





Abstract
Wolbachia are obligate intracellular bacteria which commonly infect various nematode and arthropod species. Genome sequences have been generated from arthropod samples following enrichment for the intracellular bacteria, and genomes have also been assembled from arthropod whole-genome sequencing projects. However, these methods remain challenging for infections that occur at low titers in hosts. Here we report the first Wolbachia genome assembled from host sequences using 10× Genomics linked-reads technology. The high read depth attainable by this method allows for recovery of intracellular bacteria that are at low concentrations. Based on the depth differences (714× for the insect and 59× for the bacterium), we assembled the genome of a Wolbachia in the parasitoid jewel wasp species Nasonia oneida. The final draft assembly consists of 1,293, 06 bp in 47 scaffolds with 1,114 coding genes and 97.01% genome completeness assessed by checkM. Comparisons of the five Multi Locus Sequence Typing genes revealed that the sequenced Wolbachia genome is the A1 strain (henceforth wOneA1) previously reported in N. oneida. Pyrosequencing confirms that the wasp strain lacks A2 and B types previously detected in this insect, which were likely lost during laboratory culturing. Assembling bacterial genomes from host genome projects can provide an effective method for sequencing bacterial genomes, even when the infections occur at low density in sampled tissues.
Introduction
Wolbachia, alphaproteobacterial endosymbionts, are widespread and common in arthropods and filarial nematodes, either as reproductive parasites or mutualists (Werren 1997; Fenn and Blaxter 2006; Werren et al. 2008). About half of arthropods are infected with Wolbachia (Hilgenboecker et al. 2008; Zug and Hammerstein 2012) due to horizontal movement of the bacteria between species, although the routine mode of transmission of these bacteria is vertical through the egg cytoplasm. The jewel wasp genus of Nasonia has been an excellent model for Wolbachia research (Breeuwer and Werren 1993; Perrot-Minnot et al. 1996; Bordenstein et al. 2001,, 2003; Raychoudhury et al. 2009). Eleven Wolbachia have so far been identified in the four species of Nasonia (Raychoudhury et al. 2009). These are often maintained as multiple infections within individual wasps of each species and have diverse evolutionary origins, indicating horizontal transfers from divergent host species (Raychoudhury et al. 2009). Genomic studies of Wolbachia blossomed in the recent years since the first complete genome of the A-Wolbachia parasite of Drosophila melanogaster published in 2004 (Wu et al. 2004). Wolbachia genomes are small with a range between 0.9 and 1.7 Mb. In the jewel wasp (Nasonia) species, only two Wolbachia strains have been sequenced (wVitA; Newton et al. 2016) and wVitB (Kent et al. 2011), both from Nv. In this study, we sequenced, assembled and annotated the Wolbachia strain in Nasonia oneida (No), which will facilitate the comparative genomic and evolutionary analyses of this model system.
Materials and Methods
Sample Collection, DNA Extraction, and Sequencing
Genomic DNA sample was extracted from 24-h male adults of the N. oneida NONY strain. MagAttract DNA Mini Kit (Qiagen, MD) was used to isolate high molecular weight genomic DNA. A 10× Genomic library was constructed by using the Chromium Genome Reagent Kits v2 on 10× Chromium Controller (10× Genomics Inc., CA) and sequenced on a HiSeqX lane at the Genomic Services Lab at the HudsonAlpha Institute for Biotechnology.
Genome Assembly and Annotation
The N. oneida genome was assembled using the Supernova 2.1.1 assembler (Weisenfeld et al. 2017). The following steps were conducted to identify wOneA1 scaffolds (supplementary fig. S1, Supplementary Material online): 1) all 10× reads were aligned to the N. oneida assembly to calculate median coverage for each scaffold; 2) N. oneida scaffolds were aligned to the bacterial sequence database using BLAT v3.5 (Kent 2002) to determine percent sequence identity to known Wolbachia sequences; 3) assign the scaffolds to wOneA1 genome if they have at least 20% sequence identity with known Wolbachia sequences and a median coverage around 60×. The genome completeness was further evaluated by checkM (Parks et al. 2015) with default settings and Benchmarking Universal Single-Copy Orthologs (BUSCO) (Seppey et al. 2019) comparing to bacteria database. Gene annotation was conducted using DFAST prokaryotic genome annotation pipeline (Tanizawa et al. 2018). tRNA genes were predicted by tRNAscan_SE (Lowe and Eddy 1997).
Comparative Analysis of Wolbachia Genomes in the Nasonia Genus
To compare the genome structure among three sequenced Wolbachia genomes in Nasonia, we conducted whole genome alignment of wOneA1, wVitA (GCA_001983615.1) (Newton, et al. 2016), and wVitB (GCA_000204545.1) (Kent et al. 2011) genomes using NUCmer in the MUMmer program suite with default parameter settings (Kurtz et al. 2004). The pairwise alignments were visualized using Mummerplot (Kurtz et al. 2004). Orthologous gene sets between wOneA1 and two other Wolbachia in Nasonia were generated based on reciprocal best hits using BLAST with an E-value cutoff 10−5. 32 genes in wOneA1 genome were excluded in this analysis as the gene orthology are unclear when comparing to wVitA and wVitB.
MLST Strain Typing of Wolbachia wOneA1 Strain
The five Multi Locus Sequence Typing (MLST) genes (Baldo 2006; Jolley and Maiden 2010) were examined to further characterize the phylogenetic relationships of Wolbachia strains in Nasonia. The wOneA1 MLST genes were identified on five different genome scaffolds, including coxA on SCAFFOLD17, gatB on SCAFFOLD28, hcpA on SCAFFOLD47, ftsZ on SCAFFOLD73 and fbpA in SCAFFOLD76. Sequences of MLST genes from the following strains were downloaded from the MLST database (Baldo 2006): wNvitA, wNvitB in N. vitripennis; wNgirA1, wNgirA2, wNgirB in Nasoniagiraulti; wNlonA, wNlonBl, wNlonB2 in Nasoinalongicornis (Raychoudhury et al. 2009). Multiple sequence alignments were generated using MUSCLE with default parameters (Edgar 2004). Phylogenetic analysis was performed using the Maximum Likelihood (ML) method in MEGA 7.0 software (Kumar et al. 2016). Bootstrap tests with 1,000 replicates were used to evaluate the phylogenetic trees.
Confirmation of wOneA1 Strain Using Pyrosequencing
Wolbachia infection types were checked in NONY, and DNA samples from a recently (July 2018) collected wild-type CAR262L strain using allele-specific pyrosequencing. Pyro PCR and sequencing primers were designed to target SNP positions in coxA and gatB genes (supplementary table S3, Supplementary Material online) in A1, A2, and B Wolbachia using PyroMark Assay Design 2.0 (Qiagen, USA). The A/G SNP targeted in coxA can separate B-Wolbachia from A1/A2-Wolbachia, and the C/T SNP in gatB allowed us to distinguish A1-Wolbachia from A2/B-Wolbachia. Pyrosequencing was performed on a Pyromark Q48 instrument (Qiagen, USA) using PyroMark Q48 Advanced Reagents (Qiagen, USA). Three technical replicates were performed for each sample.
Results and Discussion
Assembly of Wolbachia Genome wOneA1
This Wolbachia project emerged from a de novo assembly of the parasitoid wasp No genome (see Materials and Methods section). Wolbachia scaffolds were separated from the No genome assembly using a custom bioinformatics pipeline (supplementary figs. S1 and S2A, Supplementary Material online). No scaffolds were BLATed against bacterial genome database (Kent 2002), and we identified Wolbachia scaffolds based on the median coverage and sequence identity to known Wolbachia sequences (supplementary fig. S2, Supplementary Material online and see Materials and Methods section). We have identified this genome to be from the wOneA1 Wolbachia with median genome coverage of 59.38×, which is significantly lower compared with 713.59× for the No genomic scaffolds (P-value < 2.2 × 10−16) and the 20,000× mitochondrial genome (supplementary fig. S2B, Supplementary Material online). The differences in coverage and guanine-cytosine (GC) content further assisted in the separation of the wOneA1 scaffolds.
The wOneA1 draft genome contains 1,293,406 nucleotides with 47 scaffolds and N50 of 128.97 kb. 1,114 genomes were annotated in the wOneA1 genome including protein coding genes, 5S, 16S, and 23S rRNA and tRNA genes. The number of contigs and scaffolds are fewer than wVitA and wVitB, and the contig and scaffold N50s are longer (table 1). The genome completeness is 97.01% accessed by and checkM, which is comparable with wVitA and wVitB, suggesting high assembly quality. wVitA and wVitB have slightly higher completeness, but at a cost of 1–2% of contamination (table 1). The BUSCO completeness is 86.5%, which is typical for complete Wolbachia genomes (Sinha et al. 2019).
wOneA1 Assembly Summary Statistics and Comparison with wVitA and wVitB Genomes
| wOneA1 | wVitA | wVitB | |
|---|---|---|---|
| Number of contigs | 65 | 142 | 509 |
| Number of scaffolds | 47 | N/A | 426 |
| Contig N50 (kb) | 35.88 | 13.38 | 5.79 |
| Scaffold N50 (kb) | 128.97 | N/A | 6.21 |
| Number of proteins | 1,114 | 1,042 | 845 |
| Assembled genome size (bp) | 1,293,406 | 1,211,929 | 1,107,643 |
| BUSCO completeness (%) | 86.5 | 87.2 | 85.1 |
| checkM completeness (%) | 97.01 | 99.79 | 99.57 |
| checkM contamination (%) | 0 | 0.64 | 1.71 |
| wOneA1 | wVitA | wVitB | |
|---|---|---|---|
| Number of contigs | 65 | 142 | 509 |
| Number of scaffolds | 47 | N/A | 426 |
| Contig N50 (kb) | 35.88 | 13.38 | 5.79 |
| Scaffold N50 (kb) | 128.97 | N/A | 6.21 |
| Number of proteins | 1,114 | 1,042 | 845 |
| Assembled genome size (bp) | 1,293,406 | 1,211,929 | 1,107,643 |
| BUSCO completeness (%) | 86.5 | 87.2 | 85.1 |
| checkM completeness (%) | 97.01 | 99.79 | 99.57 |
| checkM contamination (%) | 0 | 0.64 | 1.71 |
wOneA1 Assembly Summary Statistics and Comparison with wVitA and wVitB Genomes
| wOneA1 | wVitA | wVitB | |
|---|---|---|---|
| Number of contigs | 65 | 142 | 509 |
| Number of scaffolds | 47 | N/A | 426 |
| Contig N50 (kb) | 35.88 | 13.38 | 5.79 |
| Scaffold N50 (kb) | 128.97 | N/A | 6.21 |
| Number of proteins | 1,114 | 1,042 | 845 |
| Assembled genome size (bp) | 1,293,406 | 1,211,929 | 1,107,643 |
| BUSCO completeness (%) | 86.5 | 87.2 | 85.1 |
| checkM completeness (%) | 97.01 | 99.79 | 99.57 |
| checkM contamination (%) | 0 | 0.64 | 1.71 |
| wOneA1 | wVitA | wVitB | |
|---|---|---|---|
| Number of contigs | 65 | 142 | 509 |
| Number of scaffolds | 47 | N/A | 426 |
| Contig N50 (kb) | 35.88 | 13.38 | 5.79 |
| Scaffold N50 (kb) | 128.97 | N/A | 6.21 |
| Number of proteins | 1,114 | 1,042 | 845 |
| Assembled genome size (bp) | 1,293,406 | 1,211,929 | 1,107,643 |
| BUSCO completeness (%) | 86.5 | 87.2 | 85.1 |
| checkM completeness (%) | 97.01 | 99.79 | 99.57 |
| checkM contamination (%) | 0 | 0.64 | 1.71 |
Due to the intracellular lifestyle and inability of media culture in Wolbachia, the purification of Wolbachia DNA from the host sample can be challenging. Different methods have been applied to purify the Wolbachia genomic DNA from the host DNA (Klasson et al. 2009; Mavingui et al. 2012; Duplouy et al. 2013; Ellegaard et al. 2013; Brelsfoard et al. 2014; Newton et al. 2016; Badawi et al. 2018). For the wVitB genome project, a high-density tiled oligonucleotide array was developed to enrich for Wolbachia gDNA (Kent et al. 2011). An alternative approach has been to extract Wolbachia reads from the host whole genome sequence data set, and then align to the reference genome of the closely related Wolbachia strains, or perform de novo assembly using the filtered reads (Darby et al. 2012; Saha et al. 2012; Siozios et al. 2013; Lindsey et al. 2016; Chung et al. 2017).
If no prior knowledge is available about the presence of specific microbes, sequencing without purification is preferred to identify other intracellular symbionts, as well as characterizing bacterial species in insect gut microbiota at the whole-genome level. In our study, we perform de novo assembly of the host genome and Wolbachia genome using the 10× Genomics linked-reads technology. The Wolbachia DNA fragments were labeled with unique 10× barcodes, therefore they are much less likely to be misassembled into the host genome scaffolds. The microbe (wOneA1) and the host (No) have a 12-fold difference in coverage (714× vs 59×). Using most sequencing technologies, it would be difficult to assemble the bacterial genome because of insufficient coverage against the host genome. However, the 10× sequencing linked read technology has permitted accurate identification of the bacterial scaffolds, despite the relatively low abundance of the bacteria DNA in the insect. This finding is similar to a recent study showing the efficacy of 10× technology in assembling high-quality microbial genome drafts in microbiome samples (Bishara et al. 2018). Therefore, the Wolbachia genome assembled with 10× linked reads was of good quality with no contamination of host nuclear and mitochondrial DNA. As the cost of PacBio sequencing decreases, the long-read platforms would be better for symbionts genome assembly when the bacteria can be enriched in the sample, or the infection occurs at a high level. However, microbial genome assembly by 10× sequencing technology will likely continue to have an advantage for some time in cases where microbial associates occur at low levels in hosts or tissue samples.
Comparative Genomic Analysis of Wolbachia Strains in Nasonia Species
When comparing the gene contents of these Wolbachia strains, a total of 645 genes were shared among genomes of wOneA1, wVitA and wVitB; 212 more genes were shared between the wOneA1 and wVitA genomes but not with wVitB genome (fig. 1D). Among the 210 wOneA1-specific genes, a large fraction belongs to hypothetical protein (N = 173) and transposon-related (N = 22) genes. Regarding the insertion elements (IS), the wOneA1 genome contains similar numbers of IS elements when compared with the genomes of wVitA and wVitB (supplementary table S1, Supplementary Material online). Although wVitA and wVitB infect the same host (Nasonia vitripennis) and wOneA1 infect a different host (N. oneida), the gene content of wVitA is closer to that of wOneA1 than wVitB, as expected by their supergroup affiliations and indicating that there is no rampant recombination between the wVitA and wVitB at genome-wide level. Taken together, the results indicate that A and B Wolbachia retain their genetic differences even when they infect the same host, which suggests that recombination among them is not common, with the exception of phage related genes (Bordenstein and Wernegreen 2004).
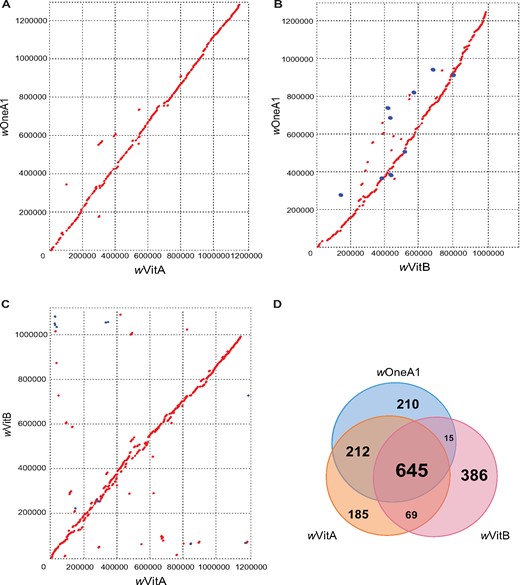
—Comparative genomic analysis of wOneA1, wVitA, and wVitB genomes. (A) Dot plot showing comparison between wOneA1 and wVitA genomes, red for a forward match and blue for a reverse match; (B) Dot plot showing comparison between wOneA1 and wVitB genomes; (C) Dot plot showing comparison between wVitA and wVitB genomes; (D) Venn Diagram showing comparison of genes and pseudogenes in wOneA1, wVitA and wVitB.
Absence of A2 and B Wolbachia in the Assembled N. oneida Strain
The whole genome alignments between wOneA1 and wVitA (fig. 1A and supplementary table S2, Supplementary Material online) and the phylogenetic analysis of MLST genes (supplementary fig. S3, Supplementary Material online) indicated the identified strain in our study is in the A supergroup. Furthermore, strain typing of Wolbachia was performed on No of our study and No genomic DNA samples that are known to be infected with all three strains (A1, A2, and B), using independent allele-specific pyrosequencing approach (supplementary table S3, Supplementary Material online). An A/G SNP in the coxA gene was used to separate B-Wolbachia from A-Wolbachia (A allele in A1/A2-Wolbachia and G allele in B-Wolbachia, supplementary fig. S4A, Supplementary Material online). In gatB gene, a C/T SNP can distinguish A1-Wolbachia allele from A2/B-Wolbachia (supplementary fig. S4B, Supplementary Material online). The pyrosequencing results confirmed the lack of A2 and B strains in the genome assembled NONY strain. All three Wolbachia infections (A1, A2, and B) were successfully identified in the CAR262L strain DNA samples (supplementary fig. S4, Supplementary Material online). Therefore, the allele-specific pyrosequencing validation experiments confirmed the absence of A2 and B-type Wolbachia infections in the lab No strain (NONY). In No DNA samples from a recently collected field strain (CAR262L), we estimate that A1 is the dominate strain and accounts for 55% of the total infection, 40% of the infection came from the B strain and only 5% from A2 strain (supplementary fig. S4, Supplementary Material online). The absence of A2 and B Wolbachia in the lab No strain is likely due to stochastic loss during laboratory maintenance and diapause.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Data deposition: This project has been deposited at DDBJ/ENA/GenBank under the accession QESS00000000.
Acknowledgments
This project is supported by an Auburn University Intramural Grant Program Award to X.W. (AUIGP180271) and a generous laboratory startup fund to X.W. from Auburn University College of Veterinary Medicine. X.W. is also supported by Alabama Agricultural Experiment Station Enabling Grant. This work is supported by the USDA National Institute of Food and Agriculture, Hatch project 1018100. X.X. and W.C. are supported by Auburn University Presidential Graduate Research Fellowship. Contributions of J.H.W. were supported by US National Science Foundation IOS 1456233 and the Nathaniel and Helen Wisch Professorship. We thank HudsonAlpha Discovery for Illumina sequencing and Ting Li for help with DNA extractions.
Literature Cited
Siozios S, et al. 2013. draft genome sequence of the Wolbachia endosymbiont of Drosophila suzukii. Genome Announc. 1(1):e00032–13.

{kind=link}