





Abstract
In prokaryotes, functionally linked genes are typically clustered into operons, which are transcribed into a single mRNA, providing for the coregulation of the production of the respective proteins, whereas eukaryotes generally lack operons. We explored the possibility that some prokaryotic operons persist in eukaryotic genomes after horizontal gene transfer (HGT) from bacteria. Extensive comparative analysis of prokaryote and eukaryote genomes revealed 33 gene pairs originating from bacterial operons, mostly encoding enzymes of the same metabolic pathways, and represented in distinct clades of fungi or amoebozoa. This amount of HGT is about an order of magnitude less than that observed for the respective individual genes. These operon fragments appear to be relatively recent acquisitions as indicated by their narrow phylogenetic spread and low intron density. In 20 of the 33 horizontally acquired operonic gene pairs, the genes are fused in the respective group of eukaryotes so that the encoded proteins become domains of a multifunctional protein ensuring coregulation and correct stoichiometry. We hypothesize that bacterial operons acquired via HGT initially persist in eukaryotic genomes under a neutral evolution regime and subsequently are either disrupted by genome rearrangement or undergo gene fusion which is then maintained by selection.
Horizontal gene transfer (HGT) plays a key role in the evolution of prokaryotic genomes, allowing acquisition of novel traits that can improve their fitness and adaptation. Numerous studies have shown that bacterial genes can be horizontally transferred to eukaryotes as well, but the extent to which entire operons—functionally linked cluster of genes—can be transferred remains poorly understood. We systematically analyzed 1,845 diverse eukaryotic genomes to shed light on the prevalence and impact of HGT of operons. Our findings indicate that, although the HGT of entire operons into eukaryotic genomes is rare, it did occur on multiple occasions, facilitating introduction of functionally cohesive sets of genes. We demonstrate that these horizontally acquired operons tend to be evolutionarily “young” and can undergo post-transfer fusion into single genes. This study provides insights into evolutionary consequences of HGT of operons in eukaryotes, highlighting their potential role in shaping genome complexity and functionality.
Introduction
Horizontal gene transfer (HGT), that is, transfer of genetic material between organisms that are not in a parent–offspring relationship, is a fundamental evolutionary process (Soucy et al. 2015). By introducing new traits to recipients, HGT facilitates adaptation to challenging and new environmental conditions. Although HGT has been primarily described in prokaryotes (Gogarten et al. 2002), where it plays a central role in evolution, the growing volume of genomic data reveals its major impact on eukaryotic evolution as well (Husnik and McCutcheon 2018; Van Etten and Bhattacharya 2020; Irwin et al. 2022). The latest comprehensive studies across various eukaryotic clades, including fungi, plants, and insects, suggest that bacteria serve as the primary donors of horizontally transferred genes (Shen et al. 2018; Li et al. 2022; Wu et al. 2024). Cross-domain HGT presents a major challenge to the recipient because eukaryotic and bacterial expression mechanisms are substantially different. Thus, for horizontally transferred genes to function effectively and to be fixed in the recipient population, they should undergo multiple molecular modifications to be integrated into their new genetic setting. These modifications include acquisition of appropriate promoters and other regulatory elements, optimization of codon usage, and adjustments to different genetic and epigenetic factors (Husnik and McCutcheon 2018). Despite these obstacles, successful HGT events equipped various groups of eukaryotes with a plethora of metabolic functions, for example, heme biosynthesis in parasitic nematodes (Wu et al. 2013), cyanide detoxification in arthropods (Wybouw et al. 2014), toxin genes for predation (Moran et al. 2012), and various genes involved in innate immunity (Chou et al. 2015; Culbertson and Levin 2023).
One of the major differences between prokaryotic and eukaryotic gene expression is that in many cases, functionally linked genes in prokaryotes are organized into distinct units known as operons (Jacob and Monod 1961). Genes in operons are transcribed as one polycistronic mRNA and have a single promoter. The selective advantage of the operon organization of genes can be attributed to a better coordination of gene expression, as all genes within operons are regulated by a shared set of regulatory elements (Price et al. 2005). As a result, operons allow prokaryotes to efficiently control multiple genes in response to environmental conditions, reducing the accumulation of toxic intermediate molecules in dosage-sensitive metabolic pathways. Moreover, this arrangement allows recipients of HGT-transferred operons to acquire new traits more efficiently, as many metabolic functions depend on the coordinated and sequential actions of multiple proteins. Indeed, multiple comparative genomic studies have shown that HGT of operons among various prokaryotes is common in nature (Omelchenko et al. 2003; Price et al. 2005) (Price et al. 2008). The much higher probability of fixation of horizontally transferred operons compared to individual genes underlies the selfish operon concept according to which the evolutionary persistence of operons and wide spread of many of them across prokaryotes are driven by HGT (Lawrence and Roth 1996; Lawrence 2003).
In contrast to the widespread horizontal transfer of operons among bacteria and archaea, there are only a few known examples of HGT from bacteria to eukaryotes involving operons. These include acquisition of thiamine biosynthesis and enterobactin biosynthesis operons by Wickerhamiella/Starmerella ascomycete fungi (Goncalves and Goncalves 2019; Kominek et al. 2019), peptidoglycan biosynthesis genes by Aspergillus spp. (Marcet-Houben and Gabaldon 2010), and pyruvate formate lyase and its activating enzyme in various groups of eukaryotes (Stairs et al. 2011). However, to our knowledge, the prevalence of horizontally captured operons in eukaryotic genomes has not been assessed systematically.
In this work, we employed comparative genomics and phylogenetic methods to systematically search for operons acquired through HGT from bacteria in more than 1,800 eukaryotic genomes. This search revealed 33 previously unknown cases of bacterial operons integrated into eukaryotic genome, primarily fungal ones, representing various functional categories. Transcriptomic data indicate that at least some of these operons are actively expressed, supporting their functionality in eukaryotes. Our findings confirm that preservation of operon organization after HGT from bacteria and eukaryotes is rare but, nevertheless, expand the known repertoire of transferred operons and provide insights into their evolutionary trajectories and how they can contribute to eukaryotic innovation and adaptation.
Results
Adjacent, Codirected Gene Pairs in Bacteria Are Functionally Linked
As a first step, we aimed to collect a comprehensive list of gene pairs that are likely to function together as components of operons in bacteria and archaea. Given that the number of experimentally validated operons is small compared to the vast amount of available genomic data, we aimed to use conserved gene pairs as a proxy for identifying operons (Omelchenko et al. 2003). For that purpose, we predicted clusters of orthologous genes (COGs) in 1,805 prokaryotic genomes, each representing the taxonomic rank of “genus” (supplementary table S1, Supplementary Material online). Under the premise that functionally linked genes tend to cluster together in bacterial genomes, forming operons (Jacob and Monod 1961; Wolf et al. 2001), we identified 45,396 COG pairs, each present in at least 5 representative genomes, that were adjacent and codirected significantly more frequently than expected by chance (P < 0.05, binomial test, Benjamini–Hochberg correction) (Fig. 1a). To verify that these COG pairs encoded functionally related genes, we examined their functional categories and roles in molecular pathways. Specifically, we assessed the tendency of genes in retained COG pairs to belong to the same functional categories and to participate in the same pathways by calculating the assortativity value. The assortativity value can range from −1 (indicating that genes in all examined COG pairs belong to different metabolic pathways or categories) to 1 (indicating that genes in all COG pairs belong to the same pathways or functional categories). The calculated assortativity values were in the range of 0.2 to 0.3, far exceeding the values for randomly shuffled COG pairs and suggesting that a large fraction of the codirected, adjacent COG pairs encoded functionally linked proteins and belonged to bona fide operons (Fig. 1b).
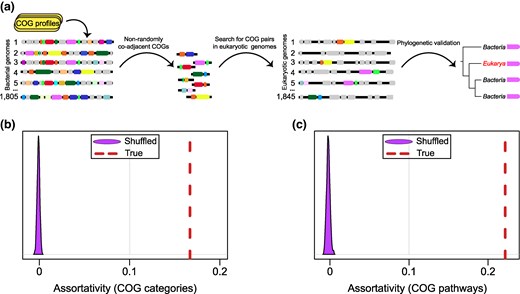
Prediction of coadjacent gene pairs using COG assignments and their functional relatedness. a) General pipeline that was used to identify HGT-transferred operons in the set of eukaryotic genomes. b) Assortativity between significantly coadjacent COG pairs and their functional category assignment. Null distribution of assortativity coefficients for 1,000 networks with randomly shuffled COG assignments is shown in purple, and the dashed red lines indicate the observed assortativity coefficient. c) Assortativity between significantly coadjacent COG pairs and metabolic pathways. Null distribution of assortativity coefficients for 1,000 networks with randomly shuffled metabolic pathways is shown in purple, and the dashed red lines indicate the observed assortativity coefficient.
To determine how extensively our data set covered validated operons, we cross-checked our list with 7,953 gene pairs with COG annotations from the OperonDB database (Okuda and Yoshizawa 2011). We found that 6,076/7,953 (76.4%) gene pairs are present in our data set, confirming that our list of COG pairs covers the majority of known operons.
Operon-Like Gene Arrays Derived from Bacteria Are Rare in Eukaryotic Genomes
To comprehensively search for operon-like gene arrays that were potentially horizontally transferred from prokaryotes to eukaryotes, we downloaded 1,845 eukaryote genomes designated as “representative species” by the RefSeq database, retaining only contigs larger than 100 kb (supplementary table S2, Supplementary Material online). This data set encompasses a broad taxonomic range, including 35 phyla, 305 orders, and 1,168 genera, with over 30,000,000 protein-coding genes. Using the compiled list of COG pairs, we identified pairs of colocalized and codirected orthologs of the respective genes in eukaryote genomes (excluding genomes of mitochondria and chloroplasts), followed by rigorous and conservative phylogenetic analysis (see Methods). To verify that operon-like gene arrays in eukaryote genomes did not represent bacterial contamination, we initially used Conterminator (Steinegger and Salzberg 2020) to detect any potential contaminants. Additionally, we checked their neighborhoods (three genes upstream and downstream) by conducting BLASTP search against the nr database. This analysis allowed us to assess the evolutionary context and confirm the eukaryotic origin of the surrounding genes, further supporting the authenticity of the operon-like arrays. In the collection of 1,845 representative eukaryote genomes, we identified only 33 distinct cases of horizontally acquired operon-like gene arrays. These “eukaryotic operons” were predominantly found in fungal genomes (16/33) and in those of various protists and encode a wide range of metabolic functions (Table 1).
Operonic gene arrays horizontally acquired by eukaryotes from bacteria
| COG pair | Representative sequence ID | Possible metabolic function | Recipient eukaryote clade | Donor bacterial clade |
|---|---|---|---|---|
| COG0413 COG0414 | XP_018895596.1a | Pantothenate biosynthesis | Bemisia | Gammaproteobacteria |
| COG0026 COG0041 | XP_638800.1a | Purine biosynthesis | Evoseab | Alphaproteobacteria |
| COG2141 COG1853 | XP_060455211.1a | Pyrimidine degradation | Fungi | Actinomycetes |
| COG1024 COG0318 | XP_031053101.1a | Fatty acid metabolism | Fungi | Actinomycetes |
| COG1539 COG0801 | XP_020610460.1a | Folate metabolism | Cnidaria | Anaerolineae |
| COG0673 COG0399 | XP_008598631.1a | Membrane biogenesis | Fungib | Alphaproteobacteria |
| COG1917 COG1850 | XP_007515483.1a | Photosynthesis | Viridiplantae | Alphaproteobacteria/Acidobacteriota |
| COG0413 COG0414 | NP_592822.1 NP_592821.1 | Pantothenate biosynthesis | Fungib | Gammaproteobacteria |
| COG0132 COG0161 | XP_013025616.1 XP_013025615.1 | Biotin biosynthesis | Fungi | Flavobacteriia |
| COG0223 COG0399 | XP_040634950.1 XP_040634951.1 | Fmt-WecE | Fungi | Gammaproteobacteria |
| COG0693 COG0451 | XP_002492919.1 XP_002492920.1 | WcaG-YajL | Fungi | Bacteroidota |
| COG0771 COG0773 | XP_011122786.1 XP_011122785.1 | Peptidoglycan metabolism | Fungib | Flavobacteriia |
| COG0812 COG0766 | XP_011119914.1 XP_011119913.1 | Peptidoglycan metabolism | Fungib | Saccharibacteria |
| COG3410 COG1694 | XP_016230087.1 XP_016230088.1 | MazG-DUF2075 | Fungi | Actinomycetes |
| COG2072 COG1063 | XP_007731551.1 XP_007731550.1 | Tdh-CzcO | Fungi | Actinomycetes |
| COG2986 COG2987 | XP_014160720.1a | Amino acid metabolism | Sphaeroforma | Betaproteobacteria |
| COG0637 COG1554 | XP_015660045.1a | Carbohydrate transport | Kinetoplastea | Gammaproteobacteria/Bacteroidota |
| COG1940 COG3010 | XP_012897763.1a | Carbohydrate metabolism | Opalinata | Chloroflexota |
| COG0637 COG1554 | XP_821775.1a | Carbohydrate metabolism | Kinetoplastea | Pseudomonadota |
| COG0367 COG1024 | XP_053036155.1a | Secondary metabolites metabolism | Fungi | Actinomycetes/Gammaproteobacteria |
| COG1917 COG1985 | XP_001330840.1a | RibD-QdoI | Trichomonas | Bacillota/Gammaproteobacteria |
| COG0673 COG0399 | XP_024735453.1a | Membrane biogenesis | Fungi | Alphaproteobacteria |
| COG0284 COG0461 | XP_001464543.1a | Pyrimidine biosynthesis | Kinetoplastea | Chloroflexota |
| COG2103 COG2971 | XP_021954652.2a | Peptidoglycan metabolism | Arthropoda | Verrucomicrobiae |
| COG0132 COG0161 | XP_002140155.1a | Biotin biosynthesis | Cryptosporidium | Alphaproteobacteria |
| COG0790 COG1187 | XP_004333747.1 XP_004333748.1 | Pseudouridine biosynthesis | Acanthamoeba | Clostridia/Myxococcia |
| COG0300 COG4221 | XP_004361637.1 XP_004361638.1 | YqjQ-YdfG | Evosea | Bacteroidota |
| COG0147 COG0512 | XP_009038158.1a | Amino acid metabolism | Pelagomonadales | Actinomycetes |
| COG0223 COG0399 | XP_009216736.1 XP_009216737.1 | Fmt-WecE | Fungi | Betaproteobacteria |
| COG0637 COG1554 | XP_010698393.1a | Carbohydrate metabolism | Kinetoplastea | Bacteria |
| COG0235 COG0329 | XP_018148101.1 XP_018148102.1 | AraD-DapA | Fungi | Pseudomonadota |
| COG0132 COG0161 | XP_018905096.1 XP_018905097.1 | Biotin biosynthesis | Bemisia | Alphaproteobacteria |
| COG0596 COG0654 | XP_056561192.1a | MenH-UbiH | Fungi | Actinomycetes |
| COG pair | Representative sequence ID | Possible metabolic function | Recipient eukaryote clade | Donor bacterial clade |
|---|---|---|---|---|
| COG0413 COG0414 | XP_018895596.1a | Pantothenate biosynthesis | Bemisia | Gammaproteobacteria |
| COG0026 COG0041 | XP_638800.1a | Purine biosynthesis | Evoseab | Alphaproteobacteria |
| COG2141 COG1853 | XP_060455211.1a | Pyrimidine degradation | Fungi | Actinomycetes |
| COG1024 COG0318 | XP_031053101.1a | Fatty acid metabolism | Fungi | Actinomycetes |
| COG1539 COG0801 | XP_020610460.1a | Folate metabolism | Cnidaria | Anaerolineae |
| COG0673 COG0399 | XP_008598631.1a | Membrane biogenesis | Fungib | Alphaproteobacteria |
| COG1917 COG1850 | XP_007515483.1a | Photosynthesis | Viridiplantae | Alphaproteobacteria/Acidobacteriota |
| COG0413 COG0414 | NP_592822.1 NP_592821.1 | Pantothenate biosynthesis | Fungib | Gammaproteobacteria |
| COG0132 COG0161 | XP_013025616.1 XP_013025615.1 | Biotin biosynthesis | Fungi | Flavobacteriia |
| COG0223 COG0399 | XP_040634950.1 XP_040634951.1 | Fmt-WecE | Fungi | Gammaproteobacteria |
| COG0693 COG0451 | XP_002492919.1 XP_002492920.1 | WcaG-YajL | Fungi | Bacteroidota |
| COG0771 COG0773 | XP_011122786.1 XP_011122785.1 | Peptidoglycan metabolism | Fungib | Flavobacteriia |
| COG0812 COG0766 | XP_011119914.1 XP_011119913.1 | Peptidoglycan metabolism | Fungib | Saccharibacteria |
| COG3410 COG1694 | XP_016230087.1 XP_016230088.1 | MazG-DUF2075 | Fungi | Actinomycetes |
| COG2072 COG1063 | XP_007731551.1 XP_007731550.1 | Tdh-CzcO | Fungi | Actinomycetes |
| COG2986 COG2987 | XP_014160720.1a | Amino acid metabolism | Sphaeroforma | Betaproteobacteria |
| COG0637 COG1554 | XP_015660045.1a | Carbohydrate transport | Kinetoplastea | Gammaproteobacteria/Bacteroidota |
| COG1940 COG3010 | XP_012897763.1a | Carbohydrate metabolism | Opalinata | Chloroflexota |
| COG0637 COG1554 | XP_821775.1a | Carbohydrate metabolism | Kinetoplastea | Pseudomonadota |
| COG0367 COG1024 | XP_053036155.1a | Secondary metabolites metabolism | Fungi | Actinomycetes/Gammaproteobacteria |
| COG1917 COG1985 | XP_001330840.1a | RibD-QdoI | Trichomonas | Bacillota/Gammaproteobacteria |
| COG0673 COG0399 | XP_024735453.1a | Membrane biogenesis | Fungi | Alphaproteobacteria |
| COG0284 COG0461 | XP_001464543.1a | Pyrimidine biosynthesis | Kinetoplastea | Chloroflexota |
| COG2103 COG2971 | XP_021954652.2a | Peptidoglycan metabolism | Arthropoda | Verrucomicrobiae |
| COG0132 COG0161 | XP_002140155.1a | Biotin biosynthesis | Cryptosporidium | Alphaproteobacteria |
| COG0790 COG1187 | XP_004333747.1 XP_004333748.1 | Pseudouridine biosynthesis | Acanthamoeba | Clostridia/Myxococcia |
| COG0300 COG4221 | XP_004361637.1 XP_004361638.1 | YqjQ-YdfG | Evosea | Bacteroidota |
| COG0147 COG0512 | XP_009038158.1a | Amino acid metabolism | Pelagomonadales | Actinomycetes |
| COG0223 COG0399 | XP_009216736.1 XP_009216737.1 | Fmt-WecE | Fungi | Betaproteobacteria |
| COG0637 COG1554 | XP_010698393.1a | Carbohydrate metabolism | Kinetoplastea | Bacteria |
| COG0235 COG0329 | XP_018148101.1 XP_018148102.1 | AraD-DapA | Fungi | Pseudomonadota |
| COG0132 COG0161 | XP_018905096.1 XP_018905097.1 | Biotin biosynthesis | Bemisia | Alphaproteobacteria |
| COG0596 COG0654 | XP_056561192.1a | MenH-UbiH | Fungi | Actinomycetes |
aFused eukaryote genes encompassing orthologs of both genes in the operon.
bOperons that were highlighted in the main text.
Operonic gene arrays horizontally acquired by eukaryotes from bacteria
| COG pair | Representative sequence ID | Possible metabolic function | Recipient eukaryote clade | Donor bacterial clade |
|---|---|---|---|---|
| COG0413 COG0414 | XP_018895596.1a | Pantothenate biosynthesis | Bemisia | Gammaproteobacteria |
| COG0026 COG0041 | XP_638800.1a | Purine biosynthesis | Evoseab | Alphaproteobacteria |
| COG2141 COG1853 | XP_060455211.1a | Pyrimidine degradation | Fungi | Actinomycetes |
| COG1024 COG0318 | XP_031053101.1a | Fatty acid metabolism | Fungi | Actinomycetes |
| COG1539 COG0801 | XP_020610460.1a | Folate metabolism | Cnidaria | Anaerolineae |
| COG0673 COG0399 | XP_008598631.1a | Membrane biogenesis | Fungib | Alphaproteobacteria |
| COG1917 COG1850 | XP_007515483.1a | Photosynthesis | Viridiplantae | Alphaproteobacteria/Acidobacteriota |
| COG0413 COG0414 | NP_592822.1 NP_592821.1 | Pantothenate biosynthesis | Fungib | Gammaproteobacteria |
| COG0132 COG0161 | XP_013025616.1 XP_013025615.1 | Biotin biosynthesis | Fungi | Flavobacteriia |
| COG0223 COG0399 | XP_040634950.1 XP_040634951.1 | Fmt-WecE | Fungi | Gammaproteobacteria |
| COG0693 COG0451 | XP_002492919.1 XP_002492920.1 | WcaG-YajL | Fungi | Bacteroidota |
| COG0771 COG0773 | XP_011122786.1 XP_011122785.1 | Peptidoglycan metabolism | Fungib | Flavobacteriia |
| COG0812 COG0766 | XP_011119914.1 XP_011119913.1 | Peptidoglycan metabolism | Fungib | Saccharibacteria |
| COG3410 COG1694 | XP_016230087.1 XP_016230088.1 | MazG-DUF2075 | Fungi | Actinomycetes |
| COG2072 COG1063 | XP_007731551.1 XP_007731550.1 | Tdh-CzcO | Fungi | Actinomycetes |
| COG2986 COG2987 | XP_014160720.1a | Amino acid metabolism | Sphaeroforma | Betaproteobacteria |
| COG0637 COG1554 | XP_015660045.1a | Carbohydrate transport | Kinetoplastea | Gammaproteobacteria/Bacteroidota |
| COG1940 COG3010 | XP_012897763.1a | Carbohydrate metabolism | Opalinata | Chloroflexota |
| COG0637 COG1554 | XP_821775.1a | Carbohydrate metabolism | Kinetoplastea | Pseudomonadota |
| COG0367 COG1024 | XP_053036155.1a | Secondary metabolites metabolism | Fungi | Actinomycetes/Gammaproteobacteria |
| COG1917 COG1985 | XP_001330840.1a | RibD-QdoI | Trichomonas | Bacillota/Gammaproteobacteria |
| COG0673 COG0399 | XP_024735453.1a | Membrane biogenesis | Fungi | Alphaproteobacteria |
| COG0284 COG0461 | XP_001464543.1a | Pyrimidine biosynthesis | Kinetoplastea | Chloroflexota |
| COG2103 COG2971 | XP_021954652.2a | Peptidoglycan metabolism | Arthropoda | Verrucomicrobiae |
| COG0132 COG0161 | XP_002140155.1a | Biotin biosynthesis | Cryptosporidium | Alphaproteobacteria |
| COG0790 COG1187 | XP_004333747.1 XP_004333748.1 | Pseudouridine biosynthesis | Acanthamoeba | Clostridia/Myxococcia |
| COG0300 COG4221 | XP_004361637.1 XP_004361638.1 | YqjQ-YdfG | Evosea | Bacteroidota |
| COG0147 COG0512 | XP_009038158.1a | Amino acid metabolism | Pelagomonadales | Actinomycetes |
| COG0223 COG0399 | XP_009216736.1 XP_009216737.1 | Fmt-WecE | Fungi | Betaproteobacteria |
| COG0637 COG1554 | XP_010698393.1a | Carbohydrate metabolism | Kinetoplastea | Bacteria |
| COG0235 COG0329 | XP_018148101.1 XP_018148102.1 | AraD-DapA | Fungi | Pseudomonadota |
| COG0132 COG0161 | XP_018905096.1 XP_018905097.1 | Biotin biosynthesis | Bemisia | Alphaproteobacteria |
| COG0596 COG0654 | XP_056561192.1a | MenH-UbiH | Fungi | Actinomycetes |
| COG pair | Representative sequence ID | Possible metabolic function | Recipient eukaryote clade | Donor bacterial clade |
|---|---|---|---|---|
| COG0413 COG0414 | XP_018895596.1a | Pantothenate biosynthesis | Bemisia | Gammaproteobacteria |
| COG0026 COG0041 | XP_638800.1a | Purine biosynthesis | Evoseab | Alphaproteobacteria |
| COG2141 COG1853 | XP_060455211.1a | Pyrimidine degradation | Fungi | Actinomycetes |
| COG1024 COG0318 | XP_031053101.1a | Fatty acid metabolism | Fungi | Actinomycetes |
| COG1539 COG0801 | XP_020610460.1a | Folate metabolism | Cnidaria | Anaerolineae |
| COG0673 COG0399 | XP_008598631.1a | Membrane biogenesis | Fungib | Alphaproteobacteria |
| COG1917 COG1850 | XP_007515483.1a | Photosynthesis | Viridiplantae | Alphaproteobacteria/Acidobacteriota |
| COG0413 COG0414 | NP_592822.1 NP_592821.1 | Pantothenate biosynthesis | Fungib | Gammaproteobacteria |
| COG0132 COG0161 | XP_013025616.1 XP_013025615.1 | Biotin biosynthesis | Fungi | Flavobacteriia |
| COG0223 COG0399 | XP_040634950.1 XP_040634951.1 | Fmt-WecE | Fungi | Gammaproteobacteria |
| COG0693 COG0451 | XP_002492919.1 XP_002492920.1 | WcaG-YajL | Fungi | Bacteroidota |
| COG0771 COG0773 | XP_011122786.1 XP_011122785.1 | Peptidoglycan metabolism | Fungib | Flavobacteriia |
| COG0812 COG0766 | XP_011119914.1 XP_011119913.1 | Peptidoglycan metabolism | Fungib | Saccharibacteria |
| COG3410 COG1694 | XP_016230087.1 XP_016230088.1 | MazG-DUF2075 | Fungi | Actinomycetes |
| COG2072 COG1063 | XP_007731551.1 XP_007731550.1 | Tdh-CzcO | Fungi | Actinomycetes |
| COG2986 COG2987 | XP_014160720.1a | Amino acid metabolism | Sphaeroforma | Betaproteobacteria |
| COG0637 COG1554 | XP_015660045.1a | Carbohydrate transport | Kinetoplastea | Gammaproteobacteria/Bacteroidota |
| COG1940 COG3010 | XP_012897763.1a | Carbohydrate metabolism | Opalinata | Chloroflexota |
| COG0637 COG1554 | XP_821775.1a | Carbohydrate metabolism | Kinetoplastea | Pseudomonadota |
| COG0367 COG1024 | XP_053036155.1a | Secondary metabolites metabolism | Fungi | Actinomycetes/Gammaproteobacteria |
| COG1917 COG1985 | XP_001330840.1a | RibD-QdoI | Trichomonas | Bacillota/Gammaproteobacteria |
| COG0673 COG0399 | XP_024735453.1a | Membrane biogenesis | Fungi | Alphaproteobacteria |
| COG0284 COG0461 | XP_001464543.1a | Pyrimidine biosynthesis | Kinetoplastea | Chloroflexota |
| COG2103 COG2971 | XP_021954652.2a | Peptidoglycan metabolism | Arthropoda | Verrucomicrobiae |
| COG0132 COG0161 | XP_002140155.1a | Biotin biosynthesis | Cryptosporidium | Alphaproteobacteria |
| COG0790 COG1187 | XP_004333747.1 XP_004333748.1 | Pseudouridine biosynthesis | Acanthamoeba | Clostridia/Myxococcia |
| COG0300 COG4221 | XP_004361637.1 XP_004361638.1 | YqjQ-YdfG | Evosea | Bacteroidota |
| COG0147 COG0512 | XP_009038158.1a | Amino acid metabolism | Pelagomonadales | Actinomycetes |
| COG0223 COG0399 | XP_009216736.1 XP_009216737.1 | Fmt-WecE | Fungi | Betaproteobacteria |
| COG0637 COG1554 | XP_010698393.1a | Carbohydrate metabolism | Kinetoplastea | Bacteria |
| COG0235 COG0329 | XP_018148101.1 XP_018148102.1 | AraD-DapA | Fungi | Pseudomonadota |
| COG0132 COG0161 | XP_018905096.1 XP_018905097.1 | Biotin biosynthesis | Bemisia | Alphaproteobacteria |
| COG0596 COG0654 | XP_056561192.1a | MenH-UbiH | Fungi | Actinomycetes |
aFused eukaryote genes encompassing orthologs of both genes in the operon.
bOperons that were highlighted in the main text.
To explore the post-HGT assimilation of operons in eukaryote genomes, we compared their properties to those of bacterial and “native” eukaryotic orthologs. Firstly, we compared lengths of genes that encode single COGs in HGT-acquired operons with homologs found in eukaryotic and bacterial genomes. We found that the gene lengths of the HGT-acquired operons, while similar to those of bacterial orthologs, are significantly shorter than those of native eukaryotic orthologs (Wilcoxon rank-sum test, P < 0.01) (Fig. 2a). Despite these differences in gene lengths, the size of the protein products of genes in HGT-acquired operons did not differ significantly from those of either bacterial or eukaryotic orthologs (Wilcoxon rank-sum test) (Fig. 2b). To explain the differences in gene lengths, we compared the intron densities in HGT-transferred operons with the intron densities in the rest of the genes in the respective contigs. Only 13 of the 46 HGT-transferred operonic genes contained introns, the intron density in these genes was significantly smaller compared to other eukaryotic genes (Fig. 2c) (Wilcoxon rank-sum test, P < 0.01). The probable cause of this difference is that these HGT events occurred relatively recently, so that the HGT-acquired genes have not yet been fully assimilated in the respective eukaryote genomes, that is, have not reached saturation with respect to intron insertion. In line with this possibility, we compared the difference in the base compositions of genes in HGT-transferred operons with those of the respective contigs as a whole, using as a negative control 20 randomly picked adjacent pairs of genes from the same contig for each operon. We found a significantly higher difference in GC content in horizontally transferred, operonized genes (Mann–Whitney U test, P < 0.01), indicating that these genes were not fully ameliorated to the eukaryotic settings.

Evaluation of various genetic properties of HGT-transferred operons. a) Gene length of horizontally transferred genes in operons and respective homologs in bacterial and eukaryotic genomes. b) Protein length of horizontally transferred genes in operons and their respective homologs in bacterial and eukaryotic genomes. c) Intron density in genes of horizontally transferred operons and other genes encoded in their contigs. d) Absolute difference in GC content relatively to other genes encoded in the contig in genes of horizontally transferred operons and randomly selected pairs of genes. e) Intergenic distances between genes of horizontally transferred operons, bacterial homologs, and other eukaryotic genes encoded in their contigs.
Notably, in 20 of the 33 horizontally transferred gene pairs, the 2 genes are fused so that a single protein product is expected to be produced. This gene fusion appears to be an adaptation to the eukaryotic translation system that relies primarily on monocistronic mRNAs. In those cases when the genes are not fused, we found that the intergenic regions between genes in the HGT-acquired operons were significantly longer than those in bacterial operons (Fig. 2d) (Wilcoxon rank-sum test, P < 0.01). Thus, the increase in intergenic distances in eukaryotes is likely partly due to the evolution of new promoters, suggesting that the adaptation of these genes to the eukaryotic setting is in progress (Kominek et al. 2019). In subsequent sections, we focus on more detailed description of four HGT-acquired operons, which include both multigene and fused loci, and for which transcriptomic data sets are publicly available.
Pantothenate Biosynthesis in Schizosaccharomyces
Pantothenate is an essential vitamin that plays a key role in multiple metabolic pathways including biosynthesis of coenzyme A (CoA) (Dansie et al. 2014). Although some organisms acquire pantothenate from the environment, many bacteria, plants, and fungi synthesize this vitamin via a series of biochemical reactions (Rueda-Mejia et al. 2023) (Fig. 3a). Our findings suggest that fungi of the genus Schizosaccharomyces, including the model organism Schizosaccharomyces pombe, have horizontally acquired two essential genes of pantothenate biosynthesis, panB and panC, that encode 3-methyl-2-oxobutanoate hydroxymethyltransferase and 4-phosphopantoate beta-alanine ligase, respectively, from bacteria of the order Enterobacterales (Fig. 3b). To confirm their transcriptional activity, we mapped the publicly available transcriptome to this region and examined its coverage. The RNA-Seq data confirmed that both panB and panC are actively transcribed with TPM values of 12.96 and 42.97, respectively (Fig. 3c), supporting their integration into the pantothenate metabolism in Schizosaccharomyces yeasts.
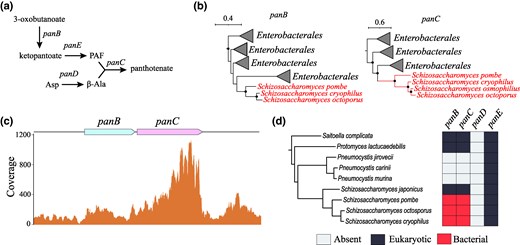
Characterization of pantothenate biosynthesis operon in Schizosaccharomyces. a) Role of panB-panD genes in the biosynthesis of pantothenate. b) Collapsed phylogenetic trees of PanB and PanC genes found in Schizosaccharomyces. Tree scale is in substitutions per site, and nodes with the support of at least 70% are denoted with black dots. c) Coverage plots of panB and panC genes based on S. pombe transcriptomic data. d) Phylogenetic profiling of panB-panD genes in Schizosaccharomyces and its sister clades.
Is pantothenate biosynthesis in Schizosaccharomyces a genuine novel trait? To address this question, we conducted phylogenetic profiling of panB, panC, panD, and panE in Schizosaccharomyces and sister clades using a previously published species tree (Li et al. 2021) (Fig. 3d). The results of this analysis suggest that ancestral eukaryotic panB and panC genes were present in the last common ancestor of Schizosaccharomyces but were later replaced by the bacterial versions in a specific clade. The panE gene, which encodes 2-dehydropantoate 2-reductase, an essential intermediary enzyme between panB and panD, was inherited vertically, indicating its eukaryotic origin. Furthermore, Schizosaccharomyces yeasts completely lack panD, despite its colocalization in the same operon with panB and panC in bacterial genomes, including in the likely donor clade of Enterobacterales. The absence of PanD suggests that these yeasts do not use the pathway of pantothenate biosynthesis from β-alanine (Fig. 3a) and that panD either was lost following HGT or was never transferred at all.
Peptidoglycan Biosynthesis in Orbiliaceae Fungi
Peptidoglycan (also known as murein) is the main component of bacterial cell walls, protecting them against environmental challenges. Thus, peptidoglycan biosynthesis is essential for bacterial survival and its biosynthesis involves multiple enzymatic reactions across three subcellular compartments (Barreteau et al. 2008) (Fig. 4a). Although fungal cells are also surrounded by a cell wall, it typically consists of chitin—a rigid, long-chain polymer of GlcNAc units (Merzendorfer 2011). Consequently, fungi generally do not need and indeed lack genes for peptidoglycan biosynthesis. Unexpectedly, however, we identified horizontally acquired peptidoglycan biosynthesis genes in species from the Orbiliaceae family, a group of saprobic sac fungi, some of which are notorious for hunting and trapping nematodes (Yang et al. 2020; Kuo et al. 2024). In particular, we found that multiple species of Orbiliaceae encode murA-murB and murC-murD gene pairs. These gene pairs are not colocalized with each other and instead are encoded on different contigs. To determine whether murA-murB and murC-murD pairs were originally part of the same operon that later was fragmented during evolution, or if they were independently transferred, we first examined their phylogenetic congruency (Fig. 4b). The results show that murA-murB were apparently horizontally acquired from Candidatus Saccharibacteria, whereas murC-murD were transferred from Flavobacteriia, supporting a scenario of independent acquisition of the two gene pairs. Furthermore, by conducting phylogenetic profiling of other peptidoglycan biosynthesis genes, we found that multiple Orbiliaceae species also encode murE gene in a separate genomic region. Additional phylogenetic analysis suggests that murE was acquired from the Abditibacteriota/Armatimonadota clade, indicating a different origin than either murA-murB or murC-murD. We assessed the transcriptional activity of peptidoglycan biosynthesis genes by analyzing RNA-Seq data from Orbilia oligospora (Fig. 4c) and identified RNA reads that clearly mapped to murA, murB, murC, and murD genes, indicating their active expression, though at markedly different levels, with TPM levels of 44.71, 4.93, 6.76, and 15.18, respectively. However, we did not find any coverage for murE gene indicating that it is either nonfunctional or is expressed only under a limited set of conditions.
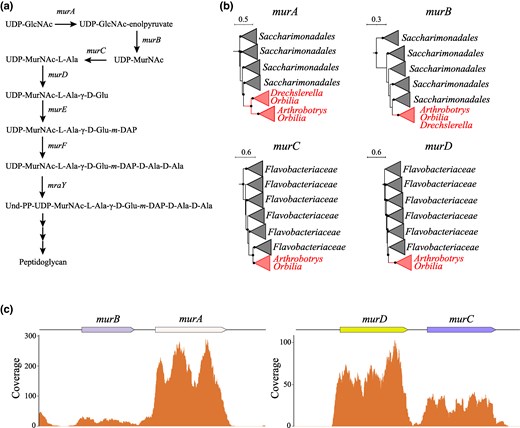
Characterization of peptidoglycan biosynthesis operon in nematode-trapping Orbiliaceae fungi. a) Role of murA-murF and mraY genes in the biosynthesis of peptidoglycan. b) Collapsed phylogenetic trees of MurA-MurD genes found in Orbiliaceae. Tree scale is in substitutions per site, and nodes with the support of at least 70% are denoted with black dots. c) Coverage plots of murA-murD genes based on Orbiliaceae oligospora transcriptomic data.
The functional role of peptidoglycan biosynthesis in Orbiliaceae remains unknown. A previous study identified the presence of horizontally transferred peptidoglycan biosynthesis genes in the genomes of Aspergillus fungi, likely originating from Firmicutes (Marcet-Houben and Gabaldon 2010). Thus, it is likely that peptidoglycan biosynthesis can benefit some clades of fungi via currently unknown molecular mechanisms. One plausible hypothesis can be that Orbiliaceae species make peptidoglycan to supply it to their endosymbionts. Indeed, multiple endosymbiotic bacteria are involved in the life cycle of the nematode-trapping Arthrobotrys musiformis (Zheng et al. 2024). Alternatively, Orbiliaceae peptidoglycan might stimulate various metabolic and physiological reactions of the fungi themselves. For instance, bacterial peptidoglycan-like molecules are known to promote hyphal growth in the opportunistic fungal pathogen Candida albicans (Xu et al. 2008).
Purine Biosynthesis in Amoeba
Purine nucleotides can be produced by two distinct metabolic routes, the salvage pathway, which recycles recovered nucleosides and bases, and the de novo pathway, which synthesizes purines from simple molecules. The latter pathway requires ten sequential enzymatic reactions to generate inosine monophosphate (Zhang et al. 2008). We observed that Eumycetozoa and Variosea amoeba encode purK and purE genes that appear to have been horizontally acquired from Alphaproteobacteria (Fig. 5a). In these amoebas, purK and purE are fused into a single hybrid gene that also encompasses purC (Fig. 5b). However, a detailed phylogenetic analysis of the purC gene in Eumycetozoa does not support its horizontal acquisition, as it is not nested within bacterial clades (Fig. 5a). Thus, a plausible scenario is that following the horizontal transfer of bacterial purK and purE genes, they fused with purC into a single coding sequence. To determine whether the purine biosynthesis pathway was functional in Eumycetozoa, we mapped transcriptome reads to the purC-purK-purE gene. All three genes were fully covered by reads, confirming their activity with an overall TPM level of 87.86 (Fig. 5c). Additionally, we used COG profiles to search for other genes necessary for de novo purine biosynthesis. Our findings indicate that Eumycetozoa species generally encode all necessary genes for this pathway, supporting its functionality. By contrast, none of the purine biosynthesis genes were found in Mastigamoebida clade that also belongs to the phylum Evosea, raising the possibility that other purine biosynthesis genes in Eumycetozoa were horizontally transferred as well. Using our conservative phylogenetic pipeline, we found that purH gene indeed is of bacterial origin, with Chloroflexota identified as the putative donor clade. Consequently, our data indicate that purine biosynthesis pathway in Eumycetozoa was assembled through multiple, independent HGT events (Fig. 5c).
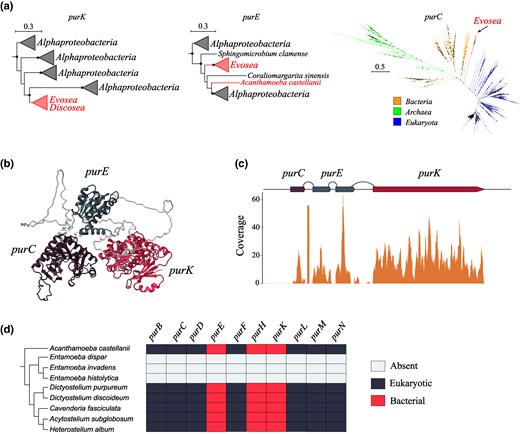
Characterization of purine biosynthesis operon in Amoeba. a) Collapsed phylogenetic trees of PurK, PurE, and PurC genes found in Amoeba. Tree scale is in substitutions per site, and nodes with the support of at least 70% are denoted with black dots. b) Tertiary structure of PurC-PurE-PurK protein predicted by AlphaFold. c) Coverage plots of purC-purE-purK gene based on D. discoideum transcriptomic data. Arcs denote introns, and solid blocks denote exons. d) Phylogenetic profiling of genes involved in de novo purine biosynthesis in various Amoeba species.
Membrane Biogenesis in Multiple Fungal Lineages
Another putative operon that was fused into a single gene in eukaryotes after HGT from bacteria encodes dehydrogenase MviM and transaminase WecE. WecE plays a role in the biosynthesis of outer membrane polysaccharides in enterobacteria (Hwang et al. 2004), whereas the function of MviM is not fully understood. To gain a better understanding of the putative function of the mviM-wecE operon, we analyzed their genomic neighborhoods across a diverse range of bacterial species (Fig. 6a). Our findings indicate that, although mviM and wecE are not always directly adjacent, they tend to be encoded within the same genomic region. This region is generally enriched in other genes associated with the biosynthesis of outer membrane sugars, suggesting that mviM and wecE function in concert with these genes in the outer membrane biogenesis. Phylogenetic analyses suggest that mviM-wecE were horizontally transferred from Alphaproteobacteria to Ascomycota but were only retained in the genera Patellaria and Beauveria (Fig. 6b). Following the HGT, mviM and wecE genes were fused into a single gene encoding a two-domain protein (Fig. 6c). Furthermore, in Beauveria, the mviM-wecE gene is colocalized with lpxA, another important gene for the outer membrane biogenesis in bacteria. The HGT of lpxA likely occurred later in the evolution of fungi because this gene was found only in Sordariomycetes, a separate class of Ascomycota (Fig. 6b). The transcriptomic data from Beauveria bassiana indicate that both genes are actively expressed (TPM value of 63.7 and 63.9 for lpxA and mviM-wecE, respectively), suggesting they play a role in membrane biogenesis of this fungus (Fig. 6d).

Characterization of mviM-wecE fused gene in fungi. a) Genetic neighborhoods of mviM and wecE genes in five bacterial species. b) Collapsed phylogenetic trees of MviM, WecE, and LpxA genes found in two fungi species. Tree scale is in substitutions per site, and nodes with the support of at least 70% are denoted with black dots. c) Tertiary structure of mviM-wecE gene predicted by AlphaFold. d) Coverage plots of lpxA and mviM-wecE genes based on B. bassiana transcriptomic data.
Discussion
In prokaryotes, genes encoding functionally linked proteins, in particular, enzymes responsible for different steps of the same biochemical pathway, are typically clustered within the same operon which is translated as a single mRNA, providing for coordinated expression of these proteins. Eukaryotes normally lack operons although operon-like organization of some functionally connected genes has been discovered in nematodes and in trypanosomatids (Bringaud et al. 1998; Pettitt et al. 2014). Given the fundamental differences in gene expression mechanisms, in general, there seems to be no reason to expect that eukaryotes would retain operons captured from bacteria via HGT for extended evolutionary spans. Nevertheless, we were interested to explore whether some cases of operonic gene organization might persist in some eukaryotic lineages.
The extensive comparative analysis of bacterial and eukaryote genomes performed in this work revealed only 33 distinct cases of identifiable HGT of operons or their fragments. How rare are these events compared to the HGT of individual genes? To address this question, we predicted HGT events for 49 COGs that were represented in the transferred operons identified here. We identified 309 likely distinct HGT events within this limited set of COGs (supplementary table S3, Supplementary Material online). Thus, fixation of HGT-acquired operons in eukaryotes is likely to occur at a rate at least an order of magnitude lower than fixation of individually transferred genes (some of which likely originate from operons split after HGT). This difference is not unexpected and might be explained by the challenges associated with the post-HGT assimilation of operons. Unlike single horizontally transferred genes, operons consist of two or more genes, each of which must acquire new promoters and other eukaryotic regulatory signals to ensure proper transcription, mRNA processing, and translation. In particular, because genes in the same operon are closely spaced in prokaryote genomes, insertion of a noncoding spacer of sufficient length between these genes is required to accommodate the necessary regulatory signals (Kominek et al. 2019). Indeed, the region between panB and panC genes in S. pombe contains CCAAT motif (McDowall et al. 2015), one of the most common promoters in eukaryotes (Kovacs et al. 2003). Furthermore, on multiple occasions, these challenges have been overcome through fusion of the genes comprising an operon into a single chimeric gene as also observed previously for horizontally acquired thiM and thiE in Wickerhamiella/Starmerella ascomycete fungi (Goncalves and Goncalves 2019). The advantages of such gene mergers are obvious with respect to maintaining the stoichiometry and coregulation of functionally linked genes and possibly providing for metabolite channeling within the emerging multifunctional enzymes. In contrast, the advantages of keeping the operon organization without fusion in eukaryotes, if any, are unclear. It cannot be ruled out that such gene arrays in eukaryotes genomes are nonadaptive heritage of HGT that persists neutrally until the respective genes are either fused or separated by processes of genome rearrangement that occur frequently during the evolution of eukaryote genomes (Seoighe et al. 2000; Crombach and Hogeweg 2007).
In terms of biology, a strong trend among the horizontally transferred operons is involvement in secondary metabolism pathways. Conceivably, these genes are relatively easily accumulated as they do not perturb basic metabolic networks of the recipient organism while conferring additional, advantageous biochemical versatility.
Indeed, the horizontally transferred operonic gene arrays in eukaryote genomes described here are likely to be relatively recent acquisitions as indicated, firstly, by their presence in relatively narrow clades of eukaryotes, and secondly, by the lack or paucity of introns in these genes compared to the genomic average in the respective organisms. Insertion of introns into eukaryotic genes appears to be under selection to enhance gene expression and nucleocytoplasmic transport of mRNA (Le Hir et al. 2003; Shaul 2017; Li et al. 2022); hence, the genes horizontally acquired from bacteria most likely are on the path to equilibration with the rest of the genes. For comparison, plant genes transferred from chloroplasts carry only faint signals of ongoing intron insertion, whereas genes of mitochondrial origin appear to be saturated with introns (Basu et al. 2008).
Altogether, our findings indicate that capture of bacterial operons by eukaryotes via HGT followed by persistence of the operonic organization is rare, but also that such events are highly lineage specific. Therefore, high-quality sequencing of eukaryote genomes, primarily those of fungi and protists, will likely lead to the discovery of additional instances of operon capture providing for exploration of evolution of gene regulation at the prokaryote–eukaryote interface.
Methods
Identification of Operonic Gene Pairs
To identify gene pairs that belong to operons, taxonomic information of 47,545 prokaryotic genomes from GenBank (Prok2311) was parsed using NCBI Taxonomy (Schoch et al. 2020) (accessed in November 2023). One genome was selected for each taxonomic level at the “genus rank,” prioritizing “complete,” “reference,” and “representative” genomes, resulting in a total of 1,805 genomes. Each genome was annotated using profiles from the COG database (Galperin et al. 2021). Because a single gene can contain multiple domains corresponding to different COGs, only COG pairs that mostly encoded in distinct genes in more than 80% of instances were included in the analysis. Then, each gene with a COG annotation that is adjacent to another codirected gene with a COG annotation was identified, extracted, and counted. To evaluate nonrandom coadjacency of COG pairs, the binomial test was applied. The expected coadjacency rate for each COG pair was defined as follows:
where is the frequency of COG A, is the frequency of COG B, and N is the total number of genes in the data set with a COG annotation. The obtained P-values were adjusted for multiple testing using the Benjamini–Hochberg correction (Benjamini and Hochberg 1995). Moreover, from the COG pairs that passed the test, only 45,396 pairs found in at least five genomes were retained, forming an “operon list.”
Information about COG categories and their metabolic pathways was extracted from the COG database (Galperin et al. 2021). The assortativity analyses were conducted using igraph package v1.3.5 in R software (Csardi and Nepusz 2006). Ambiguous COG categories such as S, function unknown, and R, general function prediction only, were removed during this analysis.
Identification of COG Pairs in Eukaryote Genomes
The 1,845 “representative” eukaryote genomes and their annotations were downloaded from the RefSeq database (O'Leary et al. 2016) (accessed in March 2024). To reduce possible contamination from short genomic fragments, only genes that are encoded on contigs larger than 100 kb were retained (Shen et al. 2018). Moreover, to reduce redundancy in genes encoding multiple isoforms, only one isoform per gene was randomly retained, resulting in a total of 30,058,376 protein-coding genes. COG profiles were used in a PSIBLAST search (e-value < 0.001) (Altschul et al. 1997) to assign COGs, resulting in the assignment of COGs to 11,141,238 genes. Consequently, adjacent and codirected genes with COG assignments were clustered in pairs, retaining only the ones that are found in “operon list.” Furthermore, genes that were found in contigs of “mitochondrial,” “chloroplast,” or “plastid’ origin were filtered out, resulting in the candidate set of 229,044 genes.
To further filter the candidate set of 229,044 genes, the previously described approach was used (Shen et al. 2018; Li et al. 2022). Briefly, retained genes were initially clustered using mmseqs2 with a minimum identity cutoff of 70% (Steinegger and Soding 2017), resulting in 80,719 representative sequences. Additionally, a BLAST database was prepared that contained all nonredundant proteins from 47,545 prokaryote genomes from GenBank and 1,845 “representative” eukaryote genomes. Each representative sequence was then run against this combined database, as well as against a database containing only proteins from the 1,845 eukaryote genomes (maximum target sequences = 500, e-value < 10E-10). Additionally, the taxonomic information for each hit was obtained using NCBI Taxonomy and ETE toolkit (Huerta-Cepas et al. 2016). Then, the Alien Index (AI) was calculated for each representative sequence, based on three values—bbhO (bitscore to the best hit in the donor lineage), bbhG (bitscore to the best hit in the group lineage [Eukarya], but outside of the recipient lineage), and maxB (bitscore of the query to itself):
Additionally, the proportion of bacterial species within the top 500 hits for each query was measured (outg_pct). One thousand and six hundred ten sequences with AI > 0 and outg_pct > 0.9 were retained, and all sequences from the respective mmseqs clusters were added to the final candidate set. Finally, sequences in the final candidate set were mapped back to their COG pairs, retaining only those COG pairs in which both sequences were included in the final candidate set. Sequences were then dereplicated at 70% of minimum sequence identity via mmseqs2 (Steinegger and Soding 2017), and the final candidate set was obtained consisting of 181 multigene loci (COGs are encoded in two distinct genes) and 201 fused loci (COGs are encoded in the same gene).
Detection of Horizontally Transferred Operons by Phylogenetic Analyses
To identify horizontally acquired operons, all COG coordinates from each sequence in the final candidate set were used in a BLASTP search (e-value < 10E-6) against the “ClusteredNR” database (Altschul et al. 1990). The search was limited to a maximum of 900 hits across all taxa, 500 hits for eukaryotes, and 100 hits for archaea (“selected hits”). Additionally, a separate BLASTP search using the same COG coordinates was performed against “ClusteredNR” database (maximum target sequences = 500, e-value <10E-6) to create a profile (“profile hits”). The median length of the “profile hits” was calculated, and only sequences within 1.4 times the median were selected and aligned using Muscle5 (Edgar 2022). The resulting profiles were used to run against “selected hits” to extract extended footprints. The extended footprints were aligned using Muscle5 (Edgar 2022), and positions with up to 90% of gaps were trimmed using ClipKIT (Steenwyk et al. 2020). The phylogenetic trees were reconstructed using IQ-Tree v2.3.4, and best substitution model was selected using ModelFinder (Kalyaanamoorthy et al. 2017) from the set of LG (Le and Gascuel 2008), WAG (Whelan and Goldman 2001), Q.pfam (Minh et al. 2021), and NQ.pfam (Dang et al. 2022). The support values for internal nodes were calculated using ultrafast bootstrap method with a hill-climbing nearest neighbor interchange search (Hoang et al. 2018). Each tree was examined in iTOL v6 (Letunic and Bork 2024), and the sequence was considered to be horizontally acquired from Bacteria if it satisfied all of the following criteria:
The sequence groups with other bacterial sequences with a bootstrap support of at least 60%.
No evidence of contamination, as checked using Conterminator (Steinegger and Salzberg 2020).
At least five out of the six genes in the immediate neighborhood should be of the eukaryotic origin, based on AI and out_pct values.
For any candidates that have bootstrap support less than 95%, the ratio of branch lengths between the putatively horizontally transferred clade and its bacterial sister clades should not exceed a factor of 2.
In cases where candidate genes have a bootstrap support less than 90% and a separate clade of eukaryotic homologs is present on the tree, ELW tests against constrained trees should not exceed a threshold of 0.7 (Strimmer and Rambaut 2002).
After manual curation of phylogenetic trees, 33 operons, in which both COGs were horizontally transferred from bacteria, were retained.
Detection of Horizontally Transferred Single Genes
Genes with the same COG assignments as those from horizontally transferred operons were extracted from eukaryotic genomes, resulting in 328,809 genes. The proteins encoded by these genes were further clustered using mmseqs2 at 60% of identity threshold (Steinegger and Soding 2017), yielding 55,691 representative protein sequences. Further search for horizontally transferred genes, including phylogenetic analyses, was performed as described above.
Transcriptome Analysis
Reads from the RNA-Seq data for S. pombe (SRR25309793), Dictyostelium discoideum (SRR032338), B. bassiana (SRR31347598), and O. oligospora (SRR30111761) were downloaded from the Sequence Read Archive database (Katz et al. 2022). Adapters were trimmed using Trim Galore v 0.6.10, and quality of the reads was assessed using FastQC. Reads were mapped to reference genomes using HISAT2 with the “sensitive” mode (Kim et al. 2019), and data were parsed in JBrowse2 (Diesh et al. 2023). TPM values were calculated using TPMCalculator (Vera Alvarez et al. 2019).
Supplementary Material
Supplementary material is available at Genome Biology and Evolution online.
Acknowledgments
The authors thank Koonin group members for their helpful discussions. The authors’ research is supported by the Intramural Research Program of the National Institutes of Health (National Library of Medicine).
Author Contributions
R.K. and E.V.K. initiated the project; R.K. performed the research; R.K., Y.I.W., and E.V.K. analyzed the data; R.K. and E.V.K. wrote the manuscript that was read, edited, and approved by all authors.
Data Availability
Genomes that were used in this study are publicly available, and their accession IDs can be found in supplementary tables S1 and S2, Supplementary Material online. Derived data, including coadjacent COG pairs, sequence alignments, phylogenetic trees, TPM calculations, and python scripts, are deposited in Zenodo under DOI: https://doi.org/10.5281/zenodo.14957726.
Literature Cited
Author notes
Conflict of Interest: The authors declare no competing interests.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}