





Abstract
The Antarctic sea urchin Sterechinus neumayeri (Echinoida; Echinidae) is routinely used as a model organism for Antarctic biology. Here, we present a high-quality genome of S. neumayeri. This chromosomal-level assembly was generated using PacBio long-read sequencing and Hi-C chromatin conformation capture sequencing. This 885.3-Mb assembly exhibits high contiguity with a scaffold length N50 of 36.7 Mb assembled into 20 chromosomal length scaffolds. These putative chromosomes exhibit a high degree of synteny compared to other sea urchin models. We used transcript evidence gene modeling combined with sequence homology to identify 21,638 gene models that capture 97.4% of BUSCO orthologs. Among these, we were able to identify and annotate conserved developmental gene regulatory network orthologs, positioning S. neumayeri as a tractable model for comparative studies on evolution and development.
The Antarctic sea urchin Sterechinus neumayeri has long served as a model system for Antarctic developmental biology, climate change, and evolution. Here, we present a chromosomal-level genome assembly as a resource for this important model system to augment the study of Antarctic systems.
Introduction
Sterechinus neumayeri (Meissner 1900) is a circumpolar, Antarctic regular sea urchin and has long been a model system for Antarctic biology research (Fig. 1a and b; Bosch et al. 1987; Brey et al. 1995; Brockington et al. 2001; Pearse and Giese 1966; Marsh et al. 2001; Lee et al. 2004; Yu et al. 2013; Kapsenberg and Hofmann 2014; Dilly et al. 2015). This species is common and often abundant on the continental shelf notably in regions close to Antarctic research stations including McMurdo Sound (Ross Sea) and along the Antarctic Peninsula (Fig. 1b). This sea urchin has free-swimming embryos and pluteus larvae similar to those found in other indirect urchins, a characteristic that makes this sea urchin an excellent model for comparison between temperate and Antarctic species (McClintock 1994). As a member of Echinidae, S. neumayeri last shared a common ancestor with the well-established developmental model systems Strongylocentrotus purpuratus and Lytechinus variegatus about 45 million years ago (Láruson 2017) and diverged from another model species, Paracentrotus lividus, about 33 MYA (Lee et al. 2004), which roughly correlated to the formation of the Antarctic Circumpolar Current that climatically isolated the Southern Ocean (Lawver and Gahagan 2003; Halanych and Mahon 2018). S. neumayeri is a sister lineage to the other four recognized Sterechinus species, some of which occur outside of Antarctic waters (Saucède et al. 2015). Nonetheless, S. neumayeri has evolved in the Antarctic environment for several millions of years. Investigations of genetic differences that underlie adaptive evolution in Antarctic waters remain understudied.
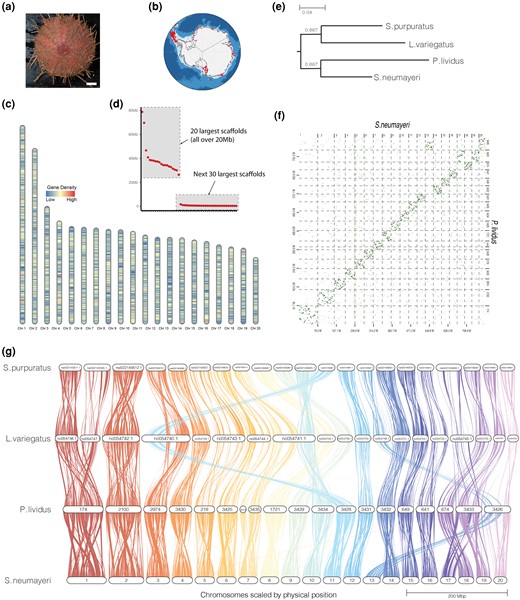
a) An adult S. neumayeri, photo credit: Kyle Donnelly, scale bar = 1 cm. b) Recorded observations of S. neumayeri in Antarctica. Data from Fabri-Ruiz et al. (2017). c) Ideogram demonstrating gene density across the 20 largest scaffolds. d) Scaffold sizes of the 50 largest scaffolds of the S. neumayeri genome assembly. Twenty chromosome-size scaffolds are observed over 20 Mb. e) Rooted species tree of the four model species inferred from gene orthogroups. f) Dot plot of whole-genome alignment between S. neumayeri and P. lividus. g) Synteny analysis of P. lividus, S. neumayeri, L. variegatus, and S. purpuratus. Sterechinus neumayeri scaffolds are named in order of size. Paracentrotus lividus scaffolds are named as they appear in the assembly in Marlétaz et al. (2023). Lytechinus variegatus and P. lividus scaffolds are named as they appear on NCBI.
Previous work on S. neumayeri focused on timing of life history stages as the development of S. neumayeri is much longer than its temperate counterparts (Pearse 1969; Bosch et al. 1987; Shilling and Manahan 1994; Stanwell-Smith and Peck 1998; Marsh et al. 2001; Pace et al. 2010). For example, when raised at their natural environmental temperature, S. neumayeri takes, on average, 6 to 7 days to reach early gastrula, whereas the temperate species, S. purpuratus, takes just 24 h (Détrée et al. 2023; Massri et al. 2023). Interestingly, development and life history stages track well between S. neumayeri and temperate sea urchins (e.g. S. purpuratus and L. variegatus) allowing for comparative studies. Such studies are augmented by the fact that temperate urchin species have been developed as exceptional model systems for studying gene regulatory networks (GRNs) and early embryonic processes (selected examples from many studies include Ransick and Davidson 1998; Davidson et al. 2002; Range et al. 2008; Range and Wei 2016; Warner et al. 2016; Erkenbrack et al. 2018). Furthermore, among Antarctic marine invertebrates, S. neumayeri is arguably the best studied developmentally (Bosch et al. 1987; Marsh et al. 2001; Pace et al. 2010; Kapsenberg and Hofmann 2014; Dilly et al. 2015), and it possesses many features that have made other sea urchin models successful, including transparent embryos and large clutches of eggs (Foltz et al. 2004). Thus, S. neumayeri presents a unique opportunity to study adaptive changes in GRNs that drive developmental timing in Antarctic benthic invertebrate species. The ability to compare Antarctic and temperate urchins allows other investigations of organismal biology, such as physiology associated with ocean acidification (Byrne et al. 2013; Sewell et al. 2013), to be examined in depth.
To support comparative genomic studies in S. neumayeri, we sequenced and annotated a high-quality whole-genome sequence for S. neumayeri, which we describe here. This genome follows several recently published high-contiguity sea urchin genomes (Davidson et al. 2020; Ketchum et al. 2022; Marlétaz et al. 2023). The S. neumayeri genome (885 Mb) is comparable to other published model sea urchin species in terms of chromosome number (putatively 20) and chromosomal organization. This high-quality S. neumayeri genome will enable comparative and functional studies at the level of genomic mechanisms and our efforts advance the understanding of adaptation to the Antarctic environment, as well as organismal responses to a rapidly changing polar marine climate.
Results and Discussion
Sterechinus neumayeri Genome Assembly and Annotation
The S. neumayeri genome assembly totals 885.29 Mb assembled into 957 scaffolds (Table 1; scaffold N50 36.75 Mb). We identified 20 chromosome-size scaffolds longer than 10 Mb, which collectively contain 88.7% of all assembled bases (Fig. 1c and d). The assembly exhibits an estimated 46.93% proportion of repetitive elements and 37.56% GC content, which is comparable to other sea urchin genome assemblies (Davidson et al. 2020; Ketchum et al. 2022; Davidson et al. 2023; Marlétaz et al. 2023). We assessed the completeness of the assembly using BUSCO, and 97.9% of BUSCO orthologs were identified in the assembly (Table 1; 91.5 single copy, 6.4 duplication, database: metazoa_odb10). When we performed the same analysis using just the 20 chromosome-size scaffolds, we recovered the same percentage of BUSCO orthologs of 97.9%, although with a slightly lower number of duplicated orthologs (91.8% single copy, 6.1% duplication) indicating that a small proportion of the unplaced scaffolds may represent duplicate haplotigs or other sequencing misassemblies and that the 20 chromosome-size scaffolds are largely complete.
Sterechinus neumayeri genome assembly statistics
| Assembly | |
|---|---|
| Size | 885,292,883 |
| # Scaffolds | 957 |
| Scaffold N50 | 36,746,519 |
| Largest scaffold | 78,747,131 |
| # Scaffolds > 10 Mb | 20 |
| % GC | 37.56 |
| % N | 0.0000013 |
| BUSCO % | metazoa_odb10 |
| Complete | 97.9 |
| Single copy | 91.5 |
| Duplicated | 6.4 |
| Fragmented | 0.4 |
| Missing | 1.7 |
| Annotation | |
| # Protein-coding genes | 21,638 |
| % Repeats | 46.93 |
| BUSCO % | metazoa_odb10 |
| Complete | 97.4 |
| Single copy | 89.1 |
| Duplicated | 8.3 |
| Fragmented | 0.8 |
| Missing | 1.8 |
| Assembly | |
|---|---|
| Size | 885,292,883 |
| # Scaffolds | 957 |
| Scaffold N50 | 36,746,519 |
| Largest scaffold | 78,747,131 |
| # Scaffolds > 10 Mb | 20 |
| % GC | 37.56 |
| % N | 0.0000013 |
| BUSCO % | metazoa_odb10 |
| Complete | 97.9 |
| Single copy | 91.5 |
| Duplicated | 6.4 |
| Fragmented | 0.4 |
| Missing | 1.7 |
| Annotation | |
| # Protein-coding genes | 21,638 |
| % Repeats | 46.93 |
| BUSCO % | metazoa_odb10 |
| Complete | 97.4 |
| Single copy | 89.1 |
| Duplicated | 8.3 |
| Fragmented | 0.8 |
| Missing | 1.8 |
Sterechinus neumayeri genome assembly statistics
| Assembly | |
|---|---|
| Size | 885,292,883 |
| # Scaffolds | 957 |
| Scaffold N50 | 36,746,519 |
| Largest scaffold | 78,747,131 |
| # Scaffolds > 10 Mb | 20 |
| % GC | 37.56 |
| % N | 0.0000013 |
| BUSCO % | metazoa_odb10 |
| Complete | 97.9 |
| Single copy | 91.5 |
| Duplicated | 6.4 |
| Fragmented | 0.4 |
| Missing | 1.7 |
| Annotation | |
| # Protein-coding genes | 21,638 |
| % Repeats | 46.93 |
| BUSCO % | metazoa_odb10 |
| Complete | 97.4 |
| Single copy | 89.1 |
| Duplicated | 8.3 |
| Fragmented | 0.8 |
| Missing | 1.8 |
| Assembly | |
|---|---|
| Size | 885,292,883 |
| # Scaffolds | 957 |
| Scaffold N50 | 36,746,519 |
| Largest scaffold | 78,747,131 |
| # Scaffolds > 10 Mb | 20 |
| % GC | 37.56 |
| % N | 0.0000013 |
| BUSCO % | metazoa_odb10 |
| Complete | 97.9 |
| Single copy | 91.5 |
| Duplicated | 6.4 |
| Fragmented | 0.4 |
| Missing | 1.7 |
| Annotation | |
| # Protein-coding genes | 21,638 |
| % Repeats | 46.93 |
| BUSCO % | metazoa_odb10 |
| Complete | 97.4 |
| Single copy | 89.1 |
| Duplicated | 8.3 |
| Fragmented | 0.8 |
| Missing | 1.8 |
The BRAKER3 pipeline (Gabriel et al. 2024) was used to predict protein-coding genes. Protein models from several urchin species including L. variegatus, Lytechinus pictus, and S. purpuratus were used as training gene sets. For evidence, we used a combination of RNA-sequencing (RNA-seq) read available from the National Center of Biotechnology Information (NCBI) at the National Institutes of Health sequence read archive (PRJNA376030, PRJNA252503, and PRJEB33967) and an in-house RNA-seq data set spanning the major stages of embryogenesis (see Materials and Methods section: RNA-seq). These analyses identified 21,638 unique protein-coding genes, which include 97.4% of BUSCO single-copy orthologs (Table 1; metazoa_odb10).
Synteny Analyses
To identify homologous chromosomes between S. neumayeri and other laboratory model sea urchin species, we compared gene orthology and gene order between S. neumayeri and the three temperate species L. variegatus, S. purpuratus, and P. lividus (Fig. 1e to g). This analysis identified orthologous putative chromosomes among each of the species. A species tree was inferred from the orthogroup analysis and recapitulates known phylogenetic positions of the study species (Fig. 1e; Dilly et al. 2015; Koch et al. 2018). Of the temperate laboratory model species, S. neumayeri is most closely related to P. lividus and consistent with the S. neumayeri genome, exhibits a high degree of macrosynteny with P. lividus (Fig. 1f). Our analysis notes the potential fusion of S. neumayeri chromosomes Sn13 and Sn20 into a single chromosome in P. lividus (Fig. 1g). We also observed a potential chromosome split of Sn 7 between S. neumayeri and P. lividus, with a portion of Sn 7 mapping to an unplaced scaffold in the P. lividus genome. As each of these species is an established model system, functional genomic comparisons, particularly in regard to developmental GRNs, have the potential to yield novel insights into Antarctic echinoderm biology and evolution.
Curation of Developmental GRN Annotation
To enable comparisons with established laboratory model sea urchins, we manually curated a list of 110 regulatory “network” genes known to be important to the early development of sea urchin embryos. These genes, which include transcription factors, developmental signaling pathway genes, and effectors, form the basis of the well-characterized GRN of cell type specification in urchins (Davidson et al. 2002; Peter and Davidson 2011). To identify orthologs of these network genes in S. neumayeri, we used orthofinder2 (Emms and Kelly 2019) to categorize orthologs between S. neumayeri, L. variegatus, P. lividus, and S. purpuratus (supplementary file S1, Supplementary Material online). We succeeded in identifying orthologs of all but three developmental GRN genes in S. neumayeri. These results reflect the “completeness” of our annotation and will support future comparative studies between these models.
This genome assembly and annotation will be a valuable resource for future studies on S. neumayeri and other polar echinoderms/invertebrates. The annotation herein identified orthologs for the majority of developmental regulatory network genes across four major model urchin species thereby enabling evolutionary comparisons between these model systems. With the recent publication of several high-contiguity sea urchin genomes, we expect S. neumayeri will play a central role in comparative genomics studies and accelerate studies of Antarctic invertebrate biology.
Materials and Methods
Tissue Collection
Sterechinus neumayeri was collected by Blake trawl (a.k.a., beam trawl) from a depth of 294 m at latitude −64 26.182 and longitude −55 49.943 on November 16, 2020 aboard the RVIB Nathaniel B. Palmer cruise NBP 20-10. This individual used herein was cataloged as Ec410.3c (Halanych lab cataloging system). At the time of collection, select intact urchins were placed in a cold room aboard the ship and later injected with 0.5 M KCl to induce spawning. Sperm was collected, flash frozen in liquid nitrogen, and stored at −80 °C. All animal tissues were collected in adherence to the Nagoya protocol.
PacBio and Dovetail Sequencing and Assembly
Frozen S. neumayeri sperm from one individual was sent to the Arizona Genomics Institute for long-read sequencing on a PacBio SEQUEL II system using CCS chemistry with SMRT cells. Returned reads were assembled with Hifiasm-0.15-1 (Cheng et al. 2021). This resulted in an 885-Mb assembly with a N50 of 1.01 Mb and 2,071 contigs.
Dovetail Genomics (now Cantata Bio) was contracted for Hi-C chromatin conformation capture sequencing using sperm and the hifiasm assembly. Dovetail performed the Omni-C library preparation, sequencing, and HiRise assembly as previously described (Warner et al. 2021).
RNA-seq
To aid in evidence-based gene annotation, we performed an embryonic RNA-seq time-series analysis. Gravid S. neumayeri was spawned by injecting 0.5 M KCl. Larval cultures were set up by combining gametes from a single male and female and proportioning embryos in 6-well cell culture plates, which were placed in a shipboard standing incubator maintained at a temperature between 0 and 1.5 °C. Embryos were staged by light microscopy and flash frozen in liquid nitrogen prior to −80 °C storage. Embryos were collected at fertilized egg, 2-cell stage, 4-cell stage, 8-cell stage, 16-cell stage, 32-cell stage, 60-cell stage, swimming blastula stage, mesenchyme blastula stage, and early gastrula stage. RNA was extracted from the staged embryos using the Qiagen RNeasy Mini kit and on-column RNase-Free DNase set. An Agilent Technologies TapeStation 2200 was used to check purity and RNA concentration and purity. Samples were then sent to Novogen for RNA-seq on an Illumina platform.
Annotation and GRN Gene Curation
Genome annotation was performed using the BRAKER3 pipeline (Gabriel et al. 2024). A repeat library was generated with RepeatModeler (Smit and Hubley 2008). The repeat families were compared to known echinoderm protein-coding gene models using BLAST and any repeat with a significant hit (e < 5e−5) was removed. The resulting repeat library was used to identify and mask repeats using RepeatMasker prior to annotation (Smit et al. 2013). BRAKER was run using protein models from S. purpuratus (available on echinobase https://download.xenbase.org/echinobase/Genomics/Spur5.0/sp5_0_GCF.gff3.gz; last accessed 2/15/24), L. variegatus (available from echinobase https://download.xenbase.org/echinobase/Genomics/Lvar3.0/Lvar3_0_GCF_proteins.fa.gz; last accessed 2/15/24), and L. pictus (available from echinobase https://download.xenbase.org/echinobase/Genomics/Lpic2.1/Lpic2_1_GCF_proteins.fa.gz; last accessed 2/15/24) along with publicly available transcriptomic data on NCBI sequencing read archive (BioProject numbers PRJNA376030, PRJNA252503, and PRJEB33967) and in-house RNA-seq data set spanning embryogenesis (see RNA-seq section). The gene models were analyzed for “completeness” with BUSCO version 4 using the metazoan gene set (Simão et al. 2015). GRN gene curation was first carried out by curating a list of GRN genes (supplementary file S1, Supplementary Material online). Then, orthofinder2 (Emms and Kelly 2019) was used to identify orthologs between S. neumayeri, L. variegatus, S. purpuratus, and recently published genome annotation for P. lividus (Emms and Kelly 2019; Marlétaz et al. 2023).
Synteny Analyses
The alignment and resulting dotplot of the S. neumayeri and P. lividus genomes were performed using D-Genies with default parameters (Cabanettes and Klopp 2018). In that analysis, the largest 20 S. neumayeri scaffolds and 19 largest P. lividus scaffolds were included as these are putative chromosomes. Synteny analysis was performed using GENESPACE (Lovell et al. 2022). Gene models for S. purpuratus, L. variegatus, and P. lividus were downloaded from NCBI (Spur 5.0, Lvar 3.0) and from the analysis by Marlétaz et al. (2023), respectively. Genome annotation files were converted to bed files using a custom script, and GENESPACE was run using default parameters.
Supplementary Material
Supplementary material is available at Genome Biology and Evolution online.
Acknowledgments
This work was supported by National Science Foundation grants OPP-1916661, OPP-1916665, OPP-2225144, OPP-2038088, and OPP-2038149. The ship, Antarctic support, and scientific crews of RVIB Nathaniel B. Palmer (cruise NBP 20-10) were invaluable in assisting field work. The authors thank Lauren Baena for her assistance in data curation.
Author Contributions
K.M.H. conceived the study. K.M.H., C.K., and A.R.M. collected the tissue samples and performed the DNA extraction. D.S.W. assembled the genome. J.F.W. and K.T.D. performed the analyses and genome annotation. K.M.H., A.R.M., J.F.W., R.C.R., and J.F., provided the funding. J.F.W., K.M.H., and K.B. wrote the manuscript.
Data Availability
The genome assembly and annotation have been uploaded to Mendeley Data and can be accessed at https://data.mendeley.com/datasets/byjrp4xns7/2. The genome and raw reads were deposited to the Sequence Read Archive (SRA) at the National Center for Biotechnology Information at the National Institutes of Health (Bio Project number = PRJNA1160214).

{kind=link}