where an exogenous recombinant plasmid-derived Ampr antisense segment is integrated (Hu et al. 2013). The type of exo-endo trans-splicing is abundant in normal human blood cells. This finding also suggests that exogenous DNA fragments, derived from recombinant plasmids or other sources, may affect cellular gene expression at both RNA and DNA levels.
Putative Mechanisms of trans-Splicing in Vertebrates
Currently, the mechanisms underlying trans-splicing in vertebrates remain largely unknown. Little is known about how the associated partner genes are physically recruited and what factors are involved in the process. Based on previous studies, we summarize several current models and propose new ones to address these issues.
tRNA-Mediated trans-Splicing Model
The tRNA sequence of two partner genes could direct their splicing reaction in a trans manner to generate a chimeric molecule in eukaryotic cells (Di Segni et al. 2008) (fig. 4A). In this model, the widespread tRNA genes in a genome or a repetitive sequence inside the coding region of an mRNA may be recognized and cleaved by the tRNA splicing endonuclease. Although experiments in vitro have shown that some mammalian mRNAs can be spliced by tRNA splicing endonuclease, tRNA-mediated trans-splicing needs to be explored further (Sidrauski et al. 1996; Deidda et al. 2003). Our recent study suggested that modern tRNAs originated from tRNA halves, potentially involving trans-splicing (Zuo et al. 2013).
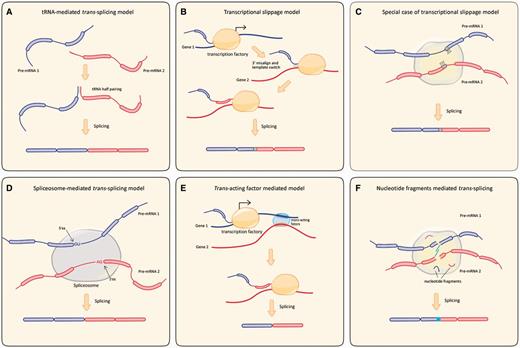
Schematic representation of proposed models of trans-splicing mechanisms. (A) tRNA-mediated trans-splicing model. Pre-tRNA halve adjacent to pre-mRNA context narrowing two associated molecules through complementary sequences, then the hybrid molecule is cleaved precisely at the sites of the tRNA intron by tRNA splicing endonuclease. (B) Transcriptional slippage model. Gray boxes represent pairing of SHSs. A pre-RNA is transcribed from Gene 1 and then misaligns to the DNA template of gene 2 via the SHSs. Transcription machinery keeps on moving on the strand of gene 2, after removal of introns, resulting in the chimeric molecule. (C) Special case of transcriptional slippage model. Both partner genes share a forward direction repeat sequence in the junction site of chimeric RNA. (D) Spliceosome mediated trans-splicing model. Like canonical cis-splicing, pre-RNA 1 and pre-RNA 2 is precisely spliced at the 5′- and 3′-splicing site and ligated as a non-linear chimeric molecule. (E) Trans-acting factor mediated model. Blurry region represent consensus DNA motif in parental gene 1 and gene 2. They can be recognized by trans-acting factor like CTCF and recruited to the shared transcription factory, and then coordinate the transcription by the same or similar transcription machinery. Transcription occurs between the Gene 1 and 2, the chimeric transcript is finally generated after intron removal. (F) Nucleotide fragments - mediated trans-splicing model. Short nucleotide fragments could induce transcription or be added into pre-mRNA. Trans-splicing could occur through base paring between two fragments. Through intermolecular splicing, this nucleotide fragments can be introduced into the chimeric molecule.
Transcriptional Slippage Model
The second possible mechanism is the “transcriptional slippage model,” which is based on a large-scale screening of chimeric RNAs in yeast, fruit fly, mouse, and human (Li, Zhao, et al. 2009). This model assumes that the transcription machinery “walks” along the primary template strand and dissociates from it in some cases, followed by “misaligning” with certain position of another locus through short homologous sequences (SHS) (fig. 4B). Thus, by continuing the transcriptional process on the new template, the chimeric RNA is generated. In this model, chimeric RNAs with classical “GU-AG” junction site only account for a small fraction (<20%), whereas the SHS type accounts for nearly 50%. Distal actively transcribed genes can frequently be corecruited to the same transcription machinery (Osborne et al. 2007), and this may be an environment promoting the occurrence of trans-splicing between two pre-mRNAs. As an example of the model, a 4-bp sequence at the junction site of chimeric Msh4-Hspa5 molecule can be exactly mapped to each of the two partner genes (Hirano et al. 2004). This homologous region between partner genes may induce transcriptional slippage and further trans-splicing (fig. 4C).
Spliceosome-Mediated trans-Splicing Model
The third model is the spliceosome-mediated trans-splicing model. It was assumed that partner genes can be corecruited to the same spliceosome (Osborne et al. 2007) and spliced at canonical “GU-AG” sites (Li, Zhao, et al. 2009) (fig. 4D). Several cases of functional trans-splicing molecules with “GU-AG” at splicing sites support this model (Sullivan et al. 1991; Robertson et al. 2007; Fischer et al. 2008). Unsaturated splice donor sites were detected in early terminated transcripts in the human MLL gene. These unsaturated splice donor sites can induce a splicing reaction. Early terminated transcripts use cryptic exons to saturate the splice donor sites, which could give rise to trans-splicing events (Kowarz et al. 2011, 2012).
Trans-Acting Factors-Mediated Model
Compared with the above models, the trans-acting factors-mediated model could be more dynamic and capable of explaining how mRNA precursors are associated with each other before splicing (Ma et al. 2012) (fig. 4E). An interesting study has identified 251 chimeric mRNAs in pig, and a considerable fraction of these molecules have the canonical “GU-AG” at junction sites (Ma et al. 2012). The study also observed four consensus DNA sequences in the genomic region of the 5′ and 3′ partner genes, which are similar to the known DNA-binding motifs of the human CCCTC-bind factor (CTCF) binding sites. In this model, it is postulated that some consensus DNA motifs, such as CTCF, that are shared by associated partner genes can be recognized and recruited by CTCF to the same transcriptional machinery. CTCF may bring distal intrachromosomal and interchromosomal regions into proximity, suggesting a role in facilitating trans-splicing events (Ling et al. 2006; Williams et al. 2008). Indeed, while CTCF is silenced in endometrial stromal cells, the trans-spliced JJAF1-JJAZ1 chimeric RNA was downregulated (Li et al. 2008; Zhang et al. 2012).
Furthermore, in line with the transcriptional slippage model, parental genes can be induced to colocalize to the same transcriptional factory so that they are coordinately transcribed to generate chimeric pre-mRNAs. After the excision of introns, exons are joined by the spliceosome to generate a mature chimeric molecule. The trans-acting factor model could be universal and sufficiently dynamic to generate trans-splicing molecules.
Nucleotide Fragments-Mediated trans-Splicing Model
Endogenous and random short fragments were observed in cells and could serve as primers for reverse transcription polymerase chain reaction (RT-PCR) without adding extra primers (Yuan, Liu, et al. 2013). These endogenous short fragments can integrate into pre-mRNAs during transcriptional or posttranscriptional processes. Homologous regions in the short fragments could serve as intermediary guides to induce trans-splicing (fig. 4F). An example for this model is chimeric ACAT-1 mRNA. Human ACAT-1 mRNA is produced from two chromosomes by trans-splicing, but a 10-bp exon Xb could not be mapped to the relevant exons. Thus, an extra nucleotide fragment was inserted into the chimeric molecule (Li et al. 1999). This model could explain the formation of chimeric RNAs without the canonical “GU-AG” junction site, as well as some chimeric molecules with a small insertion that does not exist in the pre-mRNAs.
However, none of the models completely explains the generation of all trans-splicing. Current in silico screening strategies in chimeric RNA analysis rely on the canonical splicing sites “GT/AG.” However, in real scenarios, trans-splicing could occur at some infrequent splicing sites (Herai et al. 2010). In addition, some DNA motifs, such as the GAAGAAG box in COT gene, can enhance trans-splicing frequency, suggesting a potential regulatory network (Caudevilla, Codony, et al. 2001). We are still far from a comprehensive understanding of trans-splicing mechanisms. Because of the complexity of RNA types in different cell types and different physiological conditions, there may be other mechanisms for the generation of chimeric RNAs that remain to be identified.
Challenges and Perspectives
Methodology Challenges
The identification and elimination of artificial chimeras are major challenges. Current methods utilized in gene expression analysis, such as RT-PCR, transcriptome, and cDNA library construction, typically require transcribing RNA into cDNA with RT. There are several sources of RTs. Lentiviruses (e.g., HIV-1, SIV) (Jamburuthugoda et al. 2011) and oncoretroviruses (e.g., AMV, M-MLV) encode virus RTs. In eukaryotes, both long terminal repeat (LTR) and non-LTR retrotransposable elements can encode RTs (Bibillo et al. 2002). In addition, the telomerase gene also encodes an RT to maintain telomere length. RTs lack 3′-5′ exonuclease activity and proofreading ability, and thus transcribe RNA into DNA with a high error rate (Bakhanashvili et al. 1992). The average error rate is approximately 3 × 10 −5 for M-MLV RT and approximately 6 ×10 −5 per nucleotide for AMV RT, which is one-tenth of that of HIV-1 RT (Katarzyna Bebenek 1993). The error rate of RTs encoded by LTR retrotransposable elements is similar to that of oncoretroviral RTs. The error rate for human telomerase is much higher, with approximately 2 × 10 −3 per nucleotide (Agorio et al. 2003). The error rates of viral RTs with RNA templates are consistent with retroviral mutation rates of 10−4 to 10−6. A high error rate results in the rapid evolution of viral genomes, which is essential for the virus to rapidly evade the host. However, it is difficult to avoid the introduction of many biases and artifacts when transcribing RNA into cDNA using RTs. In fact, there is a considerable amount of artificial chimeras in RNA-Seq, transcriptome, and cDNA libraries when using commercial RTs. In addition to substitution errors, it has been shown that the RT process is associated with the generation of artificial sequences due to template switching and fusions (Houseley et al. 2010). Moreover, due to different strategies in adapter ligation and fragmentation, we may generate platform-dependent biased data (Aird et al. 2011; Zheng et al. 2011). These two points may partially explain the inconsistent data generated from different RNA-Seq approaches, as observed in previous studies (Wu et al. 2014). Data retrieving and screening are also difficult. It is not easy to design an effective and reliable algorithm to identify real trans-splicing events from terabytes of data. Currently, there are some programs and databases for screening chimeric transcripts (Li, Zhao, et al. 2009; Kim et al. 2010; Al-Balool et al. 2011; Carrara et al. 2013; Frenkel-Morgenstern et al. 2013; Hoffmann et al. 2014). Further optimization, evaluation, and experimental confirmation are needed.
Even given these disadvantages, the RNA-Seq analysis and bioinformatics pipelines are still the most powerful tools for the analysis of trans-spliced chimeric RNAs. Improvement of cDNA cloning methods, for example, a new 3′-end cloning method (Yuan, Liu, et al. 2013) and other emerging technologies, will enable the discovery of more credible trans-splicing events. A new non-collinear transcript-detecting method was recently developed that can detect trans-spliced, circular, or fusion transcripts (Chuang et al. 2015). In addition, several chimeric RNA databases have been constructed and they reported appealing results (Kim et al. 2010; Abate et al. 2012; Benelli et al. 2012; Frenkel-Morgenstern et al. 2012, 2013; Bruno et al. 2013). For example, the ChiTaRS database includes comprehensive information on more than 16,000 chimeric transcripts from humans, mice, and fruit flies (Frenkel-Morgenstern et al. 2013). It is expected that the use of an optimized algorithm and filtering steps to eliminate false positives will yield more credible candidates using RNA-Seq data. The “TScan” strategy is a good example of a method of screening trans-splicing events in human embryonic stem cells (hESCs) (Wu et al. 2014). This is an integrative transcriptome sequencing technology with multiple experimental validation steps. First, the investigators acquired 0.83 million long reads (∼353.7-bp) and 230.63 million short reads (50-bp) from Roche 454 and SOLiD whole-transcriptome sequencing platforms, respectively. Then, by aligning these long reads with the public human genome database based on 454/Illumina sequencing data, 8,822 preliminary candidates were obtained. Targets validated by short-read information were extracted. The candidate group was then filtered by rules intended to identify non-trans-splicing events including: 1) chimeric junction site with SHS (McManus et al. 2010); 2) sense–antisense fusion containing a noncanonical splicing signal (Houseley et al. 2010); 3) mitochondrial–nuclear fusion events (McManus et al. 2010). Artificial products formed during the reverse-transcription (RT) process were subtracted (Houseley et al. 2010). Finally, Wu and colleagues identified and experimentally confirmed four trans-spliced RNAs (tsCSNK1G3, tsARHGAP5, tsFAT1, and tsRMST) in the hESCs (Wu et al. 2014). These trans-spliced RNAs are all highly expressed in human pluripotent stem cells and differentially expressed during hESC differentiation. tsRMST may control pluripotency through repressing lineage-specific genes, involving the pluripotency transcription factor NANOG and PRC2 complex factor SUZ12. This report not only uncovered the importance of trans-splicing as a posttranscriptional event but also established an insightful pipeline to discover trans-splicing events.
New Technologies
Direct RNA Sequencing
Direct RNA sequencing (DRS) (Ozsolak et al. 2009) can profile mRNA transcripts of interest free of interference from RT-based artificial products. Natural RNA molecules can be directly sequenced by DRS without prior conversion to cDNA (Ozsolak et al. 2011), so DRS can detect real chimeric molecules. DRS of single molecules will have practical implications in the real-time monitoring of chimeric transcripts.
Hi-C and Interactome Modeling
Two distant gene loci separated by millions of DNA base pairs can be bridged by enhancers, transcription factors, and insulator proteins, and they can interact to regulate transcription of distant genes. All these activities are carefully orchestrated in the form of the three-dimensional (3D) conformation of chromosomes, which are compartmentalized in the nucleus. Hi-C allows one to probe genome-wide individual chromatin interactions (Lieberman-Aiden et al. 2009). It was reported that active genes can be transcribed and coregulated by the same transcription machinery (Osborne et al. 2007). In this case, both Hi-C and interactome modeling (Fullwood et al. 2009) can be used to characterize the complete repertoire of chromosomal interactions and help us to probe the regulatory activity involved specifically in trans-splicing.
Molecular Labeling Techniques
Nanostring techniques can quantitatively measure the expression levels of RNA transcripts (Geiss et al. 2008). By labeling probes with specific barcode, this method can capture individual RNA transcripts and count the exact copy number with high sensitivity and a digital readout. Another technique is using the tiny molecular beacon LNA/2′-O-methyl to mark individual pre-mRNA molecules to trace dynamic mRNA activities in living cells (Catrina et al. 2012). These techniques mean that we may be able to see how the two candidate primary transcripts are recruited together and where they are processed into chimeric molecules.
Proteomic Data Analysis
Based on a comprehensive analysis of 7,424 human chimeric RNAs, Frenkel-Morgenstern et al. (2012) suggested that chimeras potentially contain common and unique domain combinations. In combination with these techniques at the protein level, the accuracy of identified trans-splicing events will be improved tremendously. In addition, multiple experimental validation steps have been shown to be efficient in the validation of trans-splicing variants (Yu et al. 2014). With continued development of the techniques mentioned above and new techniques, we will gain understanding of the nature of trans-splicing.
Conclusions
(1) Trans-splicing is evolutionarily dynamic. The discovery of trans-splicing has updated the definition of genome coding capacity. Trans-splicing may be a mechanism for cells to extend the maximum potential of limited genetic information to adapt to various physiological conditions. In prokaryotes, reprogramming events on the RNA level rely on autocatalytic group II or group I introns and may be a detour from continuous RNAs in eukaryotes (Glanz et al. 2009). Despite a limited understanding of its evolutionary origin, we realize that trans-splicing occurs more frequently in lower species than in higher vertebrates. For example, trans-splicing occurs nearly in all genes in T. brucei, while vertebrates are free of SL trans-splicing. There is an evolutionary dynamic that trans-splicing is being replaced by other mechanisms, such as alternative splicing, to adapt to intricate genomic structures through refined regulation systems in vertebrates. Nevertheless, the splicing machinery is evolutionarily conserved between lower eukaryotes and mammals. It has been observed that induced SL RNAs can be accurately trans-spliced in HeLa cells in vivo and in vitro (Bruzik et al. 1992). The SR (Ser/Arg)-rich protein is a key factor for alternative splicing. This protein has also been shown to promote trans-splicing (Bruzik et al. 1995). These data suggest a common evolutionary origin of both cis-splicing and trans-splicing.
(2) Trans-spliced chimeras could contribute to some pathological consequences, such as cancer and apoptosis, and response to external stimuli, given that trans-splicing chimeras are temporally/spatially regulated and have low expression levels in normal cells. Under specific conditions/cell types, such as in cancer cells, they are deregulated and could lead to chromosomal translocation and tumorigenesis (Li et al. 2008; Li, Wang, et al. 2009). Several models for putative mechanisms of trans-splicing in vertebrates have been proposed. Further research will elucidate the underlying mechanisms of trans-splicing and uncover the biological functions and physiological/pathological significance of trans-spliced RNAs. In addition, the development of new trans-splicing RNA technologies and their translations into clinical applications will benefit more patients.
(3) Because a considerable number of RNA chimeras are artificial products generated by RT-based technology, the question of how to identify real trans-splicing molecules remains. A global transcriptome-wide and high-throughput analysis needs to be developed with both high sensitivity and optimized algorithms to detect tissue-specific and low-copy transcripts. DRS analysis with high-throughput, high efficiency, and low cost will be the most promising technique for detection of trans-splicing events in vertebrates.
Acknowledgments
Authors thank Dr Rainer Dorn for his suggestions for the manuscript. This work was supported by the National Natural Science Foundation of China, National Key Technologies R&D Program, Hubei Science & Tech Project and the Chinese 111 Project Grant B06018.
Literature Cited
Author notes
Associate editor: Kateryna Makova

{kind=link}
{kind=link}
{kind=link}
{kind=link}