





Abstract
Adaptive radiations are characterized by the diversification and ecological differentiation of species, and replicated cases of this process provide natural experiments for understanding the repeatability and pace of molecular evolution. During adaptive radiation, genes related to ecological specialization may be subject to recurrent positive directional selection. However, it is not clear to what extent patterns of lineage-specific ecological specialization (including phenotypic convergence) are correlated with shared signatures of molecular evolution. To test this, we sequenced whole exomes from a phylogenetically dispersed sample of 38 murine rodent species, a group characterized by multiple, nested adaptive radiations comprising extensive ecological and phenotypic diversity. We found that genes associated with immunity, reproduction, diet, digestion, and taste have been subject to pervasive positive selection during the diversification of murine rodents. We also found a significant correlation between genome-wide positive selection and dietary specialization, with a higher proportion of positively selected codon sites in derived dietary forms (i.e., carnivores and herbivores) than in ancestral forms (i.e., omnivores). Despite striking convergent evolution of skull morphology and dentition in two distantly related worm-eating specialists, we did not detect more genes with shared signatures of positive or relaxed selection than in a nonconvergent species comparison. Although a small number of the genes we detected can be incidentally linked to craniofacial morphology or diet, protein-coding regions are unlikely to be the primary genetic basis of this complex convergent phenotype. Our results suggest a link between positive selection and derived ecological phenotypes, and highlight specific genes and general functional categories that may have played an integral role in the extensive and rapid diversification of murine rodents.
Introduction
Adaptive radiations provide natural experiments which allow us to characterize the diversification and convergent evolution of species in response to ecological forces (Schluter 2000; Yoder et al. 2010; Stroud and Losos 2016). Repeated phenotypic shifts and convergent evolution in response to similar environmental pressures provide indirect evidence for adaptive evolution (Losos and Ricklefs 2009; Salzburger 2009; Elmer et al. 2010; Elmer and Meyer 2011; Losos 2011). Although the evolutionary patterns that underlie adaptive radiation and diversification have been studied for many decades at a phenotypic level, advances in DNA sequencing methodologies now allow a genomic view of adaptive radiation (Loh et al. 2008; Schluter and Conte 2009; Jones et al. 2012; Losos et al. 2013; Supple et al. 2013; Berner and Salzburger 2015; Lamichhaney et al. 2015; Tollis et al. 2018; Daane et al. 2019; Li et al. 2019; Marcionetti et al. 2019; Martin et al. 2019). Despite this, many genomic studies have primarily focused on small numbers of exemplar taxa. Genome-wide data from across the taxonomic and phenotypic diversity of species-rich adaptive radiations are generally lacking (but see Lamichhaney et al. 2015; Malinsky et al. 2018), as are broad-scale links between molecular evolution and periods of rapid ecological diversification. Consequently, it remains unclear if the pronounced ecological and phenotypic shifts that are hallmarks of adaptive radiations are also associated with corresponding shifts in the pace and pattern of molecular evolution across the genome.
The opening of novel ecological niche space can facilitate adaptive radiation, and this has classically been characterized with examples of island colonization (Schluter 2000). Following colonization and subsequent adaptive radiation, nascent species face novel assemblages of biotic and abiotic factors. Particular functional categories of genes or pathways are expected to be under pervasive positive selection (i.e., positive selection in multiple lineages), as they enable adaptation to disparate, novel, and changing environments. In studies within and between species, recurrent positive selection is consistently recovered on genes associated with immune function (Castillo-Davis et al. 2004; Nielsen et al. 2005; Shultz and Sackton 2019) and reproduction (Swanson and Vacquier 2002; Swanson et al. 2003; Castillo-Davis et al. 2004; Nuzhdin et al. 2004; Zhang et al. 2004; Nielsen et al. 2005; Turner and Hoekstra 2006), respectively thought to be driven by host–pathogen evolutionary arms races and sexual selection. Additionally, signatures of positive selection across other functional categories of genes may reveal ecological drivers of adaptive diversification (e.g., Kosiol et al. 2008). Clades that have undergone adaptive radiation in geographically constrained areas (e.g., on islands) often exhibit extensive phenotypic disparity among species due to ecological character displacement (Losos 1990; Grant and Grant 2006). In these cases, positive selection presumably also acts on genes underlying ecologically relevant traits such as diet, body size, or microhabitat niche (e.g., Shultz and Sackton 2019). However, it is unclear to what extent bursts of rapid speciation, phenotypic evolution, and ecological specialization also trigger shifts in molecular evolution across the genome.
Murine rodents represent >10% of all living mammalian species (> 700 species in subfamily Murinae; Burgin et al. 2018). Their diversity is the result of a recent (ca. 12 My) radiation, and murine species have repeatedly colonized most areas of the Eastern Hemisphere (Fabre et al. 2013; Aghová et al. 2018; Rowe et al. 2019). Recurring colonization and multiple, independent adaptive radiations have led to extensive phenotypic diversity within Murinae, including a large range in body size (3–2,700 g; Denys et al. 2017), diet (omnivorous, herbivorous, and carnivorous; Rowe et al. 2016a), microhabitat niche (terrestrial, arboreal, semi-aquatic; Nations et al. 2019; Nations et al. 2021), and reproductive output (4–24 mammae; Denys et al. 2017). Given this process of repeated adaptive radiation in murines, genes associated with their ecological diversity and specialization (e.g., diet, reproduction, or microhabitat) may have been subject to pervasive positive selection.
Across the diversity of murine rodents, there are numerous examples of highly specialized morphologies, including cases of repeated convergent phenotypic evolution (Esselstyn et al. 2012; Rowe et al. 2014). One exceptional murine example of convergence is the independent evolution of vermivorous “shrew rats” on both the Indonesian island of Sulawesi (Murinae: Rattini) and the Philippine island of Luzon (Murinae: Hydromyini), with the most extreme examples among these groups being Paucidentomys vermidax (a species monotypic within its genus; Esselstyn et al. 2012) on Sulawesi, and Rhynchomys spp. (Rickart et al. 2019; Heaney et al. 2016) on Luzon. Both are nested within independent, endemic clades of carnivorous rats on the two islands, respectively (Jansa et al. 2003; Rowe et al. 2016a; Rickart et al. 2019). Most murine species are omnivores, and previous work has reconstructed the ancestral dietary state for the group as omnivorous (Rowe et al. 2016). Subsequent to their independent shifts to carnivory, species in the genera Paucidentomys and Rhynchomys have converged on a phenotype that is exceptional among Murinae, with highly elongated rostra, slender mandibles, and greatly reduced or absent molars (fig. 1; Esselstyn et al. 2012; Martinez et al. 2018; Rickart et al. 2019). These species share a common ancestor approximately 10–12 Ma, near the base of all Murinae (Rowe et al. 2016a; Aghová et al. 2018; Rowe et al. 2019), and are isolated on oceanic islands, precluding any role for gene flow. As such, this striking ecomorphological convergence may be associated with convergent changes at the genomic level. Independent fixation of shared ancestral variation could also contribute to these observations, but this seems most unlikely to bridge 12 My of independent evolution (Arendt and Reznick 2008). Although convergence at particular coding sites within genes is unlikely to be directly associated with complex convergent phenotypes (Foote et al. 2015), common sets of genes may show parallel signatures of positive or relaxed selection in convergent species (Bergey et al. 2018; Dixon and Kenkel 2019; Sahm et al. 2019). In Paucidentomys and Rhynchomys, ecological selective pressures which drove the evolution of their striking, shared phenotype may be linked to convergent shifts in selective pressures on genes associated with their derived diet and craniofacial or tooth development (Charles et al. 2013).
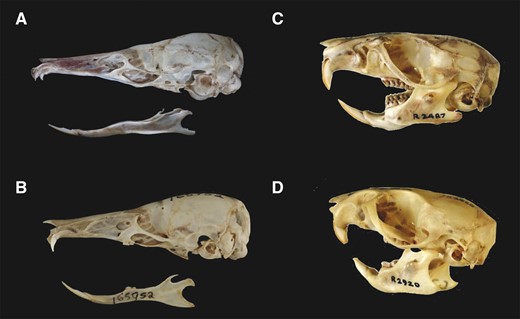
Exceptional convergence of craniofacial morphology and dentition in worm-eating specialists; (A) Paucidentomys vermidax (Muridae: Rattini) and (B) Rhynchomys labo (Muridae: Hydromyini), compared with two generalist species belonging to the same respective clades; (C) Rattus fuscipes (Muridae: Rattini), and (D) Pseudomys shortridgei (Muridae: Hydromyini). Photos by (A) D. Paul, Museums Victoria, (B) modified from Rickart et al. (2019) with permission, (C) and (D) M. Rawlinson, C. Accurso, and K. Walker, Museums Victoria.
Murine rodents are also important model organisms, both in laboratory studies and in the wild, with Mus musculus and Rattus norvegicus among the most well-studied mammalian species (Mouse Genome Sequencing Consortium 2002; Gibbs et al. 2004; Guénet 2005; Phifer-Rixey and Nachman 2015). Despite their utility as model organisms, these generalist species represent only a miniscule fraction of the ecomorphological diversity in the broader murine radiation. Comparative genomic studies have not previously examined broader Murinae, and as such there is no prior understanding of the interactions between genes, traits, and ecology in this group. Repeated, nested adaptive radiations within Murinae, extensive diversity and recurrent ecomorphological specialization make murine rodents an ideal system for testing correlates between trait evolution, convergence, and rapid molecular evolution. A broad-scale, comparative approach is warranted to begin to unlock what is largely an untapped natural system for characterizing genomic responses to ecological opportunity.
Here, we generate sequence data for >14,000 protein-coding genes from 38 species spanning the phylogenetic breadth of murine diversity, and spanning multiple adaptive radiations within the subfamily, with focused sampling from independent radiations in the Philippines and Sulawesi. Using these data, we identify genes and gene categories with signatures of pervasive positive selection across Murinae, test if heterogeneity in positive selection across lineages is associated with ecological traits (i.e., diet, microhabitat, reproductive output, and body size), and screen for evidence of convergent molecular evolution between Rhynchomys and Paucidentomys, an extreme example of ecomorphological convergence in murines.
Results
Phylogenetic Reconstruction
Using data from 1,360 phylogenetically informative exons, we inferred a consistent, well-supported species tree topology in both IQ-TREE 1.6.1 (Nguyen et al. 2015) and SVDquartets (Chifman and Kubatko 2014, fig. 2) for 38 species (supplementary table S1, Supplementary Material online). These species covered the phylogenetic breadth of subfamily Murinae, including representatives from Asian, Australian, and African radiations, and were also representative of the substantial ecomorphological variation of murine rodents, that is, dietary, microhabitat, and body size variation. Almost all nodes (n = 71) received 100% bootstrap support across all approaches implemented. Two nodes received less support in more than one analysis, but no nodes were consistently poorly supported. Across the full data set, average coverage ranged from 25 to 57×, with full mapping and coverage statistics per-sample summarized in supplementary table S2, Supplementary Material online.
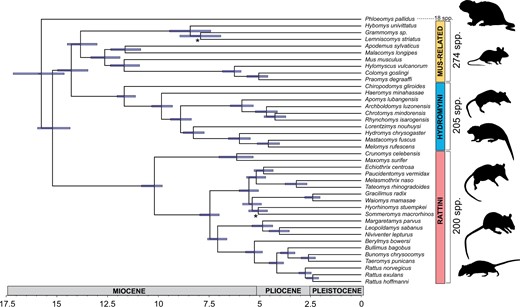
Time-calibrated phylogeny of sampled murine species generated in MCMCtree, with a consistent topology estimated in both IQ-TREE and SVDquartets. All phylogenetic analyses were based on a subset of 1,360 loci (Roycroft et al. 2020a). Nodes with <100% support using more than one branch support approach are indicated with an asterisk. Species numbers to the right of the phylogeny indicate the total number of described species in each of the three main murine clades, with Phloeomys pallidus the sole representative in this study of the tribe Phloeomyini.
Pervasive Positive Selection across Murinae
Across the murine phylogeny, site models in codeml 4.9i (Yang 2007) revealed 1,383 genes (out of 14,229 tested, supplementary table S4, Supplementary Material online) with consistent evidence for sites under positive selection (P < 0.05, using a Benjamini–Hochberg false discovery rate correction; FDR), using both individually inferred gene trees (gene tree topology data set) and the species tree (species tree topology data set). Among these, we identified 42 overrepresented Reactome pathways (Jassal et al. 2020) and 29 overrepresented KEGG pathways (Kanehisa et al. 2017) using g: Profiler (Raudvere et al. 2019; supplementary tables S5 and S6, Supplementary Material online). These pathways were largely involved in immune, digestive, taste, and reproductive functions (fig. 3A). Additionally, there were 53 “molecular function,” 116 “biological process,” and 38 “cellular component” GO category terms significantly overrepresented (supplementary table S7, Supplementary Material online). Overrepresented biological processes also broadly included terms associated with immunity, reproduction, digestion, and taste (fig. 3B). Overrepresented molecular functions included peptidase and lipase activity, taste reception, and immune receptor activity. Overrepresented cellular components included sperm morphological parts and immunity-related components, including secretory granule and cellular membranes.
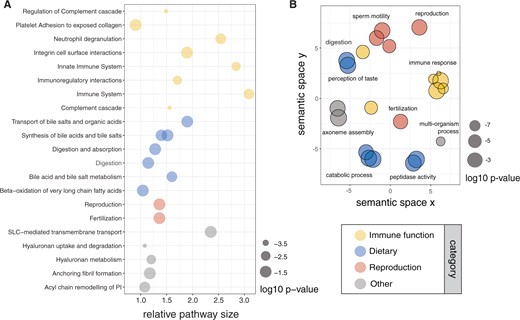
Overrepresented functions of genes under pervasive positive selection (P < 0.05) across Murinae using annotations from (A) Reactome pathways, and (B) Gene Ontology biological process categories, grouped using REVIGO semantic clustering (similarity threshold = 0.5). Circle size represents log10P-value for the significance of overrepresentation; colors indicate functions related to the immune system, dietary processes, and reproduction.
Ecological Predictors of Genome-Wide Positive Selection
Overall, branch-specific values of positive selection estimated using aBSREL (Smith et al. 2015) in HyPhy 2.5.14. (Pond et al. 2005) revealed substantial heterogeneity in the proportion of sites under selection among murine lineages (fig. 4A;supplementary table S8, Supplementary Material online), which was not explained by variation in terminal branch length (gene tree topology: R2 = 0.025, species tree topology: R2 = 0.008). This pattern of heterogeneity was consistent in analyses of the species tree topology and gene tree topology data sets (R2 = 0.70). Dietary state (carnivorous, herbivorous, or omnivorous) was a significant predictor (gene tree topology: P = 0.0026, species tree topology: P = 0.045) of mean proportion of sites under positive selection, when taking into account phylogenetic relatedness in a PGLS regression. There was also a significant difference (gene tree topology: P = 0.0094, species tree topology: P = 0.0052) between dietary states in the mean proportion of sites under positive selection in a phylogenetic ANOVA, with carnivores being higher than omnivores (fig. 4B; gene tree topology: P = 0.045, species tree topology: P = 0.012). Despite the small number of herbivores in this data set (n = 3), herbivores had significantly higher values than omnivores in the gene tree topology data set (P = 0.045) but not the species tree topology data set (P = 0.082). These patterns were also consistent using the topology-free pairwise dN/dS values estimated in codeml, where both carnivores (P = 0.003) and herbivores (P = 0.044) had significantly higher dN/dS values than omnivores. All models that jointly accounted for diet and relative population size (approximated by average heterozygosity across the whole-exome, and based only on third codon position sites) did not recover contemporary population size as a significant predictor for the mean proportion of sites under selection (whole exome estimate: species tree topology P-value = 0.21, gene tree topology P-value = 0.46, third codon estimate: species tree topology P-value = 0.25, gene tree topology P-value = 0.67).
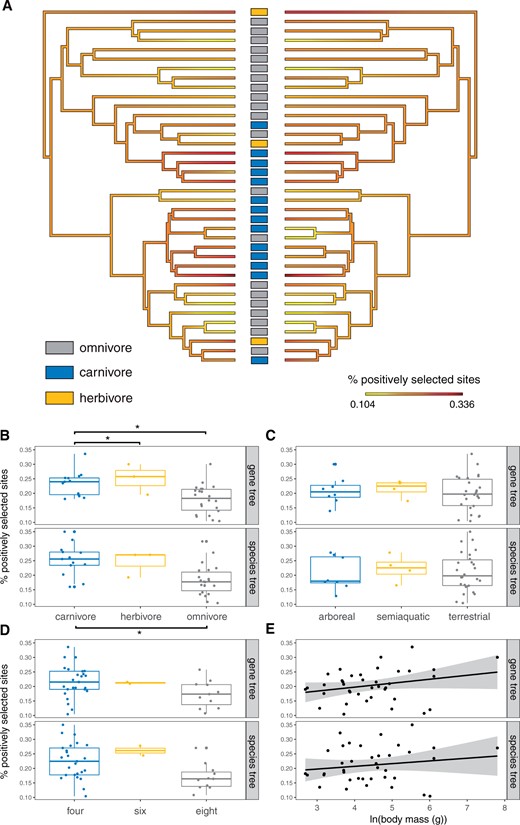
Heterogeneity in genome-wide positive selection and ecological predictors. (A) Average percentage of sites under positive selection plotted as a heat map on the species tree topology with dietary states indicated at the tips, calculated with aBSREL in HyPhy using either the species tree topology (left) or gene tree topology (right, with terminal branches matched to the species tree topology for visualization) data sets. (B) Comparison of average percent sites under positive selection across dietary states (carnivorous, herbivorous, or omnivorous), (C) microhabitats (arboreal, semiaquatic, or terrestrial), (D) number of mammae, and (E) log of body mass. Significance values are derived from phylogenetic ANOVA (* = FDR corrected P < 0.05).
There was no significant effect of microhabitat (fig. 4C), reproductive output (no. of mammae; fig. 4D), or body mass (fig. 4E) on the proportion of sites under positive selection in either PGLS or phylogenetic ANOVA analyses; however, the number of mammae was significantly correlated with the number of positively selected sites before, but not after phylogenetic correction. The proportion of positively selected sites across digestion-related genes was no more correlated with dietary state, than the proportion of positively selected sites across genes with nondigestive functions. Similarly, the proportion of positively selected sites across reproduction-related genes was no more correlated with number of mammae, than across genes with function unrelated to reproduction. Overrepresentation and functional enrichment tests of genes that were most correlated with dietary specialization (top 5% and 10%, and Spearman’s ρ values), did not yield any significant functional categories or pathways. This suggests that the increase in positive selection across genes in dietary specialists is not restricted to genes directly related to, or associated with, digestion, but potentially a suite of interacting genes in other functional categories across the genome.
Shared Positive and Relaxed Selection
Across both the species tree topology and gene tree topology data sets, 39 genes were consistently detected under shared positive selection in both Rhynchomys labo and Paucidentomys vermidax using the aBSREL test for positive selection (supplementary table S9, Supplementary Material online). For all 39 positive genes, the standard aBSREL model was a better fit (AICc) to the data than models accounting for multinucleotide mutations (MNMs; aBSREL + Double and aBSREL + Double + Triple). Among these genes was the Androgen Receptor (Ar) gene, which encodes a transcription factor known to influence bone morphogenesis through interaction with the RUNX2 transcription factor. However, the number of total genes under shared positive selection in both of these strikingly convergent vermivorous rodents was not significantly greater than expected by chance, nor greater than the number of genes under shared positive selection in a nonconvergent control comparison between Mastacomys fuscus and Echiothrix centrosa (59 convergent genes selected in the species tree topology and gene tree topology data sets). In addition, consistent signatures of relaxed selection in both Paucidentomys and Rhynchomys were detected in 14 genes across both the species tree topology and gene tree topology data sets (supplementary table S10, Supplementary Material online).
Convergent Amino Acid Profile Shifts
After filtering, 47 genes showed strong evidence (posterior probably > 0.9) for site-based, convergent amino acid profile shifts using PCOC (Rey et al. 2018; supplementary table S11, Supplementary Material online). We did not identify any significantly overrepresented functional terms among these genes, nor at lower PCOC score thresholds. Among these genes, Cdon is associated with human disease phenotype pathways (HP) “abnormality of the nasal cavity,” “cleft-lip,” “single median maxillary incisor,” and “midnasal stenosis.” In the two convergent vermivores, Cdon has undergone a significant shift in amino acid profile at site 414, where both Rhynchomys and Paucidentomys have independently experienced a shift from polar to nonpolar residues. Cdon was also under significant positive selection in Paucidentomys but not in Rhynchomys.
Discussion
We found that genes associated with immune, reproductive, and dietary processes have been subject to pervasive positive selection across the murine radiation. We also recovered a higher proportion of positively selected sites in derived dietary forms (i.e., carnivores and herbivores) than in omnivorous species (the ancestral state, Rowe et al. 2016a), suggesting a link between ecological forces of diversification and rates of putatively adaptive molecular evolution. We did not detect more genes with shared selective shifts in the strikingly convergent worm-eating species Paucidentomys and Rhynchomys, than in a non-convergent comparison. However, a subset of genes we did detect can be incidentally linked to craniofacial morphology or diet. Our results highlight functional categories of genes that may have played an integral role in the repeated radiation and extensive dietary diversification of murine rodents.
Pervasive Positive Selection on Immunity and Reproductive Genes
We found strong evidence for pervasive positive selection on genes and pathways associated with the immune system and reproduction in Murinae. Numerous immunity- and reproduction-related GO, KEGG, and Reactome terms were significantly overrepresented among genes that experienced positive selection across the radiation. Many previous studies have identified that genes associated with immune function (Schlenke and Begun 2003; Castillo-Davis et al. 2004; Nielsen et al. 2005) and reproduction (Swanson and Vacquier 2002; Swanson et al. 2003; Castillo-Davis et al. 2004; Nuzhdin et al. 2004; Zhang et al. 2004; Good and Nachman 2005; Nielsen et al. 2005; Dean et al. 2008; Turner et al. 2008) are common targets of recurrent positive selection, and on average, tend to evolve faster than other protein coding genes. More recently, comparative genomic studies at both deep and shallow taxonomic scales indicate that these patterns are consistent across all scales of animal divergence (Nielsen et al. 2005; Kosiol et al. 2008; Roux et al. 2014; Cagan et al. 2016; Cicconardi et al. 2017; Sahm et al. 2019; Shultz and Sackton 2019). The strong signal of positive selection on immune and reproduction-related genes across Murinae confirms that these pervasive patterns remain consistent during species diversification, in consort with rapid evolution of ecologically significant phenotypes.
The adaptive immune system of animals is subject to constant pressure from rapidly evolving pathogens with shorter generation times than their hosts (Woolhouse et al. 2002). This co-evolutionary “arms race” is a source of selective pressure and is thought to cause rapid adaptive evolution in immunity-related genes (Nielsen et al. 2005; Kosiol et al. 2008). Murines have likely interacted with a diverse array of both endemic and introduced pathogens (e.g. Winterhoff et al. 2020) throughout their evolutionary history, driving this rapid molecular evolution. Response to co-evolutionary change may similarly explain rapid evolution of reproductive proteins, with previous studies suggesting that sperm competition and sexual conflict are key drivers of positive directional selection (Wyckoff et al. 2000; Swanson and Vacquier 2002; Torgerson et al. 2002; Swanson et al. 2003). The set of reproductive genes under pervasive positive selection across murines in our results include a number of genes which have previously been identified as under positive selection in other mammals, including Zp3 (Swanson and Vacquier 2002; Jansa et al. 2003; Turner and Hoekstra 2006), which contains the primary species-specific sperm binding site, as well as the egg-binding proteins Adam2 and Spam1 (Torgerson et al. 2002). The coevolution of male and female reproductive proteins may be associated with the eventual development of barriers to fertilization, reproductive isolation, and subsequent speciation (Swanson and Vacquier 2002). There is substantial divergence in sperm morphology between closely related murine species (e.g., Breed 2000; McLennan et al. 2017; Pahl et al. 2018), which may contribute to the rapid evolution of prezygotic isolation between populations. Accelerated evolution, or increased positive selection, in reproductive genes in murine rodents may be linked, in part, to the rapid speciation of murines in both allopatry, and via ecological niche partitioning in spatially limited island systems. Future comparative studies may reveal whether diversifying selection, and positive selection, on immunity and reproductive genes is more intense during adaptive radiation, compared with background rates, as nascent species encounter novel pathogens, and rapidly diversify to fill available ecological niches.
Pervasive Positive Selection on Dietary and Taste-Associated Genes
We also found significant overrepresentation of functional categories associated with diet (digestion and taste), which is likely related to the exceptional ecological diversity and success of murine rodents. Recent work has also identified selection on bitter-taste genes in the desert-adapted rodent, Peromyscus eremicus (Tigano et al. 2020). Pervasive positive selection in diet-related genes has not previously been identified across a recent radiation. A study of six mammalian genomes (Kosiol et al. 2008) identified positive selection on starch digestion and bitter taste genes in primates, but not in two murine species (M. musculus and R. norvegicus). This contrast highlights the importance of taxon sampling in detecting associations between ecological diversification and genomic adaptation, with our study examining this pattern across the broad phylogenetic and ecological diversity of murine rodents. At a broader scale, previous research suggests that dietary evolution may be associated with changes in gene copy number (Feng et al. 2014; Li and Zhang 2014; Pajic et al. 2019), gene family expansions (Whiteman et al. 2012; Gloss et al. 2019; Seppey et al. 2019), or loss of gene function (Kim et al. 2016; Hu et al. 2017; Hecker et al. 2019). For example, the evolution of carnivory across mammals at a broad scale is associated with repeated loss of sweet and bitter taste receptors (Jiang et al. 2012). However, our results provide the first strong link between rapid ecological diversification of species, including repeated evolution of dietary specialization, and recurrent positive selection on multiple genes in functional categories related to dietary processes. Trophic niche is a crucial driver of phenotypic evolution (Price et al. 2012) and in the case of murine rodents, is arguably the main axis of differentiation between species, especially in island systems across the Indo-Australian Archipelago (e.g., Rowe et al. 2014, 2016a, 2016b). Pervasive positive selection on genes associated with diet, digestion, and taste in a clade with extensive dietary disparity provides a compelling link between ecological novelty, phenotypic evolution, and putatively adaptive molecular evolution.
Derived Dietary States Are Associated with Rapid Molecular Evolution
As well as triggering pervasive positive selection across dietary genes, the evolution of dietary specialization in the murine species examined in our study was significantly correlated with a genome-wide increase in the average proportion of sites under positive selection, as well as higher overall dN/dS. This pattern was most compelling in carnivores (a derived state in murines; Rowe et al. 2016a), where there was a significantly higher average proportion of positively selected sites than in omnivores (the ancestral state). This pattern was similar in herbivores, but only significant when using the gene tree topology data set or a pairwise (topology-free) contrast. Although there were only three herbivorous species in this study, these species represent three independent transitions to herbivory. The elevated dN/dS may result from long-term small effective population size (Ne), via increased fixation of deleterious mutations which are incorrectly inferred as signatures of positive selection (Ohta 1993; Deinum et al. 2015), or alternatively a large Ne resulting in increased adaptive efficacy (Gossmann et al. 2010). However, our comparative analyses found no significant effect of average heterozygosity (as a proxy for Ne). Similar patterns are evident in deeper-time comparisons among mammals, with increased signatures of molecular adaptation in carnivores (i.e., Felidae) when compared with omnivores (Hominidae) and herbivores (Bovidae; Kim et al. 2016). Our finding of an increase in positive selection at a genome-wide scale in carnivorous, and to a lesser extent in herbivorous murines, suggests that the evolution of dietary specialization may have triggered increased positive selection on a suite of interacting traits (Goldman-Huertas et al. 2015), and subsequently affected many loci in the genome.
Whether rates of molecular evolution, or positive selection, can be generally associated with the evolution of adaptive ecological traits remains an open question, and few specific examples exist. Temperate lacertid lizards were recently found to have experienced a genome-wide decrease in molecular evolution relative to tropical- and desert-adapted species (Garcia-Porta et al. 2019). Body size is a consistent predictor of neutral molecular evolutionary rate across broad taxonomic scales, with larger species expected to have slower rates due to longer generation times (Bromham 2002; Berv and Field 2018). A recent study suggested an extension of this generalization to positive selection in birds, finding that body size was linked to variation in the proportion of positively selected sites (Shultz and Sackton 2019). In contrast to diet, there was no significant correlation between positive selection and any other traits tested in our comparative phylogenetic analyses, including body size. Although the murine species examined here vary by two orders of magnitude in body size (20–∼2,000 g), differences in generation time may be insufficient to affect relative evolutionary rates.
A Genomic Basis for Convergent Evolution of Worm-Eating Rodents?
There were not more genes under shared selective shifts (positive or relaxed) in the convergent worm-eating rodents Paucidentomys and Rhynchomys when compared with the nonconvergent control comparison, Mastacomys and Echiothrix. These results are consistent with a recent study of shared positive selection in the convergent marsupial thylacine and eutherian canid (Feigin et al. 2018), suggesting that positive selection has not acted on the same genes in phenotypically convergent species more often than in general forms. However, comparing the number of genes under shared positive selection may be a relatively conservative benchmark for detecting molecular convergence. As such, it remains possible that the genes we recovered are linked to the evolution of the convergent phenotypes of Paucidentomys and Rhynchomys.
For example, we found shared positive selection on the Androgen Receptor (Ar) gene, which encodes a transcription factor known to influence bone morphogenesis through interaction with the RUNX2 transcription factor (Baniwal et al. 2009). Variation between species in the number and ratio of short repeats in RUNX2 has previously been associated with variation in mammalian cranial length (Fondon and Garner 2004; Sears et al. 2007; Pointer et al. 2012; Ritzman et al. 2017), and RUNX2 also shows signatures of an ancient selective sweep after the divergence of anatomically modern humans from other archaic lineages (Greenet al. 2010). Given shared signatures of positive selection and its pivotal role in mammalian bone metabolism (Kawanoet al. 2003), the Ar transcription factor represents a potential candidate gene contributing to the evolution of elongated craniofacial morphology in Paucidentomys and Rhynchomys.
Additionally, shared positive selection and amino acid shifts in taste-receptor genes Tas2r113 and Tas2r114, and relaxed selection in the glucose transporter gene Slc2a2, (part of the Reactome pathway “Intestinal absorption”), recapitulate the evolution of dietary specialization across Murinae at a broad scale. We also detected convergent shifts in amino acid profile in the gene Cdon, associated with craniofacial and tooth development. However, genes involved in patterning and development of morphology are often highly pleiotropic (Sivakumaran et al. 2011), and changes at the coding level likely have consequences for the function of the gene in many different contexts. As such, parallel amino acid changes are thought to rarely be directly associated with phenotypic convergence (Foote et al. 2015). Although the genes listed above can be incidentally linked to either craniofacial morphology or diet, the majority of genes we detected with convergent selective signatures in Paucidentomys and Rhynchomys do not have obvious links to their convergent phenotype.
Increasing evidence implicates regulatory elements controlling pleiotropic genes in the evolution of complex traits (Prud’homme et al. 2006; Kvon et al. 2016; Feigin et al. 2018; Roscito et al. 2018), especially in loss-of-function phenotypes, such as limb loss in snakes (Kvon et al. 2016) and eye degeneration in subterranean mammals (Roscito et al. 2018). In such cases, changes in the timing and level of gene expression via evolution in regulatory regions may underlie the evolution of convergent phenotypes. Expansion or contraction of gene families also likely contributes to patterns of convergent evolution (e.g., Hoffmann et al. 2010; Whittington et al. 2010). Given the restricted genomic scope of whole exome data, future work examining whole genomes from across Murinae may shed light on the contribution of gene family evolution, noncoding regions, and regulatory elements. More broadly, information about the function of genes in unique morphological and ecological contexts may not be captured by model species, from which their functional annotations are derived. As such, any functional relevance for the majority of genes under convergent selection in Paucidentomys and Rhynchomys remains unclear. Inclusion of species representing extreme morphological adaptation in laboratory studies, including developmental studies, may reveal novel gene function and gene interactions previously unknown from classic model species.
Conclusion
Multiple, nested adaptive radiations within Murinae have resulted in repeated and convergent ecological specializations, and we recover evidence for this at the genomic level. Pervasive positive selection on diet-related genes across the radiation, and an increase in positive selection in dietary specialists, suggests a link between ecological drivers of diversification and molecular evolution. We highlight both categories of genes, and specific genes, which may have played an integral role in the repeated invasion by murine rodents of novel ecological niches, and in the convergent evolution of worm-eating specialists. Our findings demonstrate the utility and opportunity for leveraging murine rodents as an emerging model system for understanding adaptive processes. Given the enormous phenotypic and species diversity of Murinae, and their existing genomic resources, murine rodents represent a largely untapped resource for studies of evolutionary processes.
Materials and Methods
Taxon Sampling
We selected 38 representatives of rodents from the subfamily Murinae, including representatives from Asian, Australian, and African radiations. We additionally included the model murine species M. musculus (genome assembly GRCm38) and R. norvegicus (genome assembly Rnor6), with final sampling including ten species from tribe Hydromyini, 20 species from tribe Rattini, nine species from the Mus-related clade (tribes Apodemini (1), Arvicanthini (3), Murini (1), Malacomyini (1), and Praomyini (3)), and one species of Phloeomyini. Together, these species are representative of the substantial ecomorphological variation of murine rodents, including dietary, microhabitat, and body size variation. In this comparative framework, we assume that individual samples are representative of species-specific adaptations and acknowledge that some signatures could reflect local adaptation within species. Tissues were obtained from museum collections (see supplementary table S1, Supplementary Material online for details), where vouchers are permanently curated. These specimens were collected according to the relevant legal and ethical requirements of each country.
Sample Preparation, Whole-Exome Capture and Sequencing
Total genomic DNA was extracted from liver or muscle tissue using a Qiagen DNeasy Blood and Tissue Kit, following the manufacturer protocol. DNA library preparation followed the Meyer and Kircher (2010) protocol. Target regions were enriched using two NimbleGen SeqCap EZ 1 mouse whole-exome capture reactions (Fairfield et al. 2011), targeting 54.3 Mb of exonic regions based on the M. musculus reference genome (NCB137/mm9). These 203,225 target loci represent exons from nearly all protein-coding regions in M. musculus excluding known pseudogenes, and highly similar multi-copy gene families including olfactory receptor genes (a large paralogous gene family in murines). The use of M. musculus whole-exome enrichment probes has proven efficient across approximately 7.5 My divergence (Sarver et al. 2017). Enriched libraries were sequenced across two lanes of Illumina NextSeq 550 paired-end, two lanes of Illumina NextSeq 550 single-end, one lane of MiSeq, and one lane of HiSeq 4000.
Obtaining a Database of Putatively Single-Copy Loci among Murinae
To generate an initial reference set of putatively single-copy exons across Murinae, we first used liftOver (Hinrichs 2006) to convert M. musculus (mm9) nucleotide target regions from the whole-exome bait-design (Fairfield et al. 2011) to orthologous co-ordinates in the R. norvegicus (Rn5) genome. The final reference set excluded any loci that could not be aligned between both the mm9 and Rn5 genomes, spanning ∼12 My of murine evolution. We also removed any exons from the reference set which had more than one internal hit of >95% amino acid identity within either the mm9 or Rn5 genomes, which would suggest they represent recent duplications. This filtering resulted in a final set of 162,566 exons from 18,797 genes and was used as the reference for all subsequent analyses.
Sequence Assembly and Alignment
We processed raw sequence data using ECPP v1.1.0, largely following the workflow described in Roycroft et al. (2020a), but with some modifications. Briefly, raw reads were deduplicated using FastUniq v1.1 (Xu et al. 2012) and quality trimmed using Trimmomatic (Bolger et al. 2014). Cleaned reads for assembled de novo using TRINITY 2.4 (Grabherr et al. 2011; Haas et al. 2014) to generate a sample-specific contig file for each of 38 sequenced species. Using the filtered, putatively single-copy murine loci described above, we identified the best matching contigs in each assembly using TBLASTN. Using BLAST coordinates, we extracted local matches from assembled contigs to create a sample-specific reference for mapping. We then mapped the cleaned reads to the sample-specific reference using BBmap (version 35.82, Bushnell B. 2015, sourceforge.net/projects/bbmap/) with minid = 0.8. Mapping and coverage statistics per-sample are summarized in supplementary table S2, Supplementary Material online. Consensus sequences and variants were called using the mpileup2cns command in VarScan v2.3.7 (Koboldt et al. 2012). Consensus sequences were then collated across all samples for each exon and were aligned using MAFFT v7.310 (Katoh and Standley 2013).
Data Filtering and Post Hoc Paralog Detection
We only included exons in the final data set which were successfully captured and mapped for at least 27 of 38 samples. To screen for lineage-specific paralogs that were not detected in initial filtering, we calculated average heterozygosity for each sample in each alignment. Alignments with two or more samples with > 3% average heterozygosity (Teasdale et al. 2016; Roycroft et al. 2020a) were excluded, as these may represent loci with pervasive paralogy. We assumed that cases where only one sample had >3% average heterozygosity represented lineage-specific duplications and removed only that sample from the alignment. A total of 89,621 exons were retained, that were concatenated into 14,229 gene alignments for analysis.
Phylogenetic Analyses
For phylogenetic analysis, we reduced the full data set to alignments to a previously qualified, murine-specific set of 1,360 phylogenetically informative single-copy exons (Roycroft et al. 2020a) and estimated the maximum likelihood (ML) phylogeny in IQ-TREE 1.6.1 (Nguyen et al. 2015) from a concatenated supermatrix partitioned by codon position (i.e., three global partitions). We used ModelFinder (Kalyaanamoorthy et al. 2017) to determine the best substitution model for each partition, and executed 1,000 ultrafast bootstrap replicates, using UFBoot2 (Hoang et al. 2017). We also estimated support in IQ-TREE using two-tiered resampling of genes and sites (–bspec GENESITE), an approach which we previously showed provided more accurate estimates of uncertainty in phylogenomic data sets (Roycroft et al. 2020a). To verify this inferred ML topology, we estimated the species tree topology using the coalescent approach SVDquartets (Chifman and Kubatko 2014) implemented in PAUP* v4.0a (Swofford 2002). We used MCMCtree (Yang 2007) to estimate time-calibrated branch lengths, with the ML topology inferred in IQ-TREE, a GTR+Γ substitution model, an uncorrelated Γ relaxed clock, and using the approximate likelihood calculation (Thorne et al. 1998; Reis and Yang 2011). We used three secondary calibrations from Aghová et al. (2018) that best matched our sampling of Murinae: the MRCA of Rattini (95% HPD 9.91–12.67 Ma), the MRCA of Sahul Hydromyini (95% HPD 6.48–8.34 Ma), and the MRCA Praomyini (95% HPD 5.98–7.84 Ma). Samples were drawn every 1,000 MCMC steps from a total of 107 steps, with a burn-in of 105 steps. Convergence was assessed by comparing parameter estimates from two independent runs, with all effective sample sizes >200.
Mendes and Hahn (2016) showed that estimates of positive selection derived from a fixed species tree can be subject to false positives when the individual genealogical history conflicts with the species tree. To help combat this in downstream molecular evolution analyses, we estimated individual gene trees from each alignment in IQ-TREE 1.6.1 (Nguyen et al. 2015) using ModelFinder (Kalyaanamoorthy et al. 2017) to select the single best fitting substitution model for each gene.
Detecting Genes under Pervasive Positive Selection
For all 14,229 orthologous genes, we ran two site-based models in codeml 4.9i, the M1 and M2 models (Yang 2007). The M1 model allows for two ω (dN/dS) rates across sites (ω < 1 and ω = 1), whereas M2 allows three rates (ω < 1, ω = 1 and ω > 1). Evidence of pervasive positive selection at particular sites can be inferred when the M2 model is a significantly better fit for that gene than the M1 model. Using a likelihood ratio test (LRT), we compared log-likelihood estimates for models M1 and M2 to identify genes with sites under positive selection across the murine phylogeny. These tests were performed using both the species tree and gene tree as the reference topology. We calculated LRT P-values using chi-squared distribution (d.f. = 2) and corrected for multiple tests at a P < 0.05 threshold, using a Benjamini–Hochberg FDR correction. Genes were considered to have sites under positive selection only if both the LRT was significant after correction, and at least one site was significantly selected using a Bayes Empirical Bayes (BEB) test (posterior probability > 0.95; Yang et al. 2005) against both the species tree and gene tree.
Functional Overrepresentation of Genes under Positive Selection
Using g:GOSt in gProfiler (Raudvere et al. 2019), we tested for overrepresentation of GO terms, KEGG pathways (Kanehisa et al. 2017) and Reactome pathways (Jassal et al. 2020) among genes identified as being under significant positive selection in site model tests. We used a custom background including all tested genes and applied an FDR correction for multiple comparisons (P < 0.05). To visualize overrepresented functional categories, we used REVIGO (Supek et al. 2011) to generate semantic clustering of GO biological process (GO:BP), molecular function (GO:MF), and cellular component (GO:CC) terms (allowing 0.5 term similarity).
Branch-Specific Positive Selection
Although site-based models can identify genic sites under significant positive selection across multiple lineages in a phylogeny, they do not provide information about heterogeneity in selection throughout time and across lineages. To investigate this, we used the flexible branch-site test aBSREL (Smith et al. 2015) in HyPhy 2.5.14. (Pond et al. 2005) to estimate ω values and proportion of sites under positive selection for each terminal branch in the tree. To reduce potential false positive rates due to tree misspecification (Mendes and Hahn 2016), we applied two approaches to estimating branch-specific selection in aBSREL. First, we estimated values for all terminal branches and genes using the fixed species tree topology: the species tree topology data set. Second, we inferred selection across branches and genes using each individually estimated gene tree: the gene tree topology data set.
Positive Selection and Ecomorphological Variation
For each terminal branch, we calculated the mean proportion of sites under positive selection across all genes in aBSREL, to obtain a genome-wide estimate of the proportion of sites under positive selection for each species. To first visualize heterogeneity in positive selection across the tree, we used the R function contMap in phytools (Revell 2012) to plot values from both the species tree topology and gene tree topology data sets as a heat map on the species tree. To additionally estimate the strength of positive selection for each species using a topology-free approach, we calculated average pairwise dN/dS across all species-pair comparisons using codeml (Yang and Nielsen 2000). To test whether this proportion of sites under positive selection, or strength of selection (dN/dS) were correlated with ecological factors, we obtained dietary, microhabitat, reproductive, and body mass data for each species from the literatures (Smith et al. 2003; Breed and Ford 2007; Rowe et al. 2016a, 2016b; Nations et al. 2019; Roycroft et al. 2020b). We coded species according to their diet (carnivore, omnivore, or herbivore), their microhabitat (terrestrial, arboreal, or semi-aquatic), and their reproductive output, (based on the number of mammae for each species, supplementary table S3, Supplementary Material online). Using the time-calibrated species tree inferred in MCMCtree, we performed phylogenetic generalized least squares (PGLS) regression and phylogenetic ANOVA with a Bonferroni correction in phytools (Revell 2012), to test the effects of diet, microhabitat, reproductive output, and body size on genome-wide positive selection. Further, because effective population size (Ne) can affect estimates of positive selection (Ohta 1993; Gossmann et al. 2010; Deinum et al. 2015), we jointly modeled the additive and interacting effects of average exome-wide heterozygosity, and third codon position heterozygosity (as proxies for Ne), with ecological traits in the comparative analysis.
To further determine whether there was an interaction between gene function, positive selection, and ecological traits, we used GO annotations and Gene ORGANizer (Gokhman et al. 2017) classifications to identify genes with function in the digestive (1,657 genes) and reproductive systems (2,077 genes). We then estimated the mean percent of positively selected genes across digestive and nondigestive genes, and reproductive and nonreproductive genes. Using the same approach described above, we ran PGLS and phylogenetic ANOVA with dietary state or number of mammae as the predictor, respectively. Using a binary measure of dietary state (1 = specialist; i.e., herbivore or carnivore, 0 = generalist; i.e., omnivore), we also performed a Spearman’s rank correlation test to determine which genes showed the highest correlation between positively selected sites and lineages with dietary specialization. Using g: Profiler, we tested for overrepresentation of functional categories in the top and bottom 10% and 5% of genes, and performed functional enrichment analysis using the calculated Spearman’s ρ value for each gene.
Branch-Specific Convergence in Positive and Relaxed Selection
We tested for branch-specific convergent positive selection by performing a branch-site test in aBSREL across all genes, with two phenotypically convergent vermivorous rodents, Paucidentomys vermidax and Rhynchomys labo, set as foreground branches. All analyses were repeated using both the species tree topology and gene tree topology data sets. A recent study showed that MNMs may cause false inferences in branch-site tests of positive selection (Venkat et al. 2018). For genes where we detected positive selection in both Paucidentomys and Rhynchomys, we accounted for this by applying models that allow double and triple MNMs using the --multiple-hits Double and --multiple-hits Double+Triple options in HyPhy 2.5.14. As MNM models include additional parameters compared with the standard aBSREL model, we compared AICc scores from standard aBSREL, aBSREL + Double and aBSREL + Double + Triple, and retained results from the model with the lowest AICc score. To further determine whether there were more shared genes under positive selection in Paucidentomys and Rhynchomys than in other nonconvergent murine forms, we repeated all analysis using a nonconvergent “control” comparison, that is, by comparing genes under positive selection in the graminivorous Australian rodent Mastacomys fuscus (tribe Hydromyini), and the carnivorous Sulawesi shrew rat Echiothrix centrosa (tribe Rattini) as the foreground test branches. These control species are phylogenetically equidistant to the Paucidentomys (tribe Rattini) and Rhynchomys (tribe Hydromyini) comparison and occur along comparable terminal branch lengths in the tree.
To test for genes with evidence for shared relaxation of selection in Paucidentomys and Rhynchomys, we ran RELAX in HyPhy 2.5.14 using both the species tree topology and gene tree topology data sets. For comparison, relaxation analyses were also repeated using the same nonconvergent species pair as above, M. fuscus and E. centrosa. All P-values were corrected for multiple tests at a P < 0.05 threshold, using a Benjamini–Hochberg FDR correction.
Detecting Convergence Site-Based Functional Shifts
To detect potential convergence of positively selected sites in Paucidentomys and Rhynchomys, we tested all genes for evidence of convergent amino acid shifts using PCOC (Rey et al. 2018). This approach applies a CAT model (Quang et al. 2008) of protein evolution in a species-tree context to detect convergent shifts in amino acid profile along branches with convergent phenotypes. To filter for only sites with strong evidence for convergent profile shifts, we set a posterior probability threshold of >0.9 for all PCOC, OC, and PC output.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Acknowledgments
This study was supported by the U.S. Government National Science Foundation (grant numbers DEB-1457654, DEB-1754393, DEB-1754096, DEB-1441634, and OISE-0965856), National Institutes of Health (grant numbers R01-HD073439, R01-HD094787), National Geographic Society (9025-11), Australia and Pacific Science Foundation (12-6). E.R. was supported by an Australian Government Research Training Program Scholarship, the Dame Margaret Blackwood Soroptimist Scholarship and the Alfred Nicholas Fellowship. We are grateful to Gregg Thomas for providing feedback on an earlier version of this manuscript, Jonathan Nations for advice on murine trait data and comparative analyses, and comments from two anonymous reviewers that improved the final version. We are indebted to the Indonesian Institute of Sciences (Lembaga Ilmu Pengetahuan Indonesia, LIPI) for access to material from Sulawesi, and to Museums Victoria, the Australian Biological Tissue Collection, Louisiana State University Museum of Natural Science, University of Kansas Biodiversity Research Center, and the Field Museum of Natural History for access to specimens. We also thank Melbourne Bioinformatics and Research Platform Services at the University of Melbourne for access to high-performance computing resources.
Data Availability
Raw sequence reads are available via the NCBI Sequence Read Archive under BioProject ID PRJNA705792, BioSample accession numbers SAMN18102763–SAMN18102800, SRA accession numbers SRR13848278–SRR13848315. Code used to process sequence data is available at https://github.com/Victaphanta/ECPP/. Processed sequence alignments underlying the analyses in this manuscript are available via FigShare via https://doi.org/10.6084/m9.figshare.14658762.

{kind=link}
{kind=link}
{kind=link}
{kind=link}