





Abstract
Biodegradation of the phenylurea herbicide linuron appears a specialization within a specific clade of the Variovorax genus. The linuron catabolic ability is likely acquired by horizontal gene transfer but the mechanisms involved are not known. The full-genome sequences of six linuron-degrading Variovorax strains isolated from geographically distant locations were analyzed to acquire insight into the mechanisms of genetic adaptation toward linuron metabolism. Whole-genome sequence analysis confirmed the phylogenetic position of the linuron degraders in a separate clade within Variovorax and indicated that they unlikely originate from a common ancestral linuron degrader. The linuron degraders differentiated from Variovorax strains that do not degrade linuron by the presence of multiple plasmids of 20–839 kb, including plasmids of unknown plasmid groups. The linuron catabolic gene clusters showed 1) high conservation and synteny and 2) strain-dependent distribution among the different plasmids. Most of them were bordered by IS1071 elements forming composite transposon structures, often in a multimeric array configuration, appointing IS1071 as a key element in the recruitment of linuron catabolic genes in Variovorax. Most of the strains carried at least one (catabolic) broad host range plasmid that might have been a second instrument for catabolic gene acquisition. We conclude that clade 1 Variovorax strains, despite their different geographical origin, made use of a limited genetic repertoire regarding both catabolic functions and vehicles to acquire linuron biodegradation.
Introduction
Linuron [3-(3,4-dichlorophenyl)-1-methoxy-1-methyl urea] is a phenylurea herbicide that has been widely used for weed control in agriculture. Biodegradation is the major route of linuron dissipation in the environment (Bers et al. 2011). Bacteria belonging to the genus Variovorax were isolated from geographically distant locations either as single strains (Sørensen et al. 2005; Breugelmans et al. 2007; Satsuma 2010) or as members of consortia (Dejonghe et al. 2003; Breugelmans et al. 2007) that have the ability to mineralize and utilize the herbicide for growth. Single strains convert linuron to CO2 and cell material, whereas in consortia Variovorax perform particularly the initial hydrolysis of linuron into the primary metabolite 3,4-dichloroaniline (DCA). The metabolic pathway of linuron degradation in Variovorax sp. WDL1 and SRS16 are well studied. The linuron hydrolases HylA (identified in WDL1) (Bers et al. 2013) and LibA (identified in SRS16) (Bers et al. 2011) perform the hydrolysis of linuron into DCA and N,O-dimethylhydroxylamine (N,O-DMHA) (Bers et al. 2011, 2013). In both strains, a multicomponent chloroaniline dioxygenase DcaQTA1A2B converts DCA to 4,5-dichlorocatechol, whereas chlorocatechol is further metabolized to oxo-adipate by enzymes encoded by the ccdCFDE gene cluster (Boon et al. 2001). PCR analysis has shown that other linuron-degrading Variovorax share the same catabolic genes. Interestingly, based on 16S rRNA gene phylogeny, the linuron-degrading Variovorax strains appear to belong to a clade of Variovorax strains that separates from the main bulk of strains including most of the type strains (Breugelmans et al. 2007). The ability to degrade and/or grow on linuron is unique for those strains within the Variovorax genus, indicating that they must have genetically adapted by acquiring the catabolic genes by horizontal gene transfer (HGT). This is supported by the observation that in SRS16 and WDL1, the catabolic genes are physically linked with mobile genetic elements (MGE). In SRS16, the DCA catabolic genes are bordered by multiple insertion sequence (IS) elements (Bers et al. 2011). The same applies to hylA, the dca cluster and the ccd cluster in strain WDL1 (Albers, Lood, et al. 2018). Moreover, the three catabolic gene clusters in Variovorax sp. WDL1 reside on a large extrachromosomal element that shows several plasmid features including gene functions for conjugation (Albers, Lood, et al. 2018). However, how the genetic composition and context of the catabolic genes and their linkage with MGEs vary between different linuron-degrading Variovorax strains and how these relate to the geographic origin of the strains and their phylogeny is yet unknown. Such knowledge will provide insight in the mechanisms that govern the functional evolution of genomes and especially those of organic xenobiotic degraders and more specifically of the genus Variovorax. This organism inhabits a wide variety of environments suggesting that it is prone to adaptation to new environmental constraints. To this end, we sequenced the complete genomes of six different linuron-degrading Variovorax strains isolated from distantly located geographical areas. We 1) reanalyzed the phylogenetic relationship between the strains and their phylogenetic position within the Variovorax genus, 2) examined how their genomes differ with those of Variovorax strains that do not degrade linuron, emphasizing on the occurrence and types of MGEs, and 3) compared the structure and surrounding context of the gene clusters involved in linuron metabolism.
Materials and Methods
Genome Sequencing, Assembly, and Annotation
The details of the biomass and library preparation for genome sequencing are given in the supplementary text S1, Supplementary Material online. Genome assembly was performed based on the PacBio reads by means of the RS_HGAP_Assembly.3 protocol included in SMRT Portal version 2.3.0 applying target genome sizes of 5 Mb (PBL-E5), 15 Mb (WDL1), and 10 Mb (others). All assemblies showed one chromosomal contig, several extrachromosomal contigs, and several artificial contigs. Artificial contigs were removed from the assembly. The remaining contigs were circularized and assembly redundancies at the ends of the contigs were removed. ORFs on the replicons were ordered using dnaA (chromosome) or repA/parA (plasmids) as the first ORF. Error correction was performed by mapping the Illumina short reads onto finished genomes using bwa v. 0.6.2 in paired-end (sampe) mode using default settings (Li and Durbin 2009) with subsequent variant and consensus calling using VarScan v. 2.3.6 (Parameters: mpileup2cns –min-coverage 10 –min-reads2 6 –min-avg-qual 20 –min-var-freq 0.8 –min-freq-for-hom 0.75 –P value 0.01 –strand-filter 1 –variants 1 –output-vcf 1) (Li and Durbin 2009). A consensus concordance of QV60 was reached. Automated genome annotation was performed using Prokka v. 1.8 (Seemann 2014).
Phylogenomic Analysis of Variovorax sp. Plasmids and Genomes
The accession numbers of the genome and plasmid sequences used to construct the phylogenetic trees are listed in supplementary table S1, Supplementary Material online. First, a phylogenomic analysis of the whole-genome data set was conducted at the nucleotide (nt) level using the truly whole-genome-based Genome-BLAST Distance Phylogeny method (GBDP) (Meier-Kolthoff et al. 2013, 2014; Meier-Kolthoff and Göker 2019). Briefly, GBDP infers accurate intergenomic distances between pairs of genome sequences and subjects resulting distances matrices to a distance-based phylogenetic reconstruction under settings recommended for the comparison of prokaryotic genomes (Meier-Kolthoff et al. 2013). The method is used by both the Genome-to-Genome Distance Calculator 2.1 (Meier-Kolthoff et al. 2013) and the Type Strain Genome Server (Meier-Kolthoff and Göker 2019).
A second phylogenetic analysis based on the plasmids’ amino acid sequences was conducted using GBDP as well, except that GBDP distance calculations were done under settings recommended for the analysis of bacteriophage sequences (Meier-Kolthoff and Göker 2017). The reason is that, compared to bacteriophages, the plasmids had similar sequence lengths (Meier-Kolthoff and Göker 2017) and thus promised an equally good performance of the GBDP method when applied to plasmid data. The publicly available plasmid sequences included in this study were selected based on their relatedness to the newly sequenced plasmids, in order to allocate them into known plasmid groups. In both analyses, a balanced minimum evolution tree was inferred using FastME v2.1.4 with SPR postprocessing each (Lefort et al. 2015). Hundred replicate trees were reconstructed in the same way and branch support was subsequently mapped onto the respective tree (Meier-Kolthoff et al. 2014). For the 16S rRNA gene sequence-based phylogeny, the whole 16S rRNA gene sequences were retrieved from the SILVA database (Quast et al. 2012), and aligned with the SINA aligner (Pruesse et al. 2012). The phylogenies were inferred on the GGDC web server (Meier-Kolthoff et al. 2013) using the DSMZ phylogenomics pipeline (https://ggdc.dsmz.de/phylogeny-service.php, last accessed April 05, 2020). The Maximum likelihood (ML) tree was inferred from the alignment with RAxML (Stamatakis 2014). Rapid bootstrapping in conjunction with the autoMRE bootstopping criterion (Pattengale et al. 2010) and subsequent search for the best tree was used. The sequences were checked for a compositional bias using the χ2 test as implemented in PAUP* (Cummings 2014). All phylogenetic trees were visualized with iTOL (Letunic and Bork 2019).
Analysis of Variovorax sp. Plasmids
The codon usage of chromosomes and plasmids was calculated with CompareM v. 0.0.23 (Parks 2017). Subsequently, Principal Component Analysis (PCA) was conducted and the principle components were hierarchically clustered using the Ward’s criterion with FactoMineR v. 1.36 (Le et al. 2008). The replication origins as well as the type IV secretion systems were predicted with oriTfinder v. 1.1 (Li et al. 2018). The relaxase protein sequences were classified into MOB families using MOBScan (Garcillán-Barcia et al. 2009; Garcillan-Barcia et al. 2020). Genes shared between pPBL-E5-2, pSRS16-3, and the chromosomes of the linuron-degrading Variovorax were determined with Proteinortho (Lechner et al. 2011). SimpleSynteny (Veltri et al. 2016) was used to determine the positions of the catabolic clusters and associated ORFs, and draw the catabolic cluster illustrations.
Results and Discussion
General Genome Features of Linuron-Degrading Variovorax Strains
The full-genome sequences of six linuron-degrading Variovorax sp. strains, that is, WDL1 (Dejonghe et al. 2003), SRS16 (Sørensen et al. 2005), PBL-H6, PBL-E5, PBS-H4 (Breugelmans et al. 2007), and RA8 (Satsuma 2010) were obtained. Their general genomic features are listed in table 1. Strain WDL1 was recently found to consist of two subpopulations that only deviate in the presence of the hylA or dca locus (Albers, Lood, et al. 2018). In this study, the genome of one of these two subpopulations, that is, the one carrying the hylA locus was resequenced. The new sequence deviated slightly from the one reported by Albers, Lood, et al. (2018). The 5,400- and 1,240-kbp replicons formed one chromosome of 6.7 Mb, whereas the 1,380-kbp plasmid-like extrachromosomal replicon consisted of two replicons, that is, pWDL1-1 (800 kbp) carrying the linuron catabolic genes and pWDL1-2 (540 kbp).
General Properties of the Genome Sequences of Linuron-Degrading Variovorax Strains, along with Their Phenotypes and Geographic Origins
| Strain | Replicon | Size (kb) | GC% | Number of | Degradation Genes | |||
|---|---|---|---|---|---|---|---|---|
| ORFs | tRNA Genes | Transposases | IS1071 | |||||
| WDL1 (linuron to CO2) Belgium | Chromosome | 6,724.9 | 67.2 | 6,376 | 56 | 64 | ||
| pWDL1-1 | 825.1 | 62.5 | 862 | 44 | 29 | 5 | hylA or dca and ccd | |
| pWDL1-2 | 565.9 | 63.5 | 543 | 27 | 24 | |||
| pWDL1-3 | 207.9 | 63.7 | 229 | 42 | ||||
| pWDL1-5 | 20.1 | 62.6 | 25 | |||||
| pWDL1-4 | 25.0 | 62.6 | 23 | 8 | 1 | |||
| Total | 8,368.8 | 8,058 | 127 | 167 | 6 | |||
| PBL-H6 (linuron to CO2) Halen, Belgium | Chromosome | 5,990.2 | 66.8 | 5,557 | 54 | 6 | ||
| pPBL-H6-1 | 839.2 | 62.4 | 883 | 50 | 36 | 5 | libA, dca, ccd | |
| pPBL-H6-2 (PromA) | 42.1 | 63.5 | 49 | 1 | 1 | |||
| Total | 6,871.6 | 6,489 | 43 | 6 | ||||
| PBS-H4 (linuron to DCA) Halen, Belgium | Chromosome | 6,429.8 | 66.9 | 6,031 | 57 | 7 | 2 | |
| pPBS-H4-1 | 117.4 | 64.9 | 95 | 2 | ||||
| pPBS-H4-2 (PromA) | 104.9 | 62.7 | 112 | 14 | 5 | hylA, ccd | ||
| Total | 6,652.1 | 6,238 | 161 | 23 | 7 | |||
| RA8 (linuron to DCA) Japan | Chromosome | 6,501.6 | 67.2 | 6,129 | 52 | 7 | ||
| pRA8-1 | 429.0 | 64.9 | 443 | 8 | 2 | ccd | ||
| pRA8-2 | 425.3 | 64.2 | 419 | 23 | 19 | 2 | ||
| pRA8-3 (IncP-1) | 68.4 | 61.2 | 63 | 30 | 3 | libA | ||
| Total | 7,424.2 | 482 | 23 | 64 | 7 | |||
| SRS16 (linuron to CO2) Simmelkær, Denmark | Chromosome | 5,763.0 | 67.3 | 5,469 | 50 | 13 | ||
| pSRS16-1 | 801.4 | 62.5 | 852 | 50 | 19 | 2 | dca, ccd | |
| pSRS16-2 | 560.6 | 64.5 | 555 | 31 | ||||
| pSRS16-3 | 478.8 | 67 | 469 | 0 | ||||
| pSRS16-4 (IncP-1) | 71.1 | 61.9 | 66 | 12 | 3 | libA | ||
| Total | 7,674.9 | 7,411 | 100 | 75 | 5 | |||
| PBL-E5 (linuron to CO2) Simmelkær, Denmark | Chromosome | 5,660.8 | 67.3 | 5,421 | 47 | 10 | ||
| pPBL-E5-1 | 801.5 | 62.5 | 844 | 50 | 20 | 1 | dca, ccd | |
| pPBL-E5-2 | 553.0 | 67.1 | 550 | 4 | ||||
| pPBL-E5-3 (IncP-1) | 71.0 | 61.3 | 66 | 11 | 3 | libA | ||
| Total | 7,086.4 | 6,881 | 97 | 45 | 4 | |||
| Strain | Replicon | Size (kb) | GC% | Number of | Degradation Genes | |||
|---|---|---|---|---|---|---|---|---|
| ORFs | tRNA Genes | Transposases | IS1071 | |||||
| WDL1 (linuron to CO2) Belgium | Chromosome | 6,724.9 | 67.2 | 6,376 | 56 | 64 | ||
| pWDL1-1 | 825.1 | 62.5 | 862 | 44 | 29 | 5 | hylA or dca and ccd | |
| pWDL1-2 | 565.9 | 63.5 | 543 | 27 | 24 | |||
| pWDL1-3 | 207.9 | 63.7 | 229 | 42 | ||||
| pWDL1-5 | 20.1 | 62.6 | 25 | |||||
| pWDL1-4 | 25.0 | 62.6 | 23 | 8 | 1 | |||
| Total | 8,368.8 | 8,058 | 127 | 167 | 6 | |||
| PBL-H6 (linuron to CO2) Halen, Belgium | Chromosome | 5,990.2 | 66.8 | 5,557 | 54 | 6 | ||
| pPBL-H6-1 | 839.2 | 62.4 | 883 | 50 | 36 | 5 | libA, dca, ccd | |
| pPBL-H6-2 (PromA) | 42.1 | 63.5 | 49 | 1 | 1 | |||
| Total | 6,871.6 | 6,489 | 43 | 6 | ||||
| PBS-H4 (linuron to DCA) Halen, Belgium | Chromosome | 6,429.8 | 66.9 | 6,031 | 57 | 7 | 2 | |
| pPBS-H4-1 | 117.4 | 64.9 | 95 | 2 | ||||
| pPBS-H4-2 (PromA) | 104.9 | 62.7 | 112 | 14 | 5 | hylA, ccd | ||
| Total | 6,652.1 | 6,238 | 161 | 23 | 7 | |||
| RA8 (linuron to DCA) Japan | Chromosome | 6,501.6 | 67.2 | 6,129 | 52 | 7 | ||
| pRA8-1 | 429.0 | 64.9 | 443 | 8 | 2 | ccd | ||
| pRA8-2 | 425.3 | 64.2 | 419 | 23 | 19 | 2 | ||
| pRA8-3 (IncP-1) | 68.4 | 61.2 | 63 | 30 | 3 | libA | ||
| Total | 7,424.2 | 482 | 23 | 64 | 7 | |||
| SRS16 (linuron to CO2) Simmelkær, Denmark | Chromosome | 5,763.0 | 67.3 | 5,469 | 50 | 13 | ||
| pSRS16-1 | 801.4 | 62.5 | 852 | 50 | 19 | 2 | dca, ccd | |
| pSRS16-2 | 560.6 | 64.5 | 555 | 31 | ||||
| pSRS16-3 | 478.8 | 67 | 469 | 0 | ||||
| pSRS16-4 (IncP-1) | 71.1 | 61.9 | 66 | 12 | 3 | libA | ||
| Total | 7,674.9 | 7,411 | 100 | 75 | 5 | |||
| PBL-E5 (linuron to CO2) Simmelkær, Denmark | Chromosome | 5,660.8 | 67.3 | 5,421 | 47 | 10 | ||
| pPBL-E5-1 | 801.5 | 62.5 | 844 | 50 | 20 | 1 | dca, ccd | |
| pPBL-E5-2 | 553.0 | 67.1 | 550 | 4 | ||||
| pPBL-E5-3 (IncP-1) | 71.0 | 61.3 | 66 | 11 | 3 | libA | ||
| Total | 7,086.4 | 6,881 | 97 | 45 | 4 | |||
General Properties of the Genome Sequences of Linuron-Degrading Variovorax Strains, along with Their Phenotypes and Geographic Origins
| Strain | Replicon | Size (kb) | GC% | Number of | Degradation Genes | |||
|---|---|---|---|---|---|---|---|---|
| ORFs | tRNA Genes | Transposases | IS1071 | |||||
| WDL1 (linuron to CO2) Belgium | Chromosome | 6,724.9 | 67.2 | 6,376 | 56 | 64 | ||
| pWDL1-1 | 825.1 | 62.5 | 862 | 44 | 29 | 5 | hylA or dca and ccd | |
| pWDL1-2 | 565.9 | 63.5 | 543 | 27 | 24 | |||
| pWDL1-3 | 207.9 | 63.7 | 229 | 42 | ||||
| pWDL1-5 | 20.1 | 62.6 | 25 | |||||
| pWDL1-4 | 25.0 | 62.6 | 23 | 8 | 1 | |||
| Total | 8,368.8 | 8,058 | 127 | 167 | 6 | |||
| PBL-H6 (linuron to CO2) Halen, Belgium | Chromosome | 5,990.2 | 66.8 | 5,557 | 54 | 6 | ||
| pPBL-H6-1 | 839.2 | 62.4 | 883 | 50 | 36 | 5 | libA, dca, ccd | |
| pPBL-H6-2 (PromA) | 42.1 | 63.5 | 49 | 1 | 1 | |||
| Total | 6,871.6 | 6,489 | 43 | 6 | ||||
| PBS-H4 (linuron to DCA) Halen, Belgium | Chromosome | 6,429.8 | 66.9 | 6,031 | 57 | 7 | 2 | |
| pPBS-H4-1 | 117.4 | 64.9 | 95 | 2 | ||||
| pPBS-H4-2 (PromA) | 104.9 | 62.7 | 112 | 14 | 5 | hylA, ccd | ||
| Total | 6,652.1 | 6,238 | 161 | 23 | 7 | |||
| RA8 (linuron to DCA) Japan | Chromosome | 6,501.6 | 67.2 | 6,129 | 52 | 7 | ||
| pRA8-1 | 429.0 | 64.9 | 443 | 8 | 2 | ccd | ||
| pRA8-2 | 425.3 | 64.2 | 419 | 23 | 19 | 2 | ||
| pRA8-3 (IncP-1) | 68.4 | 61.2 | 63 | 30 | 3 | libA | ||
| Total | 7,424.2 | 482 | 23 | 64 | 7 | |||
| SRS16 (linuron to CO2) Simmelkær, Denmark | Chromosome | 5,763.0 | 67.3 | 5,469 | 50 | 13 | ||
| pSRS16-1 | 801.4 | 62.5 | 852 | 50 | 19 | 2 | dca, ccd | |
| pSRS16-2 | 560.6 | 64.5 | 555 | 31 | ||||
| pSRS16-3 | 478.8 | 67 | 469 | 0 | ||||
| pSRS16-4 (IncP-1) | 71.1 | 61.9 | 66 | 12 | 3 | libA | ||
| Total | 7,674.9 | 7,411 | 100 | 75 | 5 | |||
| PBL-E5 (linuron to CO2) Simmelkær, Denmark | Chromosome | 5,660.8 | 67.3 | 5,421 | 47 | 10 | ||
| pPBL-E5-1 | 801.5 | 62.5 | 844 | 50 | 20 | 1 | dca, ccd | |
| pPBL-E5-2 | 553.0 | 67.1 | 550 | 4 | ||||
| pPBL-E5-3 (IncP-1) | 71.0 | 61.3 | 66 | 11 | 3 | libA | ||
| Total | 7,086.4 | 6,881 | 97 | 45 | 4 | |||
| Strain | Replicon | Size (kb) | GC% | Number of | Degradation Genes | |||
|---|---|---|---|---|---|---|---|---|
| ORFs | tRNA Genes | Transposases | IS1071 | |||||
| WDL1 (linuron to CO2) Belgium | Chromosome | 6,724.9 | 67.2 | 6,376 | 56 | 64 | ||
| pWDL1-1 | 825.1 | 62.5 | 862 | 44 | 29 | 5 | hylA or dca and ccd | |
| pWDL1-2 | 565.9 | 63.5 | 543 | 27 | 24 | |||
| pWDL1-3 | 207.9 | 63.7 | 229 | 42 | ||||
| pWDL1-5 | 20.1 | 62.6 | 25 | |||||
| pWDL1-4 | 25.0 | 62.6 | 23 | 8 | 1 | |||
| Total | 8,368.8 | 8,058 | 127 | 167 | 6 | |||
| PBL-H6 (linuron to CO2) Halen, Belgium | Chromosome | 5,990.2 | 66.8 | 5,557 | 54 | 6 | ||
| pPBL-H6-1 | 839.2 | 62.4 | 883 | 50 | 36 | 5 | libA, dca, ccd | |
| pPBL-H6-2 (PromA) | 42.1 | 63.5 | 49 | 1 | 1 | |||
| Total | 6,871.6 | 6,489 | 43 | 6 | ||||
| PBS-H4 (linuron to DCA) Halen, Belgium | Chromosome | 6,429.8 | 66.9 | 6,031 | 57 | 7 | 2 | |
| pPBS-H4-1 | 117.4 | 64.9 | 95 | 2 | ||||
| pPBS-H4-2 (PromA) | 104.9 | 62.7 | 112 | 14 | 5 | hylA, ccd | ||
| Total | 6,652.1 | 6,238 | 161 | 23 | 7 | |||
| RA8 (linuron to DCA) Japan | Chromosome | 6,501.6 | 67.2 | 6,129 | 52 | 7 | ||
| pRA8-1 | 429.0 | 64.9 | 443 | 8 | 2 | ccd | ||
| pRA8-2 | 425.3 | 64.2 | 419 | 23 | 19 | 2 | ||
| pRA8-3 (IncP-1) | 68.4 | 61.2 | 63 | 30 | 3 | libA | ||
| Total | 7,424.2 | 482 | 23 | 64 | 7 | |||
| SRS16 (linuron to CO2) Simmelkær, Denmark | Chromosome | 5,763.0 | 67.3 | 5,469 | 50 | 13 | ||
| pSRS16-1 | 801.4 | 62.5 | 852 | 50 | 19 | 2 | dca, ccd | |
| pSRS16-2 | 560.6 | 64.5 | 555 | 31 | ||||
| pSRS16-3 | 478.8 | 67 | 469 | 0 | ||||
| pSRS16-4 (IncP-1) | 71.1 | 61.9 | 66 | 12 | 3 | libA | ||
| Total | 7,674.9 | 7,411 | 100 | 75 | 5 | |||
| PBL-E5 (linuron to CO2) Simmelkær, Denmark | Chromosome | 5,660.8 | 67.3 | 5,421 | 47 | 10 | ||
| pPBL-E5-1 | 801.5 | 62.5 | 844 | 50 | 20 | 1 | dca, ccd | |
| pPBL-E5-2 | 553.0 | 67.1 | 550 | 4 | ||||
| pPBL-E5-3 (IncP-1) | 71.0 | 61.3 | 66 | 11 | 3 | libA | ||
| Total | 7,086.4 | 6,881 | 97 | 45 | 4 | |||
Chromosome sizes (ranging from 5.99 to 8.36 Mb) and GC content (ranging from 66.24% to 66.86%) of the linuron-degrading Variovorax strains were comparable to those of Variovorax isolates that do not degrade linuron and for which the genome sequences were available. Sizes of reported nonlinuron-degrading Variovorax genomes range between 4.31 and 9.24 Mb (median: 7.2 Mb) with GC contents of 64.6–69.6 (median: 67.4) (supplementary table S1, Supplementary Material online). All linuron-degrading Variovorax strains contained a high number of extrachromosomal elements (two to six), including smaller obvious plasmid replicons (20–70 kb) but also larger replicons of >500 kb. The GC content and codon usage of most of those larger extrachromosomal elements substantially differed from those of the chromosome (supplementary fig. S1, Supplementary Material online). They also did not contain any essential genes for cell viability, categorizing them rather as plasmids than as a second chromosome or chromid (Harrison et al. 2010). Replicons pPBL-E5-2 and pSRS16-3, however, showed GC-contents and codon usage similar to those of the chromosome, carried plasmid-like replication modules, and shared genes with the chromosomes of other linuron-degrading Variovorax strains (67 genes for pSRS16-3, and 65 genes for pPBL-E5-2) classifying them as chromids according to Harrison et al. (2010). In contrast to the genomes of the linuron degraders, none of the genomes of Variovorax isolates that do not degrade linuron carried plasmids. IncP-1 plasmids were however reported in three not phylogenetically classified Variovorax isolates with unknown genome sequences. Plasmids pHB44 (Zhang et al. 2015) and pBS64 (Zhang et al. 2015) were identified in Variovorax strains associated with the mycorrhizal fungus Laccaria proxima. They carry genes that increase the Variovorax host fitness by enabling metal ion transport and bacitracin resistance (Zhang et al. 2016). Plasmid pDB1 (Kim et al. 2013) in Variovorax sp. DB1, carries genes for the biodegradation of the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D). Another feature that distinguished the linuron-degrading strains from the nondegraders is the occurrence of a high number of IS1071 IS elements varying from four to seven copies in the degraders, whereas nondegraders did not carry any IS1071. IS1071 is an IS that was first described bordering the 3-chlorobenzoate catabolic genes of the Comamonas testosteroni BRC60 plasmid pBRC60 (Fulthorpe and Wyndham 1992) and was classified as a class II (Tn3-like) transposon (Nakatsu et al. 1991). Since then it has been frequently associated with catabolic genes in various organisms, primarily β-proteobacteria (Dunon et al. 2018). It often flanks the catabolic genes at both sites, forming a composite transposon structure (Fulthorpe and Wyndham 1992). The element has been suggested to play a primary role in the acquisition and subsequent distribution of adaptive genes and especially catabolic functions in bacteria (Providenti et al. 2006; Dunon et al. 2013, 2018).
Phylogenetic Analysis of Linuron-Degrading Variovorax Strains
The phylogenetic relatedness between the linuron-degrading Variovorax strains was determined by digital DNA:DNA hybridization values (dDDH) (supplementary table S2, Supplementary Material online). With the exception of PBL-E5 and SRS16, which represent the same species (dDDH value of 86%), all linuron-degrading Variovorax were designated as distinct species, and none of them belonged to any type species. Their phylogenetic divergence strongly indicates that the five degraders acquired linuron degradation genes independently as opposed to being derived from one common ancestral linuron degrader. Whole genome-based phylogeny showed that the Variovorax species sequenced to date separated into two clades (clade 1 and clade 2), and that the linuron degraders are closely related species, all belonging to clade 1 (fig. 1). The tree topology remained the same when only the linuron degrader chromosomes were used (supplementary fig. S2, Supplementary Material online). This separation largely replicated the 16S rRNA gene sequence-based phylogeny (supplementary fig. S3, Supplementary Material online), with the exception of Variovorax soli and Variovorax sp. OV329. The low dDDH values of the V. soli genome with the degrader genomes (24.7–25.7%) however confirmed that they are distantly related.
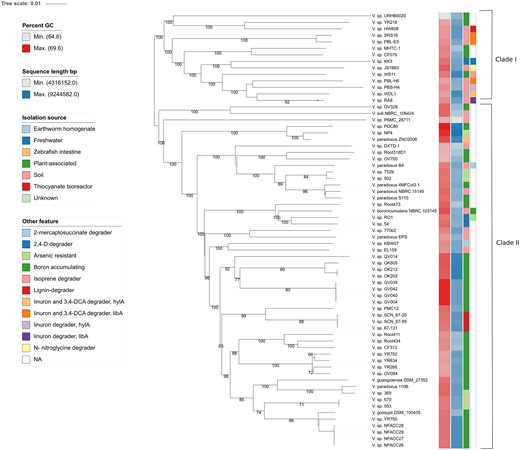
—GBDP phylogenomic analysis of the Variovorax whole-genome sequence data set. The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values from 100 replications, with an average branch support of 80.6%. Leaf labels are further annotated by their genomic G + C content, genome sequence length, phenotypic attributes, and origin of isolation. The two Variovorax clades are indicated.
In addition to the linuron-degrading Variovorax strains, clade 1 included various other isolates but no type species. From those, a closed genome sequence was only available for the lignin-degrading soil isolate strain HW608 (Woo et al. 2017). The phylogenetic position of the linuron-degrading strains was independent of either the geographic origin, the capacity to degrade linuron completely or partially to DCA, or the presence of specific catabolic genes involved in linuron biodegradation. Clade 2 contained the majority of the Variovorax strains, including the species Variovorax boronicumulans, V. soli, Variovorax paradoxus, Variovorax gossypi, and Variovorax guangxiensis. Interestingly, in contrast to the clade 2 strains, nonlinuron-degrading strains from clade 1 were often associated with the catabolism of natural and anthropogenic organic compounds such as Variovorax sp. WS11 (an isoprene-degrading phyllosphere isolate; Crombie et al. 2018), KK3 (a 2,4-D-degrading freshwater isolate; Wang et al. 2017), and JJ 1663 (an N-nitroglycine-degrading activated sludge isolate; Mahan et al. 2017). As such, including the linuron degraders, eight of the 14 clade 1 strains were degraders of anthropogenic compounds. Clade 2, however, included only one xenobiotic-degrading isolate (one in 55 strains), that is, V. boronicumulans J1 (Satola et al. 2013) that degrades the neonicotinoid thiacloprid but only cometabolically (Zhang et al. 2012). These results indicate that the linuron-degrading strains belong to a Variovorax clade or originates from a common ancestor that is/was more prone to genetic adaptation and hence specialization toward the biodegradation of anthropogenic compounds. The clade separation of strains with and without biodegradation capacity has not been observed before in other genera (Tabata et al. 2016; Kim et al. 2018).
Plasmids Hosted by Linuron-Degrading Variovorax sp
Phylogenetic analysis of the entire plasmid sequences clustered the plasmids identified in the linuron-degrading Variovorax sp. strains, into both known and novel plasmid groups (fig. 2). Some of the plasmids occur in multiple strains in which they are highly conserved (supplementary fig. S2, Supplementary Material online). Table 2 shows an overview of the replication and conjugation systems encoded on these plasmids. Twelve plasmids have type IV secretion system genes (T4SS) which facilitate conjugative transfer, but the origin of transfer (oriT) could not always be determined.
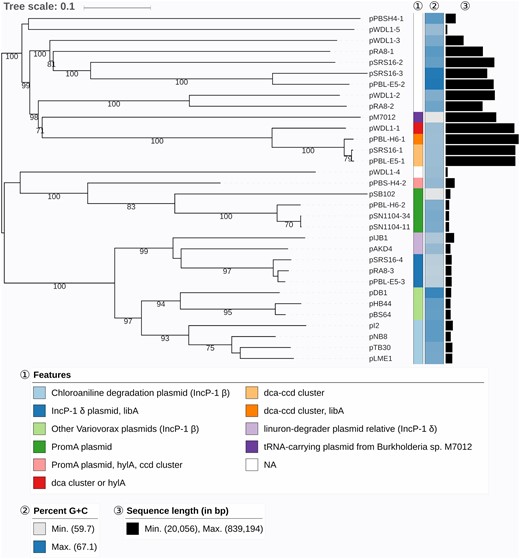
—GBDP phylogenomic analysis of Variovorax plasmids and relevant relatives. The branch lengths are scaled in terms of GBDP distance formula d6. The numbers above branches are GBDP pseudo-bootstrap support values from 100 replications, with an average branch support of 91.3%. Leaf labels are further annotated by their genomic G + C content, length as well as special attributes. NCBI accession numbers of previously reported plasmid sequences: pM7012 (NC_022995.1), pSB102 (AJ304453.1), pSN1104-34 (AP018708.1), pSN1104-11 (AP018707.1), pIJB1 (JX847411.1), pAKD4 (GQ983559.1), pDB1 (JQ436721.1), pHB44 (KU356988.1), pBS64 (KU356987.1), pL2 (JF274989.1), pNB8 (NC_019264.1), pLME1 (NC_019263.1), and pTB30 (NC_016968.1).
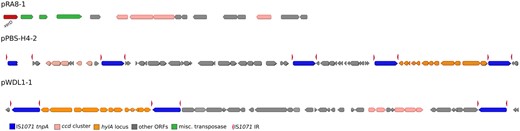
—Genomic context of linuron-catabolic genes in strains WDL1, RA8, and PBS-H4. The hylA and ccd loci on the plasmids pPBS-H4-2, pWDL1-1, and pRA8-1, as well as flanking genes are illustrated. Broken arrows indicate ORFs which were truncated by immature stop codons, due to point mutations. IS1071 inverted repeats (IR) are indicated by the red arrow points. Non-catabolic ORFs as well as hypothetical genes are indicated in gray.
Overview of Plasmid Replication and Conjugation Systems Identified in Linuron-Degrading Variovorax Strains
| Plasmid | Relaxase | T4CP | T4SS | oriT | Rep/Par Module |
|---|---|---|---|---|---|
| n.f. | 34% YP_001911165 | TraALBFHJDNUW, TrbC, VirB4 | n.f. | RepB-ParAB |
| pSRS16-2 | TraI 49% YP_195891 (MOBP) | 72% NP_990928.1 | TrbLJIHGFEDCB, TraJ, VirB1 | P | RepB-ParAB |
| n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| TraI 100% YP_006965894.1 (MOBP) | 100% NP_990928.1 | TrbBCDEFGHIJKLN, TraXF | P | RepB-ParAB |
| TraS 79% CAC79161 (MOBP) | 67% YP_001672044 | VirB123456891011, VirD4 | P | RepA |
| pRA8-1 | Rel 81% SDZ72275.1 (MOBP) | 45% WP_010895213 | VirB23456891011, VirD4 | n.f. | RepB-ParAB |
| pRA8-2 | n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| pPBS-H4-1 | TraA 37% AAV52093 (MOBF) | n.f. | n.f. | n.f. | RepA |
| pWDL1-2 | n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| pWDL1-3 | Rel 84% WP_093180082.1 (MOBP) | 45% WP_010895213 | VirB23456891011, VirD4, TrbB | n.f. | RepAB |
| pWDL1-4 | n.f. | n.f. | n.f. | n.f. | ParAB |
| pWDL1-5 | n.f. | 38% AEY63616 | VirB568, TraD | n.f. | RepA |
| chrWDL1 | TrbBCDEJLFGI, VirD4, TraF | ||||
| chrSRS16 | TrbBCDEJLFGI, VirD4 | ||||
| chrRA8 | n.f. | ||||
| chrPBL-H6 | TrbBCDEJLFGI, VirD4 | ||||
| chrPBL-E5 | TrbBCDEJLFGI, VirD4 | ||||
| chrPBS-H4 | TrbBCDEJLFGI, VirD4 |
| Plasmid | Relaxase | T4CP | T4SS | oriT | Rep/Par Module |
|---|---|---|---|---|---|
| n.f. | 34% YP_001911165 | TraALBFHJDNUW, TrbC, VirB4 | n.f. | RepB-ParAB |
| pSRS16-2 | TraI 49% YP_195891 (MOBP) | 72% NP_990928.1 | TrbLJIHGFEDCB, TraJ, VirB1 | P | RepB-ParAB |
| n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| TraI 100% YP_006965894.1 (MOBP) | 100% NP_990928.1 | TrbBCDEFGHIJKLN, TraXF | P | RepB-ParAB |
| TraS 79% CAC79161 (MOBP) | 67% YP_001672044 | VirB123456891011, VirD4 | P | RepA |
| pRA8-1 | Rel 81% SDZ72275.1 (MOBP) | 45% WP_010895213 | VirB23456891011, VirD4 | n.f. | RepB-ParAB |
| pRA8-2 | n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| pPBS-H4-1 | TraA 37% AAV52093 (MOBF) | n.f. | n.f. | n.f. | RepA |
| pWDL1-2 | n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| pWDL1-3 | Rel 84% WP_093180082.1 (MOBP) | 45% WP_010895213 | VirB23456891011, VirD4, TrbB | n.f. | RepAB |
| pWDL1-4 | n.f. | n.f. | n.f. | n.f. | ParAB |
| pWDL1-5 | n.f. | 38% AEY63616 | VirB568, TraD | n.f. | RepA |
| chrWDL1 | TrbBCDEJLFGI, VirD4, TraF | ||||
| chrSRS16 | TrbBCDEJLFGI, VirD4 | ||||
| chrRA8 | n.f. | ||||
| chrPBL-H6 | TrbBCDEJLFGI, VirD4 | ||||
| chrPBL-E5 | TrbBCDEJLFGI, VirD4 | ||||
| chrPBS-H4 | TrbBCDEJLFGI, VirD4 |
Note.—For the relaxase and T4SS, the % identity on amino acid level to the nearest relative as well as the accession number are given. The plasmids were classified into MOB classes if applicable. T4SS, type IV secretion system; T4CP, type IV coupling protein; n.f., not found; P, present.
Overview of Plasmid Replication and Conjugation Systems Identified in Linuron-Degrading Variovorax Strains
| Plasmid | Relaxase | T4CP | T4SS | oriT | Rep/Par Module |
|---|---|---|---|---|---|
| n.f. | 34% YP_001911165 | TraALBFHJDNUW, TrbC, VirB4 | n.f. | RepB-ParAB |
| pSRS16-2 | TraI 49% YP_195891 (MOBP) | 72% NP_990928.1 | TrbLJIHGFEDCB, TraJ, VirB1 | P | RepB-ParAB |
| n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| TraI 100% YP_006965894.1 (MOBP) | 100% NP_990928.1 | TrbBCDEFGHIJKLN, TraXF | P | RepB-ParAB |
| TraS 79% CAC79161 (MOBP) | 67% YP_001672044 | VirB123456891011, VirD4 | P | RepA |
| pRA8-1 | Rel 81% SDZ72275.1 (MOBP) | 45% WP_010895213 | VirB23456891011, VirD4 | n.f. | RepB-ParAB |
| pRA8-2 | n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| pPBS-H4-1 | TraA 37% AAV52093 (MOBF) | n.f. | n.f. | n.f. | RepA |
| pWDL1-2 | n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| pWDL1-3 | Rel 84% WP_093180082.1 (MOBP) | 45% WP_010895213 | VirB23456891011, VirD4, TrbB | n.f. | RepAB |
| pWDL1-4 | n.f. | n.f. | n.f. | n.f. | ParAB |
| pWDL1-5 | n.f. | 38% AEY63616 | VirB568, TraD | n.f. | RepA |
| chrWDL1 | TrbBCDEJLFGI, VirD4, TraF | ||||
| chrSRS16 | TrbBCDEJLFGI, VirD4 | ||||
| chrRA8 | n.f. | ||||
| chrPBL-H6 | TrbBCDEJLFGI, VirD4 | ||||
| chrPBL-E5 | TrbBCDEJLFGI, VirD4 | ||||
| chrPBS-H4 | TrbBCDEJLFGI, VirD4 |
| Plasmid | Relaxase | T4CP | T4SS | oriT | Rep/Par Module |
|---|---|---|---|---|---|
| n.f. | 34% YP_001911165 | TraALBFHJDNUW, TrbC, VirB4 | n.f. | RepB-ParAB |
| pSRS16-2 | TraI 49% YP_195891 (MOBP) | 72% NP_990928.1 | TrbLJIHGFEDCB, TraJ, VirB1 | P | RepB-ParAB |
| n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| TraI 100% YP_006965894.1 (MOBP) | 100% NP_990928.1 | TrbBCDEFGHIJKLN, TraXF | P | RepB-ParAB |
| TraS 79% CAC79161 (MOBP) | 67% YP_001672044 | VirB123456891011, VirD4 | P | RepA |
| pRA8-1 | Rel 81% SDZ72275.1 (MOBP) | 45% WP_010895213 | VirB23456891011, VirD4 | n.f. | RepB-ParAB |
| pRA8-2 | n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| pPBS-H4-1 | TraA 37% AAV52093 (MOBF) | n.f. | n.f. | n.f. | RepA |
| pWDL1-2 | n.f. | n.f. | n.f. | n.f. | RepB-ParAB |
| pWDL1-3 | Rel 84% WP_093180082.1 (MOBP) | 45% WP_010895213 | VirB23456891011, VirD4, TrbB | n.f. | RepAB |
| pWDL1-4 | n.f. | n.f. | n.f. | n.f. | ParAB |
| pWDL1-5 | n.f. | 38% AEY63616 | VirB568, TraD | n.f. | RepA |
| chrWDL1 | TrbBCDEJLFGI, VirD4, TraF | ||||
| chrSRS16 | TrbBCDEJLFGI, VirD4 | ||||
| chrRA8 | n.f. | ||||
| chrPBL-H6 | TrbBCDEJLFGI, VirD4 | ||||
| chrPBL-E5 | TrbBCDEJLFGI, VirD4 | ||||
| chrPBS-H4 | TrbBCDEJLFGI, VirD4 |
Note.—For the relaxase and T4SS, the % identity on amino acid level to the nearest relative as well as the accession number are given. The plasmids were classified into MOB classes if applicable. T4SS, type IV secretion system; T4CP, type IV coupling protein; n.f., not found; P, present.
Known Conjugative Plasmids and Their Role in Linuron Degradation in Linuron-Degrading Variovorax Strains
Plasmids pPBL-E5-3, pRA8-3, and pSRS16-4 were classified as IncP-1δ plasmids. The three plasmids were 99% identical at the nt level over the entire plasmid sequence and carry the libA locus between trfA and oriV, a known insertion hot spot for accessory genes in IncP-1 plasmids (Dunon et al. 2018). Catabolic IncP-1δ plasmids have been reported before either isolated by means of exogenous isolation (Sen et al. 2010) or from isolates (Xia et al. 1998). They all carry 2,4-D catabolic genes between trfA and oriV. The above-mentioned Variovorax plasmids pDB1 (Kim et al. 2013), pHB44 (Zhang et al. 2015), and pBS64 (Zhang et al. 2015) are also IncP-1 plasmids but belong to the IncP-1 β group.
Instead of IncP-1 plasmids, PBL-H6 and PBS-H4 carried PromA plasmids. PromA plasmids form a group of self-transmissible broad host range plasmids. Plasmid pPBS-H4-2 carries hylA and the ccd gene cluster each of them bordered at both ends by an IS1071 element, whereas pPBL-H6-2 carries an isolated IS1071 element. In addition, between the hylA and ccd loci, the accessory gene region of pPBS-H4-2 carries genes related to plasmid replication and maintenance such as repAB and parAB and a fifth IS1071. These extra parAB genes are unrelated to the pPBS-H4-2 backbone parAB genes. Plasmids pPBL-H6-2 and pPBS-H4-2 are most closely related to the PromA γ plasmids pSN1104-11 and pSN1104-34 (Yanagiya et al. 2018) that were isolated by exogenous isolation and hence represent the first PromA γ plasmids obtained from isolates. Their presence in Variovorax extends the host range of PromA γ plasmids, as is the case for PromA α and PromA β plasmids, to β-proteobacteria. Plasmid pPBS-H4-2 is the first catabolic PromA plasmid, and is one of the few noncryptic PromA plasmids (Mela et al. 2008). The often cryptic character of PromA plasmids has been the subject of a debate because it might harness their stability as they do not benefit the host fitness. It was suggested that they mainly support the conjugative transfer of other mobilizable replicons (Mela et al. 2008). The finding that PromA plasmids carry catabolic genes shows that they, as is the case for IncP-1 plasmids, can indeed acquire and distribute genes beneficial for the host. The location of cargo genes in both pPBS-H4-2 and pPBL-H6-2 (near virD2) differs from this in other PromA plasmids (near parA) (Van der Auwera et al. 2009) and identifies the virD locus as an alternative hot spot for insertion of accessory genes in PromA plasmids.
Novel Putatively Conjugative Plasmids in Linuron-Degrading Variovorax Strains
Other plasmids than IncP1 and PromA plasmids were identified that carry homologs of TS44 genes (table 2), that is, pWDL1-3 and pWDL1-5, pRA8-1 and pSRS16-2. None of those plasmids categorized into a known plasmid incompatibility group. Although they carry T4SS genes, the origin of transfer (oriT) and the gene encoding for the type IV coupling protein (T4CP) could not always be identified. The relaxase, which is required for conjugative transfer (Smillie et al. 2010), could only be identified in pWDL1-3 and was assigned to the MOBP family (Garcillán-Barcia et al. 2009). Plasmid pWDL1-3 carries a remarkably high number of 41 putative ISs but no IS1071, and several gene clusters for xenobiotic degradation. Among these is a gene cluster that encodes for homologs (40–43% amino acid identity) of proteins encoded by the tphA1A2A3BR–gene cluster for terephthalate degradation in Comamonas sp. E6 (Sasoh et al. 2006) as well as of benzoate 1,2-dioxygenase subunits. pRA8-1 is distantly related to pWDL1-3 and carries the ccdCFBD operon. In addition, it contains homologs of genes encoding for the biodegradation of nonchlorinated catechols, as well as for cation efflux proteins CusABF (Franke et al. 2003) and the cadmium transport protein CadA (Tsai et al. 1992), flanked by IS1071. The small pWDL1-5 carries no cargo genes. A highly similar plasmid (99% nt identity, 72% coverage), also without cargo, is present in the chlorobenzene-degrading Pandoraea pnomenusa strain MCB032 (Baptista et al. 2008). The finding of this plasmid group in two different genera/families of the same bacterial order indicates its transferability within Burkholderiales. The Trb homologs encoded by pSRS16-2 as well as the oriT are highly similar (75–80%) to those encoded by IncP-1 plasmids. However, unlike IncP-1 plasmids, pSRS16-2 does not carry trfA, and its size (560 kb) is much larger than IncP-1 plasmids. Plasmid pSRS16-2 encodes for a broad range of functions, including 37 transport-related proteins, 18 proteins related to aromatic degradation, and 31 ISs (but without IS1071). We conclude that this plasmid represents a novel plasmid group, with conjugative transfer machinery similar to that of IncP-1 plasmids.
Chromids pSRS16-3 and pPBL-E5-2
The closely related replicons pPBL-E5-2 and pSRS16-3 carrying the repB-parAB replication-partitioning module do not contain homologs of genes related to conjugal transfer, suggesting that they are not self-transmissible. Both replicons carry distantly related homologs of catabolic genes such as tfdA encoding conversion of 2,4-D (33% amino acid identity) in Cupriavidus necator JMP134 (Plumeier et al. 2002), and the dmpKLMNOPQBCDEFGHI gene cluster for phenol degradation (45–66% amino acid identity) in Pseudomonas sp. CF600 (Shingler et al. 1992), as well as the phnCDEGHIJKLMN gene cluster for phosphonate uptake and degradation (45–62% amino acid identity) in Eschericia coli K12 (Jiang et al. 1995).
tRNA-Carrying Megaplasmids in Linuron-Degrading Variovorax sp
Pairwise alignment showed that pPBL-H6-1, pSRS16-1, pPBL-E5-1, and pWDL1-1 are highly identical to one another. They show 99% nt identity over the entire aligned sequence length including the putative replication/partitioning module repB-parAB (Supplementary fig. S4, Supplementary Material online). Plasmid pPBL-H6-1 carries all three gene clusters required to convert linuron to 3-oxoadipate, whereas pSRS16-1 and pPBL-E5-1 only carry dcaQTA1A2BR and ccdCFDE. The pWDL1-1 variant sequenced in this study carries the ccdCFDE genes and the hylA gene. The proteins encoded by the repB-parAB module show only slight similarity to their nearest relatives (27%, 48%, and 39% amino acid similarity for RepB, ParA, and ParB, respectively) and hence the four mega-plasmids might represent a new plasmid group, potentially specific for Variovorax. All four plasmids carry traALBFHJDNUWG and trbCG homologs, suggesting that they might be transferrable. They however have low identity at amino acid level (30–43%) to the nearest relativesinvolved in conjugative transfer. No putative relaxase was found.
Strikingly, the four megaplasmids carry tRNA genes that encode for all the proteinogenic amino acid codons. Unlike the scattered appearance of the tRNA genes located on the chromosome, the plasmid-encoded tRNA genes are concentrated in one array. Except for the tRNA encoding for codons glutamine (CAG) and arginine (CGC), all tRNAs on these plasmids are also present on the chromosome. In all four hosts, these two codons are preferred by both plasmids and the chromosome, however, multiple other tRNAs encode for these amino acids on the chromosome, suggesting that although the plasmid-encoded tRNAs may enhance gene expression, they are not essential for expression of plasmid genes. pSRS16-1 further lacks the tRNA for valine (CAC), which is preferred by both the chromosome and plasmids; however, this tRNA is present in pSRS16-2 as well as in the chromosome.
Other plasmids that carry tRNA genes in the linuron-degrading strains are pRA8-2 and pWDL1-2. These plasmids are distantly related to pPBL-H6-1, pSRS16-1, pPBL-E5-1, and pWDL1-1 and also carry the tRNA genes in an array. However, their tRNAs do not encode for all proteinogenic amino acids and are all redundant. The two plasmids neither have functions for conjugal transfer nor carry catabolic genes but encode for putative heavy metal resistance, like the cobalt–zinc–cadmium efflux system encoded by czcABCD (Rensing et al. 1997; Legatzki et al. 2003), the copper-response two-component system encoded by cusRS and cusABRS (Franke et al. 2003), and mercury resistance encoded by merACPTR (Barkay et al. 2003). Unlike pWDL1-2, pRA8-2 carries an IS1071 adjacent to a gene cluster encoding for homologs of the toxin–antitoxin system proteins DinJ–YafQ (Armalyte et al. 2012) and the antirestriction protein KlcA that plays a role in evading host restriction systems during conjugative transfer (Serfiotis-Mitsa et al. 2010).
The presence of tRNA genes on large plasmids has been reported before in other bacteria (Puerto-Galan and Vioque 2012; Sakai et al. 2014; Bottacini et al. 2015; Morgado and Vicente 2018). None of these plasmids however related to the tRNA-carrying Variovorax plasmids. In Bifidobacterium breve, the tRNA-encoding plasmid improves gene expression from both the chromosome and the plasmid (Bottacini et al. 2015), whereas in Anabaena sp. PCC 7120, it is dispensable for growth (Puerto-Galan and Vioque 2012). Other MGEs different from plasmids (Alamos et al. 2018) as well as bacteriophages (Pope et al. 2014), encode for tRNA genes. In the acidophilic, bioleaching bacterium Acidithiobacillus ferrooxidans, the tRNA genes are located on an integrative conjugative element and are likely not essential for growth (Alamos et al. 2018).
Another feature that sets plasmids pPBL-H6-1, pPBL-E5-1, pSRS16-1, and pWDL1-1 apart is the presence of a CRISPR3-Cas cassette, which is identical in all of them. The cassette consists of a CRISPR array with 15 spacers, in addition to the genes encoding for a Cas6/Cse3/CasE-type endonuclease, the Cascade subunits CasA, CasB, CasC, and CasD and the CRISPR-associated proteins Cas1 and Cas2, which is similar to the class I-E CRISPR-Cas systems (Koonin et al. 2017). The CRISPR-defense system protects bacteria and archaea against MGEs and phages (Pothier et al. 2011), and can be transferred horizontally (Godde and Bickerton 2006; Bottacini et al. 2015). Although the exact direct repeats (DRs) of the CRISPR structure were also found in the Serpentinomonas mccroryi strain B1 genome (GCA_000828915.1), the spacer sequences did not have any match in the CRISPR databases. Interestingly, no CRISPRs were found in other Variovorax genomes available in public databases, with the exception of Variovorax sp. PDC80 (GCF_900115375.1), which carries a class I–F CRISPR-Cas system with spacers unrelated to those of the megaplasmids.
Genomic Context of the Linuron-Catabolic Loci in the Linuron-Degrading Variovorax Strains
We analyzed the presence, location, and genomic context of hylA and libA genes for linuron hydrolysis, dcaQTA1A2BR genes for DCA conversion to 4,5-chlorocatechol (Król et al. 2012) and ccdCDEFR genes for 4,5-dichlorocatechol degradation (Bers et al. 2011, 2013) in each of the degraders genome. These genes were not present in any of the other publicly available Variovorax genomes.
Genomic Context of the Linuron-Hydrolysis Genes hylA and libA
The linuron hydrolysis genes hylA and libA were highly conserved among the different strains. The six linuron degraders carried either hylA or libA, but never both. hylA is present in one copy in strains PBS-H4 and WDL1. As in WDL1 (Albers, Lood, et al. 2018), hylA in PBS-H4 makes part of a larger gene cluster of 13 ORFs that is highly conserved among the two hylA hosting strains (99–100% nt identity and complete synteny). In both strains, the gene cluster is flanked at both sites by IS1071 composing a composite transposon structure (fig. 3). The functions of the hylA associated ORFs, and in particular the downstream ORFs, are currently unclear, but their conservative nature indicates that they play a role in linuron hydrolysis. Albers, Weytjens, et al. (2018) showed the transcriptional upregulation of the downstream luxRI homologs when WDL1 is degrading linuron within a consortium. The hylA-carrying composite transposon likely originated by inserting the hylA gene together with its downstream ORFs in a precursor composite transposon carrying the iorAB and dca gene clusters as suggested by the presence of iorAB and a dcaQ remnant directly upstream of hylA. Interestingly, the dcaQ gene that directly flanks hylA, is truncated at a different nt residue in WDL1 and in PBS-H4 (in PBS-H4 at nt position 749, in WDL1 at nt position 689), which suggests that the hylA gene and its associated downstream ORFs were independently acquired by the composite transposons present in the two strains. We identified exact copies of both the WDL1 and the PBS-H4 hylA-carrying composite transposon, that is, with identical hylA-dcaQ junctions in a linuron-degrading Hydrogenophaga isolate as well as in an exogenously isolated plasmid indicating that WDL1 and PBS-H4 recruited the hylA locus through an already existing composite transposon structure (Werner et al. 2020). However, no direct indication was found of the mobile character of the structure as no direct repeats (DRs) could be recognized bordering the outward inverted repeat (IR) of the flanking IS1071 ISs.
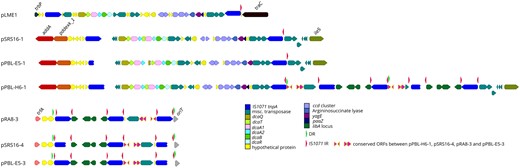
—Genomic context of linuron-catabolic genes in strains SRS16, PBL-H6, RA-8, and PBL-E5. On the upper panel, the dca and ccd clusters on plasmids pPBL-E5-1, pSRS16-1, and pPBL-H6-1 as well as the Delftia acidovorans plasmid pLME1 are illustrated, together with flanking genes and genes directly neighboring each IS1071 element. In the lower panel, the libA gene-associated IS1071 elements are illustrated for pPBL-H6-1, pPBL-E5-3, pSRS16-4, and pRA8-3. The backbone genes flanking the IS1071 insertion sites are annotated on the figure. IS1071 IRs and DRs are indicated by red and green arrow points, respectively. Broken arrows indicate ORFs which were truncated by immature stop codons, caused by point mutations.
The libA gene is present in SRS16, PBL-E5, RA8, and PBL-H6. In SRS16, PBL-E5 and RA8, libA is carried by an IncP-1 plasmid (pSRS16-4, pPBL-E5-3, and pRA8-3, respectively), whereas in PBL-H16 it is carried as two copies by the tRNA-carrying megaplasmid pPBL-H6-1. In all four strains, libA makes part of a larger highly conserved gene cluster of four ORFs flanked at both borders by IS1071. This four-ORF gene cluster contains a luxR-family transcription regulator directly adjacent to libA followed by an IS91-family IS. As such, as for the hylA locus, the libA locus is encompassed in a composite transposon structure (fig. 4). However, the two IS1071 elements are in indirect orientation hampering theoretically the mobility of the composite transposon structure as a discrete unit. Interestingly, in all four strains, directly adjacent to the libA containing composite transposon structure, a strongly conserved gene cluster was found bordered by a third IS1071 in direct orientation with the first IS1071. DRs can be found at the outward border of the first IS1071 and of the third IS1071, indicating that the libA locus is indeed mobile but that this mobility relates to a super composite transposon encompassing all three IS1071 elements and intermediate DNA. Within this super composite transposon, some of the tnpA genes appear truncated and hence not functional but IRs are always present at the outer borders and the transposon structure contains at least one complete and hence functional tnpA. As suggested above for the recruitment of the hylA locus, the remarkable conservation of the libA locus including its integration into a (super) composite transposon, might suggest that the different Variovorax strains recruited the libA locus rather through an already existing composite transposon structure carrying the libA locus then via independent recruitment events. Strains SRS16 and PBL-E5 were isolated from the same agricultural field, albeit at different times, and hence the IncP-1 plasmid carrying libA seems to contribute to the distribution of the libA locus in that field. RA8, though, was isolated in Japan, indicating that similar plasmids evolved at different locations, or that the BHR plasmid was transferred across a large geographic distance. In contrast, in PBL-H6, which originated from a Belgian agricultural field, the super composite transposon is found integrated in a replicon different from IncP-1 plasmids, that is, megaplasmid pPBL-H6-1. The plasmid contains two copies of that super composite transposon that share an outer IS1071. Likely, the second copy is due to duplication of the IS1071 flanked structure which would lead to the formation of tandem arrays (fig. 4) of the same DNA fragment interspersed with directly oriented copies of IS1071 (Harmer et al. 2014; Hudson et al. 2014; He et al. 2015). Interestingly, PBL-H6 harbors the cryptic BHR PromA plasmid pPBL6-H-2 carrying a single copy of IS1071. Plasmid pPBL6-H-2 might have been implicated in initial recruitment of the IS1071 associated linuron catabolic genes in PBLH-6 (see discussion below).
The dca and ccd Clusters of Linuron-Degrading Variovorax Strains
Similar to the hylA and libA genes, MGEs determine the genomic context of the dca and ccd genes. PBL-E5, PBL-H6, and SRS16 carry the entire dca and ccd clusters, which is consistent with their ability to degrade linuron and DCA. The ccd clusters of strains PBL-E5, PBL-H6, and SRS16 are on the 800-Mb megaplasmids, directly downstream of the dca clusters (fig. 4). In all strains, the entire dca/ccd gene cluster is bordered by IS1071 at both sides. However, the two elements lack the inward-directed IRs and one even lacks additionally the outward-directed IR. Moreover, the outer IS1071 elements are indirectly oriented. A similar composite transposon configuration including the dca and ccd genes is found in the chloroaniline degrader Delftia acidovorans LME1 (Boon et al. 2001) (fig. 4), except that PBL-H6 and SRS16 have two copies of the dcaA1A2 genes and PBL-E5 two adjacent copies of the dcaQTA1A2B genes with one intact and one truncated dcaB. Other chloroaniline degraders like C. testosteroni WDL7 and D. acidovorans B8c (Król et al. 2012) carry a similar structure but lack the dcaA1A2 duplications as well as the ccd genes (fig. 4). Amino acid-level similarity of the proteins encoded by dcaQTA1A1BR and ccdCFDE between the three Variovorax strains and LME1/WDL7/B8c is ∼99% (supplementary table S3, Supplementary Material online). As for the hylA and libA loci, we hypothesize that the dca/ccd gene clusters were obtained by being already integrated into an ancestral composite transposon but that afterwards gene rearrangements occurred that explain the observed variations. This is further supported by the observation that in the chloroaniline-degrading Comamonas/Delftia strains, the composite transposon structures carrying dca/ccd genes are located on IncP-1 plasmids, whereas in the three Variovorax strains they are located on the 800-kb tRNA-carrying megaplasmids. On the other hand, no DRs were detected bordering the dca/ccd-carrying composite transposon structure and there is no evidence for transposition. In case of PBL-H6, the dca/ccd cluster is directly adjacent to the composite transposon structure carrying libA (see above) sharing the outer IS1071, hence forming an array of multiple adjacent putative composite transposons that share a single IS1071 located between them. In all three plasmids, the location of the dca/ccd containing composite transposon is the same, marking the corresponding location as a likely accessory gene insertion hot spot in the mega-plasmid.
The dca cluster and associated ORFs present on pWDL1-1 are also bordered by two IS1071 elements, but the composite transposon does not contain a ccd cluster and the dca genes are rather related (99% nt identity) to the tadQTA1A2B encoding for conversion of aniline to catechol in Delftia tsuruhatensis AD9 (Liang et al. 2005). The ccd cluster, present on pWDL1-1, also differs substantially from those of PBL-E5, PBL-H6, and SRS16 and overall the genes are relatively distantly related to other known chlorocatechol degradation genes (supplementary table S3, Supplementary Material online). This cluster is located in the vicinity of either hylA or the dca genes, depending on the WDL1 subpopulation (Albers, Lood, et al. 2018). WDL1-1 hylA/dca clusters and ccd cluster are separated by a stretch of DNA of ∼20 kb containing 19 ORFs, forming as such another array of putative composite transposons sharing IS1071 (fig. 3). Further downstream of the ccd cluster another IS1071 is located.
The ccd gene clusters in the non-DCA degraders PBS-H4 and RA8 are identical to those of WDL1. In PBS-H4, the ccd cluster is on the PromA plasmid pPBS-H4-2, that also bears the hylA-carrying composite transposon. The ccd cluster is flanked by IS1071 at both sites and separates from the hylA locus by a stretch of ∼32 kb of DNA containing 35 noncatabolic ORFs as well as a another IS1071. As such, the cargo on pPBS-H4-2 forms another multimeric array of putative composite transposons sharing IS1071 (fig. 3). Interestingly, the noncatabolic ORFs carries the aforementioned genes related to plasmid replication and maintenance, as well as 13 hypothetical genes. No DR could be detected in this super composite transposon structure. In RA8, the ccd cluster is located on pRA8-1. This cluster is flanked by multiple IS elements but unrelated to IS1071 genes and hence is the only linuron-catabolic gene cluster without neighboring IS1071 in the linuron-degrading Variovorax strains (fig. 3).
Overall, the analysis of the genetic context of the genes involved in linuron catabolism, either upstream or downstream functions in the pathway, indicates that IS1071 played an essential role in the plasmidic acquisition of the catabolic genes and genetic adaptation of Variovorax toward the ability to degrade linuron. The phenomenon that IS1071 elements are associated with genes involved in xenobiotic degradation was previously reported using both cultivation-dependent (Boon et al. 2001; Martinez et al. 2001; Sota et al. 2003; Sen et al. 2010) and cultivation-independent (Dunon et al. 2018) methods. Subsequent inter- and intramolecular transposition of IS1071 is thought to lead to the assembly of catabolic genes into a composite transposon structure with the recruited genes flanked by IS1071 at both sites (Nakatsu et al. 1991; Di Gioia et al. 1998). The new composite transposon structure might then move as a discrete unit between different replicons (Casacuberta and González 2013) although this has never been shown for composite transposons flanked by IS1071. However, transposition would lead to the generation of DRs and this was only observed for the super composite transposon carrying the libA locus. A first step in such a transposition event, would lead to a cointegrate structure between the original replicon carrying the transposon and the receiving replicon. Interestingly, the cargo of plasmid pPBSH4-2 contains in addition to its two catabolic clusters encoding ccd and hylA, genes involved in plasmid replication/stability. The latter might belong to a plasmid that initially carried the transposons and hence be remnants of such a cointegrate structure. Another mode of transposition of IS1071 associated (catabolic) genes might involve the recent described concept of translocatable unit (TU) as recently shown for the mobility of antibiotic resistance genes associated with IS26. TUs consist of a unique DNA segment containing genes and a single copy of an IS element. The TU structure performs RecA-independent incorporation next to a second IS identical to the IS carried by the TU, resulting into structures resembling composite transposons with the two IS in direct orientation (Harmer et al. 2014; Hudson et al. 2014; He et al. 2015). Repeating this process would result into the arrays of catabolic genes we have observed with directly oriented copies of IS1071 at each end and between each unique segment. The involvement of TUs in IS associated mobility of cargo genes has however only been reported for ISs belonging to the IS6 family (like IS26) and not for ISs of the Tn3 family (like IS1071). Another explanation of the observed multimeric composite transposon array configuration with shared IS1071 elements between two adjoining composite transposons might be linked with homologous recombination between two outer IS1071 elements of two nearby newly acquired discrete composite transposons. Whatever the process of transposition of the IS1071 associated catabolic gene clusters might be, as discussed above, the strong conservative nature of the catabolic loci suggests that the recruitment of the different catabolic clusters in the linuron-degrading Variovorax strains is rather due to the recruitment of already existing catabolic composite transposon structures than by the assembly process itself.
Next to IS1071, plasmids appear to play an essential role in the genetic adaptation of Variovorax toward linuron catabolism. Broad host range plasmids such as IncP-1 are known to distribute catabolic clusters in communities and catabolic IncP-1 plasmids often contain IS1071 associated catabolic composite transposons (Boon et al. 2001; Król et al. 2012). Interestingly, each of the linuron degraders with the exception of WDL1 carries at least one plasmid of a well-known promiscuous plasmid group (IncP-1 or PromA). Their involvement in distributing linuron catabolic genes in the linuron-degrading strains is suggested from the fact that these plasmids all carry at least one of the linuron catabolic gene functions with the exception of pPBL-H6-2. However, the latter carries an IS1071 copy and hence, as mentioned above might have been involved in initial recruitment of the composite transposon carrying the linuron catabolic locus in PBL-H6-2. After transposition of the composite transposon carrying the catabolic genes onto pPBL-H6-1, deletion of the catabolic genes might have taken place due to recombination between the IS1071 ISs flanking the catabolic locus. Such an event was reported before for a similar composite transposon structure carrying atrazine catabolic genes (Devers et al. 2007). In addition, other plasmids of yet unknown type, carrying signs of conjugative features, were present. Likely, plasmids move around in a community, and pick up the composite transposons either by transposition as a discrete unit or by involving TU, for further transfer to their final hosts. As such, the BHR plasmids found in the linuron-degrading strains might have, next to IS1071, functioned as a second crucial vehicle for acquisition of the catabolic genes.
Regarding catabolic functions, only a limited reservoir of catabolic loci was utilized for integration in the linuron catabolic pathways indicating that the source of suitable catabolic functions for composing a functional linuron catabolic pathway in Variovorax is limited, even on a worldwide scale. Curiously, these genes seem only to be recruited by a specific clade 1 of Variovorax, whose members, in addition to linuron, seem to be prone to genetic adaptation and hence specialization toward the biodegradation of other anthropogenic compounds. Apparently, for unknown reasons, this clade is able to recruit and express foreign genes for xenobiotic biodegradation. Sequence analysis of other genomes within this clade, in addition to the linuron degraders, and comparison with the genome sequences of clade 2, might unravel the mechanisms involved in this special ability of catabolic gene recruitment.
Data deposition: Thegenome and plasmid nucleotide sequences have been deposited at EMBL/ENA under the accessions LR594659–LR594661 (PBL-H6), LR594662–LR594665 (RA8), LR594666–LR594670 (SRS16), LR594671–LR594674 (PBL-E5), LR594675–LR594677 (PBS-H4), and LR594689–LR594694 (WDL1).
Acknowledgments
We thank Sebastian S. Sørensen and Koji Satsuma for the donation of strains SRS16 and RA8, Simone Severitt for technical assistance, Markus Göker, Jörn Petersen, Kornelia Smalla, Masa Shintani, and Isabel Schober for valuable discussions regarding the article, and Jörg Overmann for his support with the sequencing of the strains. We acknowledge funding by Fonds voor Wetenschappelijk Onderzoek – Vlaanderen (FWO) Project G.0371.06, and the EU project METAEXPLORE (EU Grant No. 222625). J.M.K. was supported by Deutsche Forschungsgemeinschaft within “Sonderforschungsbereich TRR 51.” We acknowledge the use of de.NBI cloud and the support by the High Performance and Cloud Computing Group at the Zentrum für Datenverarbeitung of the University of Tübingen, the Federal Ministry of Education and Research (BMBF) (Grant Number 031 A535A). The open access publication of this study was funded by the Leibniz Open Access Publishing Fund (Grant No. 4055).
Literature Cited
Barkay T, Miller SM, Summers AO. 2003. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol Rev. 27(2-3):355–384.

{kind=link}
{kind=link}
{kind=link}
{kind=link}