





Abstract
The diverse array of codon reassignments demonstrate that the genetic code is not universal in nature. Exploring mechanisms underlying codon reassignment is critical for understanding the evolution of the genetic code during translation. Hemichordata, comprising worm-like Enteropneusta and colonial filter-feeding Pterobranchia, is the sister taxon of echinoderms and is more distantly related to chordates. However, only a few hemichordate mitochondrial genomes have been sequenced, hindering our understanding of mitochondrial genome evolution within Deuterostomia. In this study, we sequenced four mitochondrial genomes and two transcriptomes, including representatives of both major hemichordate lineages and analyzed together with public available data. Contrary to the current understanding of the mitochondrial genetic code in hemichordates, our comparative analyses suggest that UAA encodes Tyr instead of a “Stop” codon in the pterobranch lineage Cephalodiscidae. We also predict that AAA encodes Lys in pterobranch and enteropneust mitochondrial genomes, contradicting the previous assumption that hemichordates share the same genetic code with echinoderms for which AAA encodes Asn. Thus, we propose a new mitochondrial genetic code for Cephalodiscus and a revised code for enteropneusts. Moreover, our phylogenetic analyses are largely consistent with previous phylogenomic studies. The only exception is the phylogenetic position of the enteropneust Stereobalanus, whose placement as sister to all other described enteropneusts. With broader taxonomic sampling, we provide evidence that evolution of mitochondrial gene order and genetic codes in Hemichordata are more dynamic than previously thought and these findings provide insights into mitochondrial genome evolution within this clade.
Introduction
The genetic code, as originally described, was thought to be universal and its remarkable conservation across taxa suggests that it was established early in the evolution of life on Earth (Crick 1968). However, the genetic code has been shown to vary across organismal diversity (Crick 1968; Castresana et al. 1998; Knight et al. 2001; Ambrogelly et al. 2007). The magnitude of coding variation is most notable among mitochondrial genomes where at least 16 codon reassignment events have been described (Knight et al. 2001; Moura et al. 2010; Ling et al. 2014). Events leading to diversification of the code have been argued to be influenced by evolution toward extremely small genome sizes (e.g., mitochondrial genomes) and/or strong mutational biases (Osawa and Jukes 1989). Moreover, the vast majority of described codon reassignments involve termination, or Stop codons (e.g., UAG, UGA, and UAA codons) being reassigned to encode an amino acid (Keeling 2016). For instance, UAA Stop codons are frequently preferred over UGA and UAG in most A+T rich genomes, making the latter codons easier to reassign (reviewed by Knight et al. [2001]). Codon reassignment of amino acid coding nucleotide triplets often arise from changes in tRNAs, and some codons seem to be reassigned more frequently than others (Knight et al. 2001). For instance, the AGG codon is ancestrally assigned to serine (Ser) in all bilaterian animal mitogenomes, but it has been reassigned to glycine (Gly) in tunicates (Yokobori et al. 1993), asparagine (Asn) in a hemichordate lineage (Perseke et al. 2011), lysine (Lys) in some arthropod lineages (Abascal, Zardoya, et al. 2006,) and Stop in nonhuman vertebrates (Osawa et al. 1989; Temperley et al. 2010) (fig. 1A).
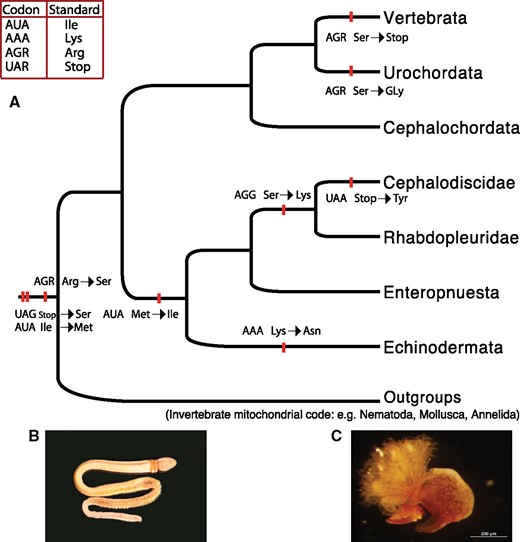
Variant codes in mitochondrial genomes within deuterostomes and photos from representatives of hemichordates. (A) Phylogenetic relationships according to the current understanding of deuterostome phylogeny based on multiple studies, red bars show codon reassignment events, the table on the top left shows standard genetic code for the affected codons. (B) Acorn worm Schizocardium braziliense collected from Mississippi (photo by Joie Cannon). (C) Cephalodiscus sp. collected from Heron Island, Queensland, Australia (photo by Kevin Kocot).
To account for codon reassignments, two major hypotheses have been proposed (Knight et al. 2001). First, the “codon capture hypothesis” suggests that a single codon simultaneously coding for two distinct amino acids (or an amino acid and a Stop) is lethal. This hypothesis argues that fluctuations of the nucleotide substitution bias that influence A+T content can eliminate certain codons from the entire genome, after which neutral evolution of nucleotide positions can allow another tRNA to use the previously lost nucleotide triplet (Osawa and Jukes 1989). Alternatively, the “ambiguous intermediate hypothesis” proposes that tRNA mutations other than at the anticodon can cause a codon to be recognized by two tRNAs (or a tRNA and a Stop), leading to gradual fixation of the reassignment if the new assignment is adaptive (Schultz and Yarus 1994). These two hypotheses are not mutually exclusive and codon reassignment may be due to factors proposed in both hypotheses. Exploring codon reassignment at the molecular level is critical for understanding genetic code evolution and provides insights to genetic code manipulation in synthetic biology (Ling et al. 2014), especially in taxa that have been underexplored.
Among deuterostomes, hemichordates share features with both echinoderms and chordates (e.g., gill slits) and play a central role in understanding deuterostome evolution. Phylogenetic analyses strongly support hemichordates as sister to echinoderms, which together form a clade called Ambulacraria (Halanych 1995; Swalla and Smith 2008; Cannon et al. 2009, 2014; Halanych et al. 2013). To date, ∼130 recognized species have been described within the two classes of Hemichordata: Enteropneusta (fig. 1B) (commonly called acorn worms) and Pterobranchia (fig. 1C) (Cannon et al. 2009; Tassia et al. 2016). Both enteropneusts and pterobranchs display a tripartite body plan, with a proboscis, collar, and trunk (Hyman 1959). Acorn worms, which comprise the vast majority of extant hemichordate taxa (∼85% of described species), possess numerous pharyngeal gill slits homologous to those found in vertebrates and live as burrowing or epibenthic deposit feeders. In contrast, pterobranchs (∼20 described species) have a reduced number of gill slits, or lack them all together, and are colonial filter feeders (Tassia et al. 2016). Although recent molecular analyses have greatly improved of our understanding of evolutionary relationships within hemichordates (Cannon et al. 2009, 2013, 2014), the phylogenetic position of some important taxa remains contentious. In particular, rhabdopleurid pterobranchs and the enteropneust Stereobalanus have been difficult to place in part due to long branches even in phylogenomic data sets. Further, despite the evolutionary importance of pterobranchs, genomic resources from this clade are extremely scarce in comparison to other major deuterostome groups.
Mitogenomics has proven useful in resolving phylogenetic relationships and investigating genome evolution across a wide range of metazoans (Boore 1999; Osigus et al. 2013; Li et al. 2015). Mitochondrial genomes have been well characterized in major chordate and echinoderm clades (e.g., Asakawa et al. 1995; Gissi et al. 2004; Perseke et al. 2010, 2011), but data on hemichordate mitochondrial evolution are notably scarce. In contrast to echinoderm and chordate mitochondrial genomes, only three enteropneust and one pterobranch mitochondrial genomes have been characterized (Castresana et al. 1998; Perseke et al. 2011). Moreover, variability of codon reassignments has been discovered in a variety of lineages within deuterostomes (fig. 1A). Notably, the AAA codon, which is assigned to Lys in most metazoans and Asn in echinoderms, is absent in two Balanoglossus genomes (Castresana et al. 1998). Absence of the AAA codon has been suggested to support for the “codon capture hypothesis” (Castresana et al. 1998). Moreover, the codon reassignment of AGG from Ser to Lys was described in the mitochondrial genome of the pterobranch Rhabdopleura compacta and has not been identified in other deuterostomes (Perseke et al. 2011); however, this is consistent with the AGG coding transitions seen among other vertebrates (e.g., fig. 1A). Notably, the mitochondrial genome of R. compacta has an extremely G+T rich frequency on the main-coding strand (Perseke et al. 2011). Variations have also been found in the gene orders of these four hemichordate mitochondrial genomes (fig. 2). However, the three hemichordate mitochondrial genomes characterized to date represent just two recognized families.
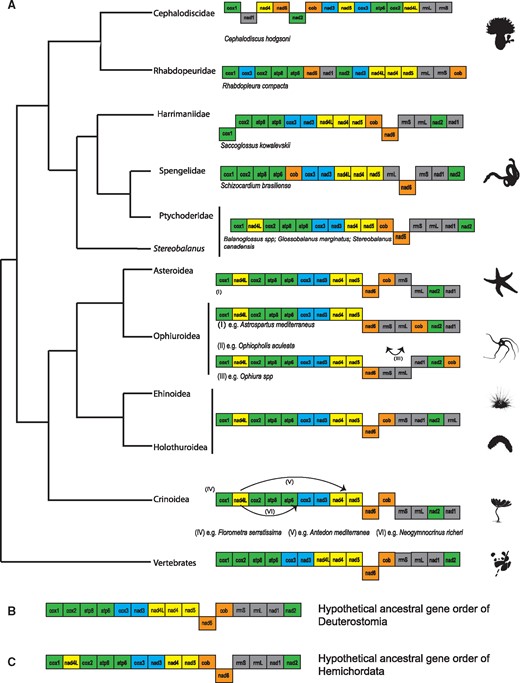
—Mitochondrial gene order within deuterostomes. (A) Current understanding of relationships within Deuterostomia and mitochondrial gene order for each taxon. Deuterostome phylogeny is depicted based on Cannon et al. (2014). Only protein-coding genes and ribosomal RNA genes of available mitochondrial genomes are included. Gene orders of tunicates were excluded. Genes are not scaled to real length and are indicated by standard abbreviations. Genes in a different line indicate different coding strand and colors indicate by protein complexes, and arrows indicate alternative gene arrangements in the corresponding species. (B) Hypothetical ground pattern of deuterostome mitochondrial genome. (C) Hypothetical ground pattern of hemichordate mitochondrial genome.
Previous analyses attempting to reconstruct the ancestral deuterostome gene order sampled a limited number of taxa, especially in the case of hemichordates (Bourlat et al. 2009). Moreover, earlier studies investigating the ancestral deuterostome mitochondrial genome organization included Xenoturbella as sister to Ambulacraria (see Bourlat et al. 2009). However, recent phylogenomic analyses have shown that Xenoturbella is not a deuterostome (Cannon et al. 2016; Rouse et al. 2016). To further explore evolution of mitochondrial genomes of deuterostomes, with particular interest in codon reassignments and gene rearrangements, we sequenced one pterobranch and three enteropneust mitochondrial genomes, as well as the transcriptomes of two additional pterobranchs to supplement data sets available in public repositories. We also explored the phylogenetic position of hemichordate taxa (e.g., Stereobalanus and Rhabdopleura) that have previously been shown to be unstable in their phylogenetic placement in previous studies (Cannon et al. 2009, 2013, 2014).
Materials and Methods
Specimen Collection and Mitochondrial Genome Sequencing
Specimen collection data are shown in supplementary table S1, Supplementary Material online. All specimens were preserved frozen at −80 °C, in 80–100% nondenatured ethanol or in RNAlater following collection. Total genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen) following manufacturer’s protocols. For Cephalodiscus hodgsoni, sequencing of genomic DNA was performed by The Genomic Services Lab at the Hudson Alpha Institute in Huntsville, AL using 2 × 100 paired-end reads on an Illumina HiSeq 2000 platform (San Diego, CA). For Glossobalanus marginatus, Schizocardium brasiliense, and Stereobalanus canadensis, sequencing libraries were prepared from total genomic DNA using Illumina’s Nextera DNA kit and run on an Illumina MiSeq sequencer with 2 × 250 paired-end reads Department of Biological Sciences, Auburn University.
Because of a limiting amount of tissue from Cephalodiscus sp. and Rhabdopleura sp., which were collected on Heron Island, QLD, Australia, only RNA was extracted as mitochondrial protein-coding and ribosomal RNA genes are usually recovered from transcriptome sequencing (e.g., Li et al. 2015). Unfortunately, information on noncoding regions and mitochondrial gene order are lacking for these taxa. Total RNA was extracted using the Ambion RNAqueous Micro Kit with DNase digestion following the manufacturer’s instructions. Total RNA was sent to Macrogen (Seoul, South Korea) for Clontech SMART-Seq v4 Ultralow RNA Input cDNA synthesis and Illumina TruSeq sequencing library preparation. Libraries were sequenced using ∼1/12 of an Illumina HiSeq 2500 lane with 2 × 101 bp paired-end reads.
Mitochondrial Genome Assembly and Annotation
For genomic data, paired-end reads were assembled de novo using Ray 2.2.0 (Boisvert et al. 2010) with k-mer = 31. For G. marginatus, Sc. brasiliense, and S. canadensis, mitochondrial contigs were identified using BlastN (Altschul et al. 1997) and the previously published hemichordate mitochondrial genome from Balanoglossus carnosus (Castresana et al. 1998) as bait. For C. hodgsoni, the mitochondrial contigs were identified using the R.compacta (Perseke et al. 2011) mitochondrial genome as bait for a TBlastX (Altschul et al. 1997) which was used to cope with the highly biased nucleotide composition in comparison to enteropneusts (see Results and Discussion). Annotation of mitochondrial genomes was conducted initially with the MITOS web server (Bernt et al. 2013), followed by manual validation of start and stop positions of each gene using Artemis (Rutherford et al. 2000).
In addition to mitochondrial and transcriptomic data generated in this study, we included seven more hemichordate transcriptomic data sets (Cannon et al. 2014) acquired from NCBI SRA Database (supplementary table S2, Supplementary Material online). Transcriptome data were assembled using Trinity 2.2.0 with the –trimmomatic and –normalize_reads flags (Grabherr et al. 2011). Mitochondrial protein-coding genes were identified by TBlastX and BlastN using the mitochondrial genomes identified that have been described above as queries. The full-length sequences of complete mitochondrial genomes and transcripts used for this study can be accessed through GenBank (supplementary table S2, Supplementary Material online).
Comparison of Codon Usage, Frequency, and tRNA Structure
The genetic code of all complete hemichordate mitochondrial genomes was predicted using the GenDecoder server v1.6 (Abascal, Zardoya, et al. 2006). Positions in the alignment with more than 20% gaps or with Shannon entropy (S) higher than 2 were interpreted as highly variable or poorly aligned, and are excluded from further analyses as suggested by Abascal, Posada, et al. (2006). The relative synonymous codon usage (RSCU) values of protein-coding genes from each hemichordate mitochondrial genome were calculated using GCUA V1.2 (McInerney 1998) to evaluate the codon usage bias in a single-codon family (supplementary table S3, Supplementary Material online). If the RSCU is close to 1, synonymous codons are used without apparent biases. When the RSCU value is greater or less than 1, codons are used either more or less frequently than expected, respectively, when compared with synonymous codons. Sequence and structural alignment of hemichordate mitochondrial tRNATyr was conducted using LocARNA web server (Smith et al. 2010).
Mitochondrial Gene Order Comparison
Pairwise comparisons of gene order among all available ambulacrarian mitochondrial genomes were performed with CREx (Bernt et al. 2007) using the common intervals parameter, which considers events of transpositions, inverse transpositions, inversions, and tandem duplication random loss (supplementary fig. S1, Supplementary Material online). To reconstruct a parsimonious scenario of gene order evolution within deuterostomes and putative ancestral gene order at the internal nodes, the program TreeREx (Bernt and Middendorf 2011) was used with -s (strong consistency method) -w (weak consistency method) -W (parsimonious weak consistency method) options as suggested in the manual. The fully bifurcated and rooted reference tree used for mapping gene order was derived from previous phylogenomic studies (e.g., Cannon et al. 2014; O’Hara et al. 2014). For all rearrangement analyses, all tRNAs and gene orders of tunicates and C. hodgsoni were excluded due to vastly different arrangement of genes within their mitochondrial genomes. In the TreeREx analysis, consistent codes shown in green are considered to be the most reliable, the k-consistent nodes shown in yellow exhibit an intermediate level of certainty, and the fallback nodes shown in red have the highest level of uncertainty.
Phylogenetic Analysis
Twenty taxa were included in the phylogenetic analysis (supplementary table S2, Supplementary Material online). The echinoderms Strongylocentrotus purpuratus (Echinoidea), Acanthaster planci (Asteroidea), and Cucumaria miniata (Holothuroidea) were selected as the outgroup for our phylogenetic analysis based on current understanding of ambulacrarian evolutionary relationships (Cannon et al. 2014). Prior to alignment, nucleotide sequences were translated to amino acids using the appropriate genetic translation code followed by manual correction. Each gene was individually aligned in MAFFT V7.2.3 (Katoh and Standley 2013) and trimmed using Gblocks (Talavera and Castresana 2007) to discard ambiguously aligned sites with default parameters. Gene alignments were then concatenated into a final supermatrix data set using FASconCAT (Kück and Meusemann 2010).
Maximum-likelihood (ML) phylogenetic analysis was conducted using RAxML v8.2.7 (Stamatakis 2014). Prior to ML analyses, PartitionFinder v2 (Lanfear et al. 2017) was used to evaluate best-fit partition schemes and associated substitution models for each partition using 20% relaxed clustering. Each ML analysis employed best-fit models and partitions indicated by PartitionFinder using a gamma (Γ) distribution to model site-substitution rate heterogeneity. Nodal support for ML analyses was evaluated with 1,000 fast-bootstrapping replicates. Competing phylogenetic hypotheses were evaluated using the approximately unbiased (AU) test (Shimodaira 2002) in CONSEL (Shimodaira and Hasegawa 2001). Per-site likelihood values were determined using RAxML with the same evolutionary models. Given that pterobrachs appear to have higher substitution rates in mitochondrial genomes, a Bayesian inference (BI) analysis was also conducted using PhyloBayes v1.6j (Lartillot and Philippe 2004) with the site-heterogeneous CAT+GTR+ Γ substitution model. Each data set was run with four parallel chains for 20,000 generations each. A burn-in of 20% was determined with trace plots as viewed in Tracer (Rambaut et al. 2014). Chains were considered to have reached convergence when the maxdiff statistic among chains was below 0.1 (as measured by bpcomp) and the effective sample size was >100 for each parameter (as measured by tracecomp). A 50% majority rule consensus tree was computed with bpcomp, and nodal support was estimated by posterior probability (fig. 3).
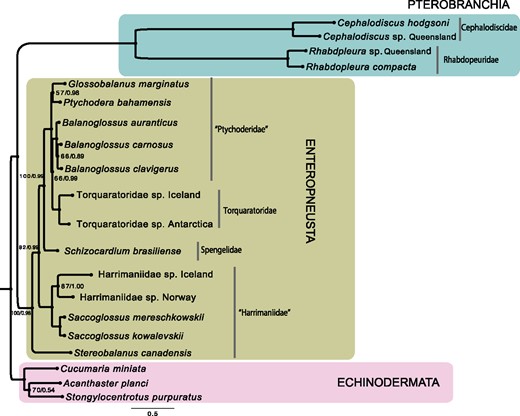
—Phylogenetic reconstruction of Hemichordata based on 13 mitochondrial protein-coding genes. Majority rule (50%) consensus phylogram from the Bayesian analysis of the concatenated data matrix is shown. Values are shown next to nodes with ML bootstrap support values left and posterior probabilities right. All nodes are supported with 100% bootstrap value or posterior probabilities of 1.0 unless otherwise noted.
Results and Discussion
General Mitochondrial Genome Features
All four newly sequenced mitochondrial genomes range in size from 15,416 to 17,366 bp (supplementary table S2, Supplementary Material online), similar to other deuterostomes. Mitochondrial genomes of G. marginatus, Sc. brasiliense, and S. canadensis contain all genes typical of bilaterian mitochondrial genomes, including 13 protein-coding, 2 rRNA, and 22 tRNA genes, whereas ATP8 is lacking in C. hodgsoni. Noticeably, the ATP8 gene in R. compacta also shows an unusual feature with about 20 residues of the C-terminal missing with respect to other hemichordates (Perseke et al. 2011). AT/GC-skew and third nucleotide codon usage of all available hemichordate mitochondrial genomes are shown in supplementary table S4, Supplementary Material online. The general mitochondrial gene features from transcriptomic data of Cephalodiscus sp. Queensland, Rhabdpeura sp. Queensland, Ptychodera bahamensis, Balanoglossus auranticus, Torquaratoridae sp. Iceland (missing atp8), Torquaratoridae sp. Norway (missing atp8), Harrimaniidae sp. Iceland (missing atp8), Harrimaniidae sp. Norwayand Sacoglossus mereschkowskii are largely similar to available mitochondrial genomes mentioned above. Consistent with the previously sequenced R. compacta (Perseke et al. 2011) mitochondrial genome, C. hodgsoni also shows an unusual nucleotide bias toward higher content of G (29.24%) and T (42.78%) than A (19.32%) and C (8.64%) on the main-coding strand (supplementary table S4, Supplementary Material online). All mitochondrial genes on the main-coding strands in C. hodgsoni also contain higher content of G+T (68.06%; supplementary table S4, Supplementary Material online), whereas nad1 and nad2 genes that are transcribed from the alternative strand show the higher amount of A+C (67%). Moreover, C. hodgsoni possesses a substantially lower amount of T but higher amount of G overall and at third codon positions compared with the mitochondrial protein-coding genes of R. compacta mitochondrial protein-coding genes (supplementary table S4, Supplementary Material online). This pattern of uneven nucleotide frequencies seems to be specific only to pterobranch mitochondria among all deuterostomes, and our findings are consistent with those described in the R. compacta mitochondrial genome (Perseke et al. 2011).
Reassignment of UAA from Stop to Tyr in Cephalodiscus
The UAA codon, which typically encodes a Stop codon in most nuclear and mitochondrial codes, was abundant in all open reading frames of the 12 protein-coding genes in the mitochondrial genome of C. hodgsoni (N = 109 UAA codons). Here, we found that UAA is reassigned to Tyr in the C. hodgsoni mitochondrial genome, with 56 conserved sites discovered in GenDecoder (supplementary table S5, Supplementary Material online). This finding was subsequently verified in mitochondrial gene alignments of C. hodgsoni and Cephalodiscus sp. transcriptomic data (e.g., see cox1 gene alignment in fig. 4), strongly supporting the fixation of the UAA codon reassignment in the Cephalodiscus lineage. This is not the first reported case of UAA codon reassignment in nature, as UAA has been reassigned from Stop to glutamine (Gln) in the nuclear genomes of several lineages of ciliates (Knight et al. 2001). Reassignment of UAA from Stop to tyrosine (Tyr) was previously reported in planarian flatworms (Bessho et al. 1992), ultimately to be disputed in a subsequent study (Telford et al. 2000). However, a later study found a similar reassignment in the mitochondrial genome of a parasitic nematode species (Jacob et al. 2009). Thus, it appears that the UAA codon reassignment from Stop to Tyr in mitochondria occurred in two independent lineages, although no obvious similarity of tRNATyr was observed due to truncated tRNA sequences in the mitochondrial genome of Radopholus similis (Jacob et al. 2009). Interestingly, the Tyr codon evolved from the UAG stop codon in the sponge species Clathrina clathrus (Lavrov et al. 2013).

Partial alignment of conserved region of translated cox1 gene sequences (100 amino acid positions) indicates that UAA decodes Tyr instead of Stop in Cephalodiscus mitochondrial genomes (confirmed by mitochondrial gene in the transcriptomic data from the same species). The alignment corresponding to amino acid positions is labeled in the header of each sequence. Codon UAA is translated into either Stop (*) based on Echinodermata codon table (Codon 9) or Tyr in Cephalodiscus mitochondrial genomes (Codon “Cepha”). The header of the sequence (Left) starting with “MT” indicates that the corresponding sequence is from mitochondrial genome assemblies, whereas “TRI” indicates that the sequence is extracted from transcriptomic data.
The alteration of first anticodon position of tRNATyr of C. hodgson could lead to this reassignment. For example, the tRNATyr in C. hodgsoni possesses a GUA anticodon (supplementary figs. S2 and S3, Supplementary Material online), which should only pair with the Tyr codons UAC and UAU based on pairing rules given the code’s degenerate nature. The tRNASer of sea star and squid mitochondrial genomes also possesses a G at the first anticodon position of tRNASer (anticodon GCU), which should only pair with AGG and AGA. However, the conversion from a G at the first anticodon position to 7-methylguanosine allows the first anticodon G to decode any nucleotide (Hyouta et al. 1987; Matsuyama et al. 1998; Tomita et al. 1998). Another possibility of the reassignment is due to tRNA modifications at locations other than anticodon positions. For example, although most animal mitochondrial Tyr tRNA genes (Osawa et al. 1992) possess a U nucleotide at position 33 (adjacent to the first anticodon position), both starfish and C. hodgsoni mitochondrial tRNATyr genes have a C (supplementary figs. S2 and S3, Supplementary Material online). The substitution in position 33 is thought to be involved in a special anticodon loop structure, which may facilitate G–A paring in the first anticodon position. Moreover, in many bacterial and eukaryotic translation systems, the G at the first anticodon position is modified to queuosine (Q) and further hypermodified into a variety of Q34-derivatives of tRNAs specific for Asp, Asn, His, and Tyr (Harada and Nishimura 1972; Morris and Elliott 2001). Q at the first anticodon position is known to only pair with U and C, but not with A and G. Unmodified G in the first anticodon position should pair with C, U, and A in the third codon position in Drosophila melanogaster tRNASer(GUU) for decoding AGU/AGG/AGA codons and echinoderm tRNAAsn(GψU) for decoding AAA codons (Tomita et al. 1998, 1999; Watanabe and Yokobori 2011). Interestingly, the echinoderm mitochondrial tRNAAsn(GUU) with C33 does not have Q at their anticodon first position and is able to pair with A at the third codon position (Tomita et al. 1999). The absence of Q34 is also thought to be how UAG codon is decoded by the tRNATyr (Grosjean et al. 2010; Watanabe and Yokobori 2011).
With the exception of the nad1 gene in the antisense strand, the sole Stop codon UAG is inferred to be used by all protein-coding genes in the C. hodgsoni mitochondrial genome. Although multiple UAA codons are detected in the nad1 gene sequence, the Stop codon UAG could not be identified (codon UAG is only present with 600 nucleotides overlapping with cox1 gene in the sense strand). The incomplete stop codons (U or UA) can form a complete canonical UAA stop codon after polyadenylation in other animal mitochondrial genomes (Boore 2006; Bobrowicz et al. 2008), but this could not be achieved in the case for Cephalodicscus due to the codon reassignment of UAA. Noticeably, the nad1 gene of C. hodgsoni may be terminated using UAA instead of UAG codon with four nucleotides overlapping with the cox1 gene in the main-coding strand, indicating that the UAA codon in Cephalodiscus mitochondria was potentially translated via an intermediate stage (see below).
Nucleotide bias can influence the frequency of Stop codon usages (Ivanova et al. 2014). For enteropneust mitochondrial genomes, which contain a slightly elevated AT content (52–60%), the UAA Stop codon is used to a much higher extent than UAG (RSCU values of UAA are higher than 1; supplementary table S3, Supplementary Material online). However, although the A+T content of pterobranchs (60–64%) is higher than that of enteropneust mitochondrial genomes because T is used in a greater extent (supplementary table S4, Supplementary Material online), pterobranch mitochondrial genomes are more G+T rich (supplementary table S4, Supplementary Material online) and G is used more frequently than A in both main-coding strand and third codon positions (supplementary table S4, Supplementary Material online). Indeed, UAG is used more commonly than UAA as a Stop codon in Rhabdopeura (9 out of 13, RSCU= 1.64, supplementary table S5, Supplementary Material online), and UAA has even been reassigned to an amino acid in Cephalodiscus mitochondrial genes.
UAA and UAG Stop codons are assumed to be decoded by the release factor (RF) mtRF1 in animal mitochondrial genomes (Lind et al. 2013). To reassign UAA from a Stop to a sense codon, the activity or mutation of the corresponding RF has to be modified or stopped based on the “codon capture hypothesis,” or mtRF1 has more functional flexibility than currently realized. Moreover, UAU-encoded Tyr is used more frequently (RSCU of UAU = 1.7) in the R. compacta mitochondrial genome, whereas UAA coded for Tyr (RSCU = 1.97) is more dominant than UAC and UAU (RSCU = 0.87) in the C. hodgsoni mitochondrial genome. This result suggests that reassignment of UAA to Tyr might provide a strong selective advantage over UAU and UAC given that C. hodgsoni’s mitochondrial genome contains a higher T content but a lower amount of adenine in the third codon position, which favors the “ambiguous intermediate hypothesis.” Another explanation is that genetic codon ending with dinucleotides with identical nucleotide bases (e.g., AA, UU) occurs more often than those ending with different bases in some mitochondrial genomes (Jia and Higgs 2008). This explanation seems less likely as the C. hodgsoni mitochondrial genome uses CUU less than CUA in Leu codon family; GAA is used less than GAG in Glu codon family; etc. (supplementary table S4, Supplementary Material online).
In summary, the UAA codon in the mitochondrial genome of Cephalodiscus appears to have been reassigned through a series of hypothesized steps. First, the asymmetrical directional nucleotide bias increased the G+T content of the main-coding strand making the UAA Stop codon extremely rare. Second, a substitution to a C33 in tRNATyr potentially involved a special anticodon-loop structure associated with an unmodified G34 or modified G34 in the first anticodon position (similar to echinoderm and squid tRNASer), which may account for this unusual G–A pairing. At this stage, the UAA codon is most likely translated ambiguously by the both RF and tRNA because 1) UAA is potentially used as stop codon in the nad1 gene in Cephalodiscus; and 2) UAA is used to a higher extent than UAU and UAC, contradicting the bias of the G+T rich third codon positions in mitochondrial protein-coding genes, which strongly suggests that this new codon reassignment is more adaptive and favors the “ambiguous intermediate hypothesis.”
A Revision of Mitochondrial Codon Usage within Hemichordata
Aside from the reassignment of UAA, mitochondrial codon usage in Cephalodiscus is largely consistent with the mitochondrial code that has been suggested for Rhabdopleura (Perseke et al. 2011). The assignment of AGG to Lys rather than Ser appears to be fixed in all pterobranchs studied so far (fig. 1A). AGG, which codes for Ser in invertebrate and echinoderm mitochondrial genetic codes, was hypothesized to code for Lys in the mitochondrial genome of R. compacta (Perseke et al. 2011). Here, we found that the reassignment of AGG to Lys is also present in the C. hodgsoni mitochondrial genome, with 17 conserved sites discovered in GenDecoder (64% with Lys, no Ser). Interestingly, several mitochondrial genomes of arthropod taxa also have an assignment of AGG for Lys, and there is evidence of correlated evolution between tRNALys and tRNASer involving specific point mutations at the anticodons (Abascal, Posada, et al. 2006). All these genomes contain an unusual CUU instead of UUU anticodon sequence of tRNALys. However, mitochondrial genomes of echinoderms and the enteropneust Balanoglossus also have the derived anticodon sequence of CUU while Saccoglossus shows the usual anticodon of UUU. Noticeably, the codon AGG is missing in all enteropneust genomes (supplementary table S5, Supplementary Material online). The codon–anticodon paring between CUU and AGG is poor, and such a wobble paring at the second codon position is rare (Abascal, Posada, et al. 2006). Clearly, further research is needed to determine the details of this unusual codon reassignment.
The genetic code in echinoderm mitochondria differs from that of other deuterostomes and most protostomes in that the codon AAA encodes Asn instead of Lys and codon AUA codes for Ile instead of Met (fig. 1A). Our results are consistent with the idea that the reassignment of AUA from Met to Ile occurred in Ambulacraria (fig. 1A;supplementary table S5, Supplementary Material online) and Platyhelminthes independently. Moreover, the codon AAA is absent in the Balanoglossus (Enteropneusta) mitochondrial genome, which has been suggested as strong support for the codon capture hypothesis (Castresana et al. 1998). However, this view was later disputed because AAA would not be expected to disappear from an AT-rich mitochondrial genome (Knight et al. 2001). Indeed, although AAA is absent or occurs only once in Balanoglossus spp., G.marginatus, and Sc.brasiliense mitochondrial genomes, AAA is predicted to encode Lys in all other hemichordate mitochondrial genomes (harimaniid Saccoglossus and pterobranchs) with strong support (supplementary table S5, Supplementary Material online). Thus, our results imply that the codon reassignment of AAA to Asn only occurred independently in Echinodermata and Hemichordata retain the ancestral state, decoding AGG to Lys. In contrast with the previous hypothesis (Jose Castresana et al. 1998), the loss of the codon AAA is most likely secondary and only occurred in a clade comprising Ptychoderidae and Spengelidae within Enteropneusta (mitochondrial data of Torquaratoridae are lacking) (fig. 3 and supplementary table S5, Supplementary Material online).
Our results imply that genetic codon usage in hemichordate mitochondria is more complicated than previously understood, and the echinoderm and flatworm mitochondrial code should not be applied to hemichordates. We propose two revised mitochondrial codes (supplementary tables S6 and S7, Supplementary Material online): One (supplementary table S6, Supplementary Material online) for Enteropneusta, which differs from that of the invertebrate mitochondrial code in that ATA codes for Ile instead of Met (supplementary table S6, Supplementary Material online); and the other (supplementary table S7, Supplementary Material online) proposes a novel genetic code for Cephalodiscus, which differs from the “Pterobranchia” mitochondrial code (Perseke et al. 2011) by the reassignment of UAA to Tyr instead of Stop (supplementary table S7, Supplementary Material online). Thus, the “Pterobranchia” code implemented to date (i.e., NCBI translation table 24) should only be applied to Rhabdopleura.
Hemichordate Phylogeny
The AA data set consisted of 3,538 positions in length after trimming using Gblocks. Both ML and BI (fig. 3) analyses yielded congruent tree topologies with high bootstrap support values (bs) and posterior probabilities (pp), respectively. Pterobranchia and Enteropneusta were recovered as reciprocally monophyletic (fig. 3). As previously observed, all pterobranchs generally had longer branch lengths when compared with enteropneusts. Moreover, our mitogenomic analysis placed the deep-sea family Torquaratoridae within Ptychoderidae (fig. 3), consistent with previous phylogenomic study (Cannon et al. 2014).
Phylogenetic relationships within Enteropnuesta are largely consistent with previous analyses (Cannon et al. 2009, 2013, 2014), except for placement of Stereobalanus. The most species-rich enteropneust family Harrimaniidae, represented in this study by two species of Saccoglossus, two species of an undescribed genus, and S. canadensis, was recovered as paraphyletic with Stereobalanus sister to the remaining enteropnuests with strong support (bs = 99, pp = 0.98). Although placement of Stereobalanus was unstable due to a remarkably long branch for this species in previous phylogenetic analyses based on 18S rRNA and phylogenomic data sets (Cannon et al. 2009, 2013, 2014), we found Stereobalanus to have a similar branch length as the other enteropneusts sampled in our mitogenomic analysis (fig. 3). Although less likely than our consensus topology (fig. 3), the hypothesis of Stereobalanus as sister to remaining harrimaniids was not explicitly rejected by the AU test (supplementary table S8, Supplementary Material online). Notably, Stereobalanus is morphologically distinct among all enteropneusts in that it has four short gonad regions, or genital ridges, in the anterior-most region of the trunk, with a broad gill slit opening between the dorsoventral and ventrolateral gonads.
Mitochondrial Gene Order and Rearrangements
As mentioned above, tRNAs and gene orders of tunicates and C. hodgsoni were excluded in the reconstruction of gene order evolution due to their high positional variability. Within Hemichordata, consistent with previous hypothesis (Perseke et al. 2011), the mitochondrial gene order of Balanoglossus appears to represent the ancestral state of hemichordates, with one translocation of cob or nad6 and another translocation of nad4l from the putative ancestral deuterostome arrangement (supplementary fig. S1, Supplementary Material online). Within Enteropneusta, the gene order of G. marginatus is essentially the same as two previously sequenced Balanoglossus mitochondrial genomes and is consistent with the inferred ancestral gene order (fig. 2). The gene order of Sc. brasiliense possesses two transpositions in comparison to the hemichordate ancestral order (transpositions of cob and rrnL). Spengelidae is sister to a clade comprised Torquaratoridae and Ptychoderidae and has a mitochondrial genome that appears to be more derived based on our reconstruction (supplementary fig. S1, Supplementary Material online). However, improved taxon sampling within Spengelidae, Ptychoderidae, and Torquaratoridae is needed to determine whether this observation is generally true. Notably, Stereobalanus shares the same ancestral gene order with Balanoglossus and Glossobalanus, representing the ancestral state within hemichordates, whereas two genome rearrangements were identified in Saccoglossus (an inversion of cox1, and a transposition of nad1 and nad2).
In contrast with other deuterostomes, the two pterobranch mitochondrial genomes show a variable gene order. The mitochondrial gene order of R. compacta is more similar to enteropneusts than C. hodgsoni with at least three rearrangements between the hemichordate ancestral state and that of R. compacta (Perseke et al. 2011) (supplementary fig. S1, Supplementary Material online). However, the gene order of C. hodgsoni differs significantly from other hemichordates and deuterostomes (four rearrangements from Rhabdopleura and nine from the hypothetical deuterostome ancestral state) (supplementary fig. S1, Supplementary Material online). Extensive gene rearrangements between closely related taxa with accelerated evolutionary rates have also been identified in tunicates, especially ascidian mitochondrial genomes. Moreover, tunicates also utilize a modified genetic code with a considerably shorter atp8 gene compared with other deuterostome mitochondrial genomes. Thus, exploration of heterogeneity in mitochondrial gene order and evolutionary rates in other pterobranch species would be of great interest.
The ancestral arrangement for Ambulacraria, comprising Hemichordata and Echinodermata, also shares the same gene order with current vertebrate mitochondrial genomes. As mentioned above, gene rearrangements from the ancestral state of hemichordates (resembling the extant arrangement present in Stereobalanus and Balanoglossus) could be derived from one translocation of nad6 and cob. However, our results of the TreeREx reconstruction showed a high level of uncertainty within Echinodermata (supplementary fig. S1, Supplementary Material online). Previous analyses favor a hypothetical crinoid gene order as the ancestral state (details of the echinoderm gene order are given by Perseke et al. [2008, 2010]; supplementary fig. S1, Supplementary Material online), which is consistent with the hypothesis that Crinoidea is placed as sister to Eleutherozoa from recent phylogenomic analysis. Following this hypothesis, the transposition of nad4l, and one inversion of the fragment containing 16S, nad1 and nad2 led to the ancestral gene rearrangement of the echinoderm group. With a broader sampling of hemichordate taxa, we provide evidence that evolution of mitochondrial genomes and genetic codes are more dynamic than previously thought for this phylogenetically important clade and provide insights into the mitochondrial genome evolution within Deuterostomia.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Acknowledgments
This study was supported by awards (DEB-0816892, ANT-1043745) to K.M.H. from the U.S. National Science Foundation (NSF). Y.L. is supported by a scholarship from the China Scholarship Council (CSC) for studying and living abroad. We thank Justin Havird, Damien Waits, and Kyle David for proofreading this manuscript. We also thank Scott Santos for the helpful discussion about genetic mechanisms of codon reassignment. All bioinformatic analyses were conducted on the Auburn University Molette Laboratory SkyNet server and Auburn University Hopper HPC system. This is Molette Biology Laboratory contribution 87 and Auburn University Marine Biology Program contribution 184. The authors confirm that there are no known conflicts of interest associated with this publication.
Literature Cited
Author notes
Data deposition: This project has been deposited in GenBank under the accession MG652439, MH841935, MH841936 and MF374802. Mitochondrial genes derived from hemichordate transcriptomic data acquired from NCBI SRA Database were deposited to figshare, figshare.com (https://doi.org/10.6084/m9.figshare.7077737).

{kind=link}
{kind=link}
{kind=link}
{kind=link}