





Abstract
While typically a flea parasite and opportunistic human pathogen, the presence of Rickettsia felis (strain LSU-Lb) in the non-blood-feeding, parthenogenetically reproducing booklouse, Liposcelis bostrychophila, provides a system to ascertain factors governing not only host transitions but also obligate reproductive parasitism (RP). Analysis of plasmid pLbAR, unique to R. felis str. LSU-Lb, revealed a toxin–antitoxin module with similar features to prophage-encoded toxin–antitoxin modules utilized by parasitic Wolbachia strains to induce another form of RP, cytoplasmic incompatibility, in their arthropod hosts. Curiously, multiple deubiquitinase and nuclease domains of the large (3,841 aa) pLbAR toxin, as well the entire antitoxin, facilitated the detection of an assortment of related proteins from diverse intracellular bacteria, including other reproductive parasites. Our description of these remarkable components of the intracellular mobilome, including their presence in certain arthropod genomes, lends insight on the evolution of RP, while invigorating research on parasite-mediated biocontrol of arthropod-borne viral and bacterial pathogens.
Introduction
The ability of microbial parasites to influence the sexual reproduction of their hosts (hereafter reproductive parasitism, RP; Hurst and Frost 2015) is well known and includes such processes as male-killing, feminization, and parthenogenesis induced by parasites to drive their retention in host populations via the female germline (Werren et al. 2008). Cytoplasmic incompatibility (CI), extensively studied in Wolbachia (Rickettsiales: Anaplasmataceae) parasites, is another RP strategy which entails parasite-induced modifications to sperm that prevent paternal chromosome condensation during the first cell cycle, ultimately killing the zygote (Yen and Barr 1971). This lethal phenotype is alleviated if females are infected with a compatible parasite strain, implicating a “modification-rescue” mechanism directed by the parasite (Hurst 1991; Presgraves 2000; Clark et al. 2003; Poinsot et al. 2003). Indeed, diligent efforts to identify the genetic factors underpinning CI led to the recent discovery of Wolbachia toxin–antitoxin (TA) systems, wherein two-gene modules encode the sperm-modifying toxins and cognate rescue antitoxins (Beckmann and Fallon 2013; Beckmann et al. 2017; LePage et al. 2017). Four distinct Wolbachia TA modules (Types I–IV) were identified (LePage et al. 2017) and predominantly occur within the eukaryotic association module (EAM) of WO prophage (Bordenstein and Bordenstein 2016). However, only two distinct toxins, CI-inducing deubiquitinase (CidB, or Type I) and CI-inducing nuclease (CinB, or Type IV), were shown to bind with their cognate antitoxins, CidA and CinA, respectively, with such binding neutralizing toxicity (Beckmann et al. 2017). Whereas groundbreaking, the exact cellular functions of these TA systems remain unknown, as do their roles in other types of RP phenotypes elicited not only by Wolbachia species but also other host-associated microbes.
Aside from a very recent report on Spiroplasma male-killing (Harumoto and Lemaitre 2018) (discussed below in section Wolbachia do not walk alone: CI-like TA modules abound in the bacterial mobilome), the genetic factors underpinning CI and other RP phenotypes induced by other bacteria have not been elucidated. A Wolbachia relative, Rickettsia felis (Rickettsiales: Rickettsiaceae), provides an opportunity to identify the molecular mechanisms underpinning thelytokous parthenogenesis, a type of parthenogenesis in which females are produced from unfertilized eggs (Tucker 1958). Rickettsiafelis is typically transmitted by blood-feeding fleas (Insecta: Siphonaptera) (Adams et al. 1990), with its pathogenic potential in vertebrates the subject of contention (Labruna and Walker 2014; Blanton and Walker 2017). Importantly, R. felis is not known to induce RP in its blood-feeding vectors (Gillespie et al. 2007). However, R. felis has been identified in several non-blood-feeding insects (Reif and Macaluso 2009), including the booklouse Liposcelis bostrychophila (Insecta: Psocoptera), which is a storage grain pest found almost exclusively in nature as parthenogenetic reproducing populations. Such female L. bostrychophila harbor R. felis as an obligate mutualist required for early oocyte development and maintained 100% transovarially (Yusuf and Turner 2004; Perotti et al. 2006). Sexually reproducing L. bostrychophila are rare, and R. felis has not been identified in these populations (Mockford and Krushelnycky 2008; Yang et al. 2015). The exact role that R. felis may play in parthenogenesis induction remains to be elucidated.
Reasoning that a genetic basis underlies R. felis host specialization and possibly RP, we previously sequenced the genome of strain LSU-Lb, which is 100% present within an all-female L. bostrychophila colony (Thepparit et al. 2011), and carried out comparative genomics analysis with several genomes of flea-associated R. felis strains (Gillespie et al. 2015). Indeed, R. felis str. LSU-Lb was found to carry the unique plasmid pLbAR, which contains the signature DnaA-like protein found on all rickettsial plasmids as well as the hallmark integrative conjugative element associated with the rickettsial mobilome (Gillespie et al. 2012, 2014). pLbAR also encodes a repeats-in-toxin (RTX)-like type I secretion system (T1SS) and an adjacent recombination hot spot (RHS)-like toxin (pLbAR_38), elements not present in other Rickettsia genomes. Although different regions of the large, highly modular pLbAR_38 were found to be similar to a minimal assortment of proteins from other obligate intracellular bacteria, as well as eukaryotic RHS toxins, we also observed that pLbAR_38 and an adjacent coding sequence (pLbAR_36) had low similarity to two proteins encoded by adjacent genes in many Wolbachia genomes. Just prior to our report, a pLbAR_36-like protein from the Wolbachia endosymbiont of Culex quinquefasciatus Pel (wPip) was found in mosquito spermathecae (Beckmann and Fallon 2013). This protein and its partner have since been characterized as CidA and CidB (Beckmann et al. 2017; LePage et al. 2017), raising the possibility pLbAR_38/36 might also function in modification-rescue.
Accordingly, in this work we reexamined pLbAR_38 and pLbAR_36 by conducting robust bioinformatics, structural, and comparative genomics analysis. Despite only 4 years since our original report, the accumulated knowledge on Wolbachia CI-inducing factors, coupled with a plethora of new genome sequences from diverse intracellular species, allowed us to further strengthen the case that pLbAR encodes parthenogenesis-inducing proteins. Moreover, our analyses implicate the intracellular mobilome for dissemination of an intriguing assortment of potential RP-inducing genes across a diverse assemblage of intracellular bacteria, and quizzically, certain arthropods. Thus, our work identifies focal areas for increasing our understanding of the molecular factors triggering RP, while expanding the range of candidate RP-inducing microbes for controlling vector transmission of pathogens.
Materials and Methods
Protein Domain Prediction
All analyses utilized the original pLbAR_36 (NCBI accession no. WP_039595309) and pLbAR_38 sequences (WP_081996388) (Gillespie et al. 2015), the latter differing from a more recent NCBI gene prediction that truncates the protein at the N-terminus by 11 residues (KHO02170). All protein domains and motifs were predicted using the EMBL’s Simple Modular Architecture Research Tool (SMART) (Letunic and Bork 2017) or inferred from structural modeling using the Protein Homology/analogY Recognition Engine V 2.0 (Phyre2) (Kelley and Sternberg 2009). The specific coordinates assigned for predicted ovarian tumor (OTU) cysteine protease (Pfam OTU, PF02338) (Makarova et al. 2000), endoMS-like (Nakae et al. 2016) and NucS-like (Ren et al. 2009) PD-(D/E)XK nuclease (Pfam PDDEXK_1, PF12705) (Kinch et al. 2005), CE clan protease (Pfam Peptidase_C1, PF00112) (Pruneda et al. 2016), hemolysin-type calcium-binding repeat (Pfam HemolysinCabind, PF00353) (Economou et al. 1990), salivary gland secreted protein domain toxin (Pfam Tox-SGS, PF15651) (Zhang et al. 2012), and coiled-coil domains, as well as ankyrin (ANK) repeats, are listed in supplementary fig. S1, Supplementary Material online. Proprotein convertase cleavage sites were predicted using the ProP 1.0 Server (Duckert et al. 2004). Individual protein schemas were generated using Illustrator of Biological Sequences (Liu et al. 2015) with manual adjustment.
Database Searches
Initial Blastp searches with complete pLbAR_36 and pLbAR_38 sequences were performed using the NCBI RefSeq non-redundant (All GenBank+RefSeq Nucleotides+EMBL+DDBJ+PDB) protein database, coupled with a search against the Conserved Domains Database (Marchler-Bauer et al. 2011). Searches were performed with composition-based statistics across “bacteria (taxid: 2) excluding Wolbachia (taxid: 953)”, “Wolbachia”, and “Eukaryota (taxid: 2759)”. No filter was used. Default matrix parameters (BLOSUM62) and gap costs (Existence: 11, Extension: 1) were implemented, with an inclusion threshold of 0.005. Refined Blastp searches were performed in the same manner, but used specific pLbAR_38 domains as queries (see supplementary fig. S1, Supplementary Material online). Information pertaining to all sequences selected for further analysis is provided in supplementary table S1, Supplementary Material online.
Comparative Sequence Analyses
For subjects retrieved from the Blastp searches using the pLbAR_38 deubiquitinase (DUB) and nuclease domains, and entire pLbAR_36 proteins, individual domain predictions were carried out as described above. In some cases, one-to-one threading was performed with Phyre2 between subjects and specific protein structures (gleaned from Phyre2 analyses using pLbAR_38). All retrieved subjects were aligned using MUSCLE (default parameters) (Edgar 2004). Sequence compositions within protein active sites and other conserved regions were displayed using Weblogo (Crooks et al. 2004).
Phylogeny Estimation
Phylogenies were estimated from the pLbAR_38 central region and related sequences (for data set construction see supplementary fig. S1, Supplementary Material online). The protein alignment was masked using Gblocks 0.91b (Talavera and Castresana 2007), with both unmasked and masked alignments used in separate maximum likelihood (ML)-based phylogeny estimations with RAxML v8.2.4 (Stamatakis 2014), implementing a gamma model of rate heterogeneity and estimation of the proportion of invariable sites. Two models were utilized per alignment (WAG and LG), resulting in a total of four ML-based phylogeny estimations. Branch support was assessed with 1,000 pseudoreplications (for full details see supplementary fig. S1, Supplementary Material online).
Data Availability
All analyses were performed using published protein sequences available at NCBI, with all relevant information provided in supplementary table S1, Supplementary Material online. The authors declare that all other data supporting the findings of this study are contained within the article, including the Supplementary Information files, or available from the authors upon request.
Results and Discussion
pLbAR_38 is a Highly Modular Component of the Intracellular Mobilome
In addition to previously identified domains, that is, OTU cysteine protease, salivary gland secreted protein domain toxin (Tox-SGS) and ANK repeats, we predicted additional domains within pLbAR_38: two divergent PD-(D/E)XK nuclease domains (Pfam PDDEXK_1) (Kinch et al. 2005), a CE clan protease domain (Pfam Peptidase_C1) (Pruneda et al. 2016), and a hemolysin-type calcium-binding repeat domain (Pfam HemolysinCabind) (Economou et al. 1990) (fig. 1; supplementary fig. S1A, Supplementary Material online). Additionally, two coiled-coils and two furin-like cleavage sites were predicted, raising the possibility that pLbAR_38 is processed in the host cytosol consistent with proprotein processing of large secreted toxins. The presence of a HemolysinCabind domain, which is typically associated with T1SS toxins, agrees with pLbAR_38 being adjacent to four genes encoding an RTX-like T1SS that is evolutionarily related to T1SSs of several obligate intracellular symbionts and parasites (Gillespie et al. 2015). These analyses strongly imply that pLbAR_38 is a highly modular secreted toxin, with predicted activities tailored to the eukaryotic cytosol.

—The modular protein pLbAR_38, carried on plasmid pLbAR of Rickettsia felis str. LSU-Lb, shares similarity to a diverse assemblage of proteins from a narrow range of obligate intracellular bacteria. Protein domains of pLbAR_38 are described in more detail in the text and supplementary figure S1, Supplementary Material online. Black triangles, proprotein convertase cleavage sites (Duckert et al. 2004). A graphical depiction of a Blastp search against the NCBI nr database using pLbAR_38 (coordinates 1–3,048, which excludes the ankyrin repeats) as the query is shown (inset, color key for alignment scores). White numbers indicate % identity across significant alignments. Yellow highlighting depicts the exact region of similarity projected down from the pLbAR_38 schema at top. Subjects missing internal sequence relative to pLbAR_38 (nos 3, 11, 19, and 30) are joined by dashed lines; one subject with a large insertion relative to pLbAR_38 is adjusted accordingly (no. 22). Black regions within subjects depict sequences with no significant matches to pLbAR_38. Taxon color scheme as follows: black, Rickettsia; purple, Wolbachia; dark green, other Rickettsiales; light blue, Cardinium (Bacteroidetes); blue, Diplorickettsia and Rickettsiella (Gammaproteobacteria). Taxa colored red indicate Wolbachia genes within three arthropod genomes: clonal raider ant (Ooceraea biroi), common pillbug (A. vulgare) and little fire ant (W. auropunctata). wVulC-f refers to the f element of A. vulgare (Leclercq et al. 2016). Type I–IV cytoplasmic incompatibility toxins of Wolbachia (LePage et al. 2017) are noted. Note.—Each subject (numbered 1–35 at left) represents a distinct protein architecture, yet in some cases multiple similar proteins can be found for closely related species and strains). See supplementary table S1, Supplementary Material online for information pertaining to all sequences.
Consistent with our prior analyses, complete pLbAR_38 homologs are not present in other organisms, though certain domains are highly similar to proteins from diverse intracellular bacteria as well as some eukaryotes. This evolutionary mosaicism across proteins from diverse bacteria is accentuated by high similarity across a large portion of pLbAR_38 and proteins from three recently sequenced Wolbachia genomes: wDacB (Ramirez-Puebla et al. 2016), wStri, and wVulC-f (Leclercq et al. 2016) (fig. 1). Furthermore, over a dozen other proteins from diverse obligate intracellular species contain similarity to the pLbAR_38 central region, and phylogeny estimation suggests that these toxins have spread via lateral gene transfer (LGT; supplementary fig. S1B, Supplementary Material online). The mobile nature of elements encoding these toxins is clearly demonstrated in another recently sequenced Wolbachia genome, wFol (Faddeeva-Vakhrusheva et al. 2017); this endosymbiont possesses at least five large toxins with high similarity to pLbAR_38 (only one protein is shown for brevity: no. 9 in fig. 1) that likely depict recent proliferation within its genome (supplementary fig. S1B, Supplementary Material online).
Like pLbAR_38, many of the identified proteins are very large (fig. 1) and highly modular. Smaller proteins include Wolbachia CI-inducing toxins (Types I–IV) and other proteins encoded within the EAM of WO prophage (Bordenstein and Bordenstein 2016), as well as a myriad of proteins that have restricted similarity to specific domains of pLbAR_38. Telescopic analyses of these domains, presented below, reveal a veritable cornucopia of architecturally diverse proteins found almost exclusively in a narrow range of intracellular bacteria (i.e., species/strains of Rickettsia, Orientia, Occidentia, Wolbachia, Rickettsiella, Diplorickettsia, and Cardinium). However, reciprocal Blastp searches using these proteins as queries tends to unearth an even greater assortment and range than what we report here, accentuating the role of the mobilome in shaping intracellular bacterial diversity.
OTU Domain-Containing Proteins: Rare yet Highly Diverse Architectures
Although known from certain viruses and bacteria (Makarova et al. 2000), OTU domain-containing proteins (OTU-DCPs for brevity) are predominantly found in eukaryotes where they primarily function as DUBs in ubiquitin-dependent signaling pathways (Edelmann et al. 2009; Mevissen et al. 2013). Using the predicted OTU domain from pLbAR_38, we identified OTU-DCPs in a limited number of Rickettsiales genomes (fig. 2A). Many of these proteins are exceedingly large and highly modular, with the majority found in strains of Wolbachia, which were previously shown to carry OTU-DCPs both within and outside of the EAM of WO prophage (Bordenstein and Bordenstein 2016). Strikingly, several Wolbachia OTU-DCPs also have a region that is structurally analogous to the N-terminal half of Drosophila Apaf-1 related killer, an apoptosis effector homologous to cell-death proteins Apaf-1 (mammals) and CED-4 (Caenorhabditis elegans) (Rodriguez et al. 1999). Other eukaryotic motifs (Pfam NB-ARC, Pfam Latrotoxin_C) (van der Biezen and Jones 1998; Zhang et al. 2012) and various repeat types indicate the likelihood that rickettsial OTU-DCPs target host molecules.
![—Ovarian tumor (OTU) domain-containing proteins are not common in bacteria and are often highly modular. (A) An assortment of OTU domain-containing proteins (OTU-DCPs). The pLbAR_38 OTU domain (coordinates 19–180), used as the query in Blastp searches against the NCBI nr database, identified OTU-DCPs in Rickettsia (black), Wolbachia (purple), and arthropod host (red) genomes, and the f element of A. vulgare (wVulC-f) (Leclercq et al. 2016). Protein domains are described in the gray inset. Red inset: predicted Dark (DrosophilaApaf-1 related killer) domain of wAlbB OTU-DCP (protein number 6) modeled to a single subunit of the D. melanogaster apoptosome (PDBID: 4V4L) (Yuan et al. 2011). Note.—In some cases, multiple similar proteins can be found for closely related species and strains. See supplementary table S1, Supplementary Material online for information pertaining to all sequences. (B) Characteristics of the rickettsial OTU domain. Left, pLbAR_38 OTU domain modeled to Homo sapiens Otubain-2 (PDBID: 1TFF) (Nanao et al. 2004). Modelling done using Phyre2 (Kelley and Sternberg 2009). Green highlighting indicates the active site. Middle, sequence and linear depiction of the predicted structure of pLbAR_38 OTU domain. Highlighting shows the regions encompassing active site residues Asp and Cys (yellow) and His (gray). Red sequence indicates length variable region across rickettsial OTU domains. Blue shading shows the area of large insertions in many rickettsial OTU domains (indicated with black dots in the schemas in [A]). Right, sequence logo (Crooks et al. 2004) illustrating active site conservation across the 14 proteins depicted in (A). Amino acid coloring as follows: black, hydrophobic; red, negatively charged; green, hydrophilic; purple, aromatic; blue, positively charged. (C) Other bacterial OTU-DCPs are also highly modular. NCBI acc. nos. are in parentheses. Protein domains are described in the gray inset. Taxon color scheme as follows: brown, Deltaproteobacteria; gray, unclassified Parcubacteria group; blue, Gammaproteobacteria; green, Betaproteobacteria.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/gbe/10/9/10.1093_gbe_evy159/2/m_evy159f2.jpeg?Expires=1750203399&Signature=ndQbqDrZy6wsSt6HE3uEFoHFIp9oBC4fAia3BUQnELVwLyCnVjvkJ89fYwkAfkojRMP78moxBxF-F0ozliJXajR5W1~RjulpSN1XB9RaR7YsWrFMtyc3x6QBq0p7lbsqws8CypjfhyAWUfGy47fMBooVTumu-R1JITyuwOb5YQkdA~mEYWhM-sM0V4qB5hpafuQIXRr71szKvx2rt2d2sAMyuMQM64Yihp2moqDF93rmApkkjDmbeER0Tho4NHoiPRQ5W2FSQj6F48tpCF-~onLR7PFbM5ml0c7vfELU~t5XmNXmJ0nQZFhS60RtDHPfH570SfzQEGyV4ig2yvouYg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
—Ovarian tumor (OTU) domain-containing proteins are not common in bacteria and are often highly modular. (A) An assortment of OTU domain-containing proteins (OTU-DCPs). The pLbAR_38 OTU domain (coordinates 19–180), used as the query in Blastp searches against the NCBI nr database, identified OTU-DCPs in Rickettsia (black), Wolbachia (purple), and arthropod host (red) genomes, and the f element of A. vulgare (wVulC-f) (Leclercq et al. 2016). Protein domains are described in the gray inset. Red inset: predicted Dark (DrosophilaApaf-1 related killer) domain of wAlbB OTU-DCP (protein number 6) modeled to a single subunit of the D. melanogaster apoptosome (PDBID: 4V4L) (Yuan et al. 2011). Note.—In some cases, multiple similar proteins can be found for closely related species and strains. See supplementary table S1, Supplementary Material online for information pertaining to all sequences. (B) Characteristics of the rickettsial OTU domain. Left, pLbAR_38 OTU domain modeled to Homo sapiens Otubain-2 (PDBID: 1TFF) (Nanao et al. 2004). Modelling done using Phyre2 (Kelley and Sternberg 2009). Green highlighting indicates the active site. Middle, sequence and linear depiction of the predicted structure of pLbAR_38 OTU domain. Highlighting shows the regions encompassing active site residues Asp and Cys (yellow) and His (gray). Red sequence indicates length variable region across rickettsial OTU domains. Blue shading shows the area of large insertions in many rickettsial OTU domains (indicated with black dots in the schemas in [A]). Right, sequence logo (Crooks et al. 2004) illustrating active site conservation across the 14 proteins depicted in (A). Amino acid coloring as follows: black, hydrophobic; red, negatively charged; green, hydrophilic; purple, aromatic; blue, positively charged. (C) Other bacterial OTU-DCPs are also highly modular. NCBI acc. nos. are in parentheses. Protein domains are described in the gray inset. Taxon color scheme as follows: brown, Deltaproteobacteria; gray, unclassified Parcubacteria group; blue, Gammaproteobacteria; green, Betaproteobacteria.
Structural modeling of the pLbAR_38 OTU domain revealed a larger sequence within the typical papain fold of other OTU-DCPs (fig. 2B, red sequence). Most Wolbachia OTU-DCPs have a second region of additional sequence (fig. 2B, blue shading), making these atypically large OTU domains. Although the immediate regions encompassing the active site residues are highly conserved (fig. 2B), the larger size and lack of conservation in the intervening sequence indicate rapid rates of divergence, consistent with a previous report detecting a wMel OTU-DCP evolving under positive selection (Metcalf, Jo, et al. 2014). These characteristics challenged the search for similar OTU domains in other bacteria. For instance, pLbAR_38-based searches did not identify any of the OTU DUBs utilized by pathogenic chlamydiae during mammalian cell infection (Misaghi et al. 2006; Furtado et al. 2013; Fischer et al. 2017).
Remarkably, half of the retrieved non-rickettsial OTU-DCPs contain additional domains associated with DNA processing: reverse transcription (Pfam RVT_1), DNA mismatch repair (Pfam Exo_endo_phos) or chromosomal unwinding (Pfam HMG17, Pfam Agro_virD5) (González-Romero et al. 2015; Zhang et al. 2017), reflecting a commonality with certain eukaryotic OTU DUBs that function in histone modification and transcriptional control (Pinto-Fernandez and Kessler 2016; Keren and Citovsky 2017) (fig. 2D). These bacterial OTU-DCPs may target DNA, and possibly the host nucleus if secreted intracellularly, provided that K63 ubiquitin (and ubiquitin-like) modifications are intricately linked to various processes in genome maintenance (Walczak et al. 2012). A host nuclear function for the OTU domain of pLbAR_38 seems reasonable given that its adjacent CE clan protease and nuclease domains are similar to Wolbachia toxins CidB and CinB, respectively, which presumably target DNA during mitosis (discussed below). We did not identify any other bacterial proteins containing both the OTU and CE clan protease domains. For Wolbachia strains, the roles (if any) OTU-DCPs play in CI or other RP phenotypes remain to be determined, though selection has not favored retention of proteins with both DUB domains.
Curiously, such “double-DUBs” (dual OTU/CE clan protease-containing DUBs) were found to be encoded in the genomes of two arthropods, Armadillidium vulgare (pillbug) and Ooceraea biroi (clonal raider ant) (fig. 2A). In A. vulgare, the protein (Wxf_03240) is encoded outside of the W chromosome-encoded f element, a 3-Mb insertion of a genome from a feminizing Wolbachia strain (hereafter wVulC-f) that was recently transferred into the pillbug nuclear genome (Leclercq et al. 2016). Curiously, Wxf_03240 is divergent from the three OTU-DCPs of wVulC-f. For O. biroi, which reproduces clonally via thelytokous parthenogenesis (Tsuji and Yamauchi 1995; Ravary and Jaisson 2004), the protein appears to be encoded within a small region of WO prophage introgression (data not shown). The functional significance of double-DUBs, particularly whether they play a role in RP, remains to be determined.
CE Clan Proteases: Widespread DUBs with a Myriad of Architectures
The second predicted DUB domain of pLbAR_38 belongs to proteases of the CE clan (Barrett and Rawlings 2001), originally described to encompass the families of adenain from adenoviruses, the eukaryotic ubiquitin-like protease (Ulp) 1 (C48 peptidase), and the bacterial YopJ protease that, aside from DUB activity, also uses the same active site (His-Asp-Cys) for acetyltransferase activity (Orth et al. 2000; Zhou et al. 2005). Eukaryotic CE clan proteases typically target ubiquitin-like (UBL) modifications (e.g., NEDD8, ISG15, and SUMO) over ubiquitin (Clague et al. 2013; Ronau et al. 2016). Conversely, bacterial CE clan effectors mostly recognize ubiquitin, though several enzymes (i.e., Xanthomonas XopD, Chlamydia ChlaDUB1, and Rickettsia RickCE) have ubiquitin/UBL cross reactivity (Pruneda et al. 2016). Thus, while we refer below to CE clan proteases as DUBs, the full range of modifications for each protein, as well as their possible acetyltransferase activity, cannot be determined without experimentation.
Unexpectedly, we identified a wide array of proteins with similarity to the pLbAR_38 CE clan protease domain (fig. 3A). Like the OTU-DCPs discussed above, many of these CE clan protease domain-containing proteins (CE-DCPs for brevity) are large and highly modular, possessing eukaryotic-like domains and repeats. However, a much larger span in size was found for CE-DCPs, epitomized by the many Wolbachia proteins under 700 aa. Furthermore, a greater range of bacterial species was found to harbor CE-DCPs compared with OTU-DCPs, though nearly all are obligate intracellular species. Remarkably, aside from the A. vulgare and O. biroi double-DUBs described above, we also identified two CE-DCPs encoded on adjacent genes in the genome of Wasmannia auropunctata (the little fire ant, protein nos. 14 and 15 in fig. 3A). These proteins are most similar to NB-ARC domain-containing proteins found in the EAM of WO prophage (Bordenstein and Bordenstein 2016), but are also highly similar to counterparts that are widespread in arthropods (data not shown). As the flanking genes are strictly arthropod in nature, it is probable that genes encoding these two W. auropunctata CE-DCPs originated from WO prophage. Curiously, in some W. auropunctata populations, queens and males can arise clonally from only maternal and paternal genomes, respectively, whereas sterile workers are produced sexually (Fournier et al. 2005). Thus, for some arthropods, there is a stunning correlation between genes encoding bacterial-like DUBs and alternate modes of reproduction.
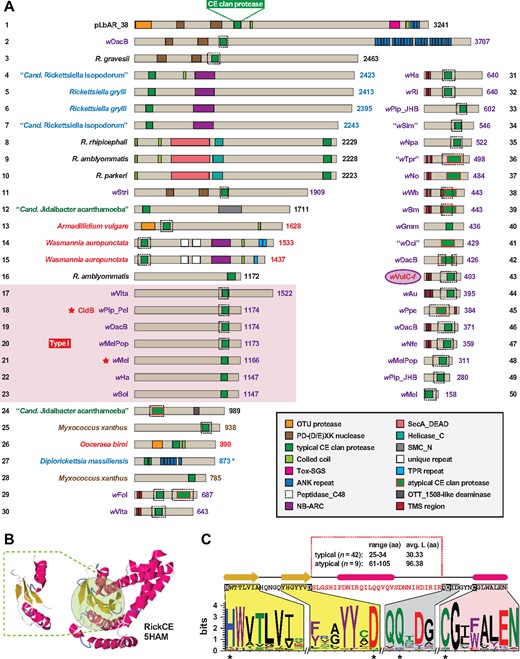
—Cryptic CE clan protease domains are present in putative deubiquitinases across diverse obligate intracellular bacterial species. (A) An assortment of CE clan protease domain-containing proteins (CE-DCPs). The pLbAR_38 CE clan protease domain (coordinates 1,095–1,173), used as the query in Blastp searches against the NCBI nr database, identified CE-DCPs in Wolbachia (purple), Rickettsia (black), Legionellales (blue), other Rickettsiales (dark green), arthropod host (red), and Myxococcus xanthus (brown) genomes, and the f element of A. vulgare (wVulC-f) (Leclercq et al. 2016). Protein domains are described in the gray inset. Dashed boxes indicate Peptidase_C48 (Pfam PF02902) predictions using SMART (Letunic and Bork 2017). Type 1 cytoplasmic incompatibility deubiquitinases (protein nos 17–23) of Wolbachia are grouped within the red box, with stars depicting previously characterized toxins (Beckmann et al. 2017; LePage et al. 2017). Note.—Each subject (numbered 1–50) represents a distinct protein architecture, yet in some cases multiple similar proteins can be found for closely related species and strains. Asterisk for Diplorickettsia massiliensis (protein no. 27) indicates split ORFs (NCBI acc. Nos WP_010597134 and WP_010597133). See supplementary table S1, Supplementary Material online for information pertaining to all sequences. (B) pLbAR_38 CE clan protease domain modeled to RickCE, a CE-DCP of Rickettsia bellii (PDBID: 5HAM) (Pruneda et al. 2016). Modeling done using Phyre2 (Kelley and Sternberg 2009). Green highlighting indicates the RickCE active site, with modeled region of pLbAR_38 within dashed lines. (C) Top, sequence and linear depiction of the predicted structure of pLbAR_38 CE clan protease domain. Highlighting shows the regions encompassing active site residues His and Asp (yellow), Gln (gray) and Cys (magenta). Red sequence indicates length variable region across CE clan protease domains shown in (A). Bottom, sequence logo (Crooks et al. 2004) illustrating active site conservation across the 50 proteins depicted in (A). See figure 2 legend for amino acid coloring scheme.
Notably, the Type I CI-inducing toxins were recovered in this analysis, consistent with prior characterization of DUB activity in the wPip_Pel CidB protein and the essentiality of its active site (Cys) for inducing CI in flies (Beckmann et al. 2017). Aside from CidB, the functions of any of these diverse proteins remain unknown. Although the majority of retrieved CE-DCPs contain a predicted Peptidase_C48 (Pfam PF02902) domain (dashed boxes, fig. 3A), pLbAR_38 does not. However, structural modeling (Kelley and Sternberg 2009) revealed a strong match between pLbAR_38 and the catalytic region of Rickettsia bellii RickCE, a Ulp with an unknown role in rickettsial biology (Pruneda et al. 2016) (fig. 3B). Despite tremendous variability in sequence length and composition across the catalytic regions of the CE-DCPs (fig. 3C), all the CE clan protease domains modeled to RickCE with high confidence (data not shown). Three distinct length variable regions were previously observed across divergent bacterial CE-DCPs (both DUBs and Ulps), and in conjunction with accessory regions outside of the CE clan protease domains, may provide these diverse enzymes with varying substrate specificities (Pruneda et al. 2016). Although modeling of the retrieved CE-DCPs (fig. 3A) indicates additional regions of variability within the CE clan protease catalytic region, the impact of these length variations on host substrate specificity, as well as contribution of accessory domains, remains to be determined.
We identified several other distinct groups of CE-DCPs in Rickettsiales (supplementary figs. S2A–S2D, Supplementary Material online). Like pLbAR_38, CidB, and RickCE, all these additional groups have a highly conserved catalytic region (supplementary fig. S2E, Supplementary Material online). Most notable is the presence of a CE clan protease domain within Rickettsia Sca15 autotransporters, members of the diverse surface cell antigens utilized by rickettsiae for host cell invasion, host actin polymerization and likely many other functions (Blanc et al. 2005; Gillespie et al. 2014). This indicates that different secretion systems mediate delivery of CE-DCPs into host cells: for example, RTX-like T1SS (pLbAR_38), autotransporters (Sca15), and the Rickettsiales vir homolog type IV secretion system (rvh T4SS) (Gillespie et al. 2009); the last was previously hypothesized to secrete CidB and other Wolbachia CI-inducing toxins as well as their cognate antitoxins (Beckmann et al. 2017). As all Rickettsiales genomes encode both the rvh T4SS and a T1SS (not RTX-like) (Gillespie et al. 2014, 2010, 2016), the majority of CE-DCPs are likely substrates of these secretion pathways.
Our analyses of both CE-DCPs and OTU-DCPs indicate that a myriad of diverse DUBs and Ulps exist within the intracellular mobilome. These secreted effectors likely mimic eukaryotic counterparts that target ubiquitin and UBL modifications to regulate processes such as protein turnover, signaling pathways and the DNA damage response (Leznicki and Kulathu 2017; Lin and Machner 2017). Revealing more specific functions of these various DUBs and Ulps, particularly their ability to induce RP in arthropods, will require focused characterization similar to that conducted for Wolbachia type I CI-inducing toxins (Beckmann et al. 2017; LePage et al. 2017).
Cryptic Nuclease Domains Hint at the Origin of RP-Inducing Toxin Architecture
Between the DUB domains of pLbAR_38 we detected two divergent nuclease domains, both belonging to the PD-(D/E)XK superfamily (Knizewski et al. 2007). pLbAR_38-like dual PD-(D/E)XK nuclease domains were subsequently identified in a modest number of bacterial proteins, many of which are large, highly modular, and possess eukaryotic-like domains and repeats (fig. 4A). Notably, the large proteins from intracellular Legionellales (protein nos. 5 and 6 in fig. 4A) contain a TcdA_TcdB_pore domain (Pfam PF02902) characteristic of Clostridium difficile Toxin A and Toxin B pore-forming regions (Barth et al. 2001). These proteins are xenologs to distantly related Gammaproteobacteria (i.e., Xenorhabdus nematophila and Photorhabdus luminescens) toxins, with the “Cand. Rickettsiella isopodorum” protein encoded within a pathogenicity island that is absent from the closely related Rickettsiella grylli (Wang and Chandler 2016). This exemplifies the mobile nature of these dual PD-(D/E)XK nuclease domains and their association with large modular toxins.
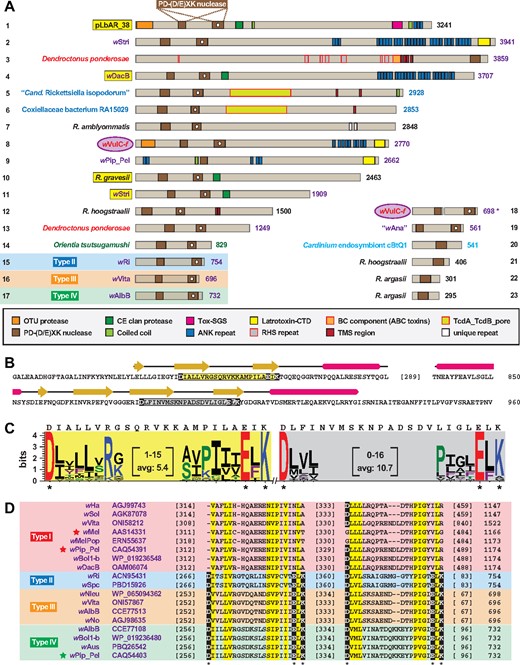
—Proteins with conserved and cryptic PD-(D/E)XK nuclease domains occur in diverse obligate intracellular bacterial species. (A) Assorted PD-(D/E)XK nuclease domain-containing proteins. The pLbAR_38 region encompassing two PD-(D/E)XK nuclease domains (coordinates 462–960), used as the query in Blastp searches against the NCBI nr database, identified PD-(D/E)XK nuclease domain-containing proteins in Wolbachia (purple), arthropod host (red), Legionellales (blue), Rickettsia (black), other Rickettsiales (dark green), and Cardinium endosymbiont cBtQ1 (light blue) genomes, and the f element of A. vulgare (wVulC-f) (Leclercq et al. 2016). Protein domains are described in the gray inset. PD-(D/E)XK nuclease domains containing a white dot indicate less conserved cryptic domains. Note.—Each subject (numbered 1–23) represents a distinct protein architecture, yet in some cases multiple similar proteins can be found for closely related species and strains. Asterisk for wVulC-f (protein no. 18) indicates split ORFs (NCBI acc. nos OJH31453 and OJH31454). See supplementary table S1, Supplementary Material online for information pertaining to all sequences. Yellow highlighting indicates proteins containing both PD-(D/E)XK nuclease and CE clan protease domains. (B) Sequence and predicted structures of pLbAR_38 PD-(D/E)XK nuclease domains. Structure at top depicts modeling of the conserved nuclease domain to Pyrococcus abyssi endoMS (PDBID: 5GKE) (Nakae et al. 2016), and the cryptic nuclease domain to P. abyssi NucS (PDBID: 2VLD) (Ren et al. 2009). Modeling done using Phyre2 (Kelley and Sternberg 2009). Catalytic regions within the endoMS- and NucS-like domains are highlighted yellow and gray, respectively, with active sites Asp, Glu and Lys in black boxes. (C) Sequence logo (Crooks et al. 2004) showing conservation across the catalytic regions of 23 endoMS-like (highlighted yellow) and 17 NucS-like (highlighted gray) domains depicted in (A). pLbAR_38 sequences are shown above logos. See figure 2 legend for amino acid coloring scheme. (D) Alignment of selected Wolbachia Type I–IV CI-inducing toxins. Stars depict previously characterized toxins (Beckmann et al. 2017; LePage et al. 2017). Only the conserved and cryptic PD-(D/E)XK nuclease domains are shown. NCBI protein acc. nos. are provided for each sequence. Yellow highlighting depicts both invariant and semiconserved residues. Active sites Asp, Glu, and Lys are in black boxes.
Strikingly, two PD-(D/E)XK nuclease domain-containing proteins were found in the genome of the mountain pine beetle (Dendroctonus ponderosae). Curiously, the larger D. ponderosae single PD-(D/E)XK nuclease domain-containing protein is probably a xenolog to the pcBtQ1 plasmid-encoded nuclease of the Cardinium endosymbiont cBtQ1 (protein nos. 3 and 20, respectively, in fig. 4A). The D. ponderosae protein is a modular RHS toxin (Busby et al. 2013) and is 57% identical to an RHS toxin that is also encoded on pcBtQ1 (NCBI acc. no. CDG50386) (Gillespie et al. 2015), indicating that the PD-(D/E)XK nuclease domain has become liberated from the large pcBtQ1 RHS toxin. A second D. ponderosae protein with dual PD-(D/E)XK nuclease domains (protein no. 16 in fig. 4A) is a likely derivative of these larger toxins (data not shown). Curiously, female sex ratio biases exist in D. ponderosae populations, with females disproportionately surviving males during overwintering and initiating live pine tree colonization (Lachowsky and Reid 2014). Thus, like the presence of bacterial-like DUBs in the A. vulgare, O. biroi, and W. auropunctata genomes, the PD-(D/E)XK nuclease domains associated with RHS toxins may correlate with alternative modes of host sexual reproduction such as sex ratio distortion. Intriguingly, the D. ponderosae and pcBtQ1 RHS toxins, as well as a few highly similar Wolbachia RHS toxins (e.g., WD0513 within the wMelPop octomom region [Chrostek and Teixeira 2015] and WP1346 of prophage WOPip5 of wPip_Pel [Bordenstein and Bordenstein 2016]) are more similar to widespread arthropod RHS toxins than other bacterial RHS toxins (data not shown), indicating substantial LGT of these toxins between eukaryotes and mobile elements such as plasmids and prophage (Klasson et al. 2009; Woolfit et al. 2009). This is reminiscent of another ANK repeat-containing protein of D. ponderosae that was shown to be xenologous to a Diplorickettsia massiliensis (Legionellales) protein (Bordenstein and Bordenstein 2016).
The second PD-(D/E)XK nuclease domain of pLbAR_38 was more difficult to detect (white dot in in fig. 4A); however, both predicted nuclease domains modeled to nuclease structures with high confidence (fig. 4B). Indeed, the more cryptic nuclease was shown to be less conserved in sequence length and composition across all the assembled PD-(D/E)XK nuclease domain-containing proteins (fig. 4C). This likely accounts for initial reports predicting only the conserved PD-(D/E)XK nuclease domain in Wolbachia CinB (and other Type IV nucleases) (Beckmann et al. 2017) and Type II nucleases (LePage et al. 2017). More recently, a robust comparative analysis by Lindsey et al. predicted two PD-(D/E)XK nuclease domains within all four types of Wolbachia CI-inducing toxins (Lindsey et al. 2018). Our analysis indicates that only Type II–IV toxins contain both the conserved and cryptic domains, with strict conservation of the active site PD-(D/E)XK signature residues in both domains (fig. 4D). This argues against Type I toxins possessing nuclease activity and agrees with the essentiality of CinB active sites for lethality in yeast (Beckmann et al. 2017). However, CidB and other Type I DUBs are conserved enough throughout these domains to superficially resemble Type II, III, and IV nucleases, indicating that Type I CI-inducing toxins probably were once chimeras that contained the dual PD-(D/E)XK nuclease domains (see Beckmann et al.’s supplemental discussion and figure S1; Beckmann et al. 2017).
In addition to pLbAR_38, we identified three other chimeric proteins harboring both the CE clan protease domain and dual PD-(D/E)XK nuclease domains (yellow highlighting in fig. 4A). These chimeras probably represent the ancient and complete form of RP-inducing toxins, serving as platforms for scaffolding of additional eukaryotic-like domains that tailor these toxins to specific host reproductive systems. This diversity of large, highly modular chimeras provides a pool for streamlining and specialization, resulting in 1) toxins retaining only DUB functions, such as CidB and other Type I proteins, 2) toxins retaining only nuclease functions, such as CinB and all other related proteins, and 3) proteins that have retained neither the protease nor nuclease domains, and may or may not function in RP (fig. 1; supplementary fig. S1B, Supplementary Material online). For the first two cases, liberation of the protease and nuclease domains coincides with codivergence of cognate protease and nuclease antitoxin-coding genes (Beckmann et al. 2017; Bonneau et al. 2018; Lindsey et al. 2018). In theory, any number of derivatives of the chimeric platforms can be present in an organism, equipping it with an arsenal to highjack the reproductive system of the host (or multiple hosts). This is typified by the multiple combinations of CI-inducing TA modules within different strains of parasitic wPip (LePage et al. 2017; Lindsey et al. 2018), which very recently have been shown to correlate with cross-compatibility phenotypes in Culex pipiens (Bonneau et al. 2018). We find this to be the most parsimonious explanation for the origin of these toxin architectures, provided that both Rickettsia and Wolbachia chimeras exist, and that a phylogeny estimated from a large region outside of the protease/nuclease domains indicates tremendous divergence from their common “scaffolding” domain (supplementary fig. S1B, Supplementary Material online).
Antitoxins to RP-Inducing Toxins Span the Host-Parasite Divide
For Wolbachia parasites, nearly all the identified CI-inducing toxins occur in modules with their cognate antitoxins. Disregarding a likely spurious ORF (pLbAR_37, NCBI locus tag KHO02169) (Gillespie et al. 2015), a gene upstream of pLbAR_38 was investigated as a possible antitoxin to pLbAR_38. Indeed, the encoded protein pLbAR_36 was found to match wPip_Pel CidA (22% ID, 55% query coverage) and CinA (21% ID, 41% query coverage) antitoxins in Blastp searches. Similarity was also found to most other Wolbachia type I–IV antitoxins, and unexpectedly, several other rickettsial proteins. Alignment of selected sequences revealed six regions of strict conservation that strongly support these proteins as antitoxin analogs (fig. 5A). In several cases, putative antitoxins from other non-Wolbachia species were encoded by genes adjacent to toxin-encoding genes (fig. 5B). For instance, a putative nuclease TA module was found in all genomes of Orientia tsutsugamushi, the agent of Scrub Typhus that induces parthenogenesis in Leptotrombidium mites (Roberts et al. 1977; Takahashi et al. 1997). Thus, this is the first instance of a potential RP-inducing TA module carried by a human pathogen.
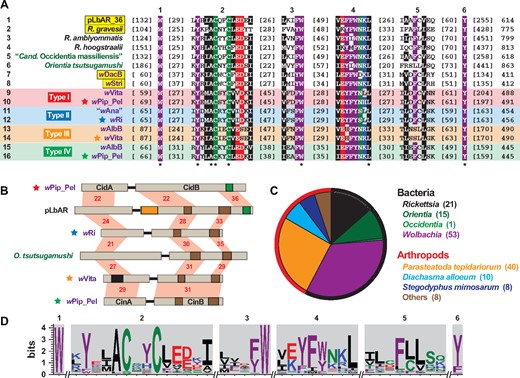
—Xenologs to Wolbachia CidA and CinA antitoxins occur in other obligate intracellular species and some arthropod genomes. Alignments generated using MUSCLE, default parameters (Edgar 2004). See figure 2 legend for amino acid coloring scheme, and supplementary table S1, Supplementary Material online for information pertaining to all sequences. (A) Alignment of CidA, CinA and related proteins from selected Rickettsiales species. Top, six conserved regions. Bottom, asterisks indicate invariant residues. Taxon color scheme as follows: black, Rickettsia; dark green, other Rickettsiales; purple, Wolbachia. Wolbachia Type I–IV antitoxins are labeled accordingly. Yellow highlighting indicates species that contain associated toxins with both CE clan protease and PD-(D/E)XK nuclease domains. (B) Selected toxin/antitoxin operon structures. Information as follows: CidA/B of wPip_Pel (CAQ54390, CAQ54391); pLbAR_36/38 (WP_039595309, WP_081996388); type II TA proteins of wRi (ACN95432, ACN95431); OTBS_1545/1546 of Orientia tsutsugamushi (CAM80637, CAM80639); type III TA proteins of wVita (ONI57866, ONI57867); CinA/B of wPip_Pel (CAQ54402, CAQ54403). Note.—Only coordinates 1–1,200 are shown for pLbAR_38, and a likely spurious ORF encoding 84 aa (KHO02169) occurs between pLbAR_36 and pLbAR_38 (Gillespie et al. 2015). Protein domains as follows: green, CE clan protease; brown, PD-(D/E)XK nuclease; orange, OTU cysteine protease; black, SMART SCOP domain: Wolinella succinogenes cytochrome c nitrite reductase (PDBID: 1fs7) (Einsle et al. 2000). Red shading and numbers indicate % identity across pairwise alignments. (C) Taxonomic breakdown of all antitoxins identified using pLbAR_36 as a query in Blastp searches against the NCBI nr protein database. Outer circle demarcates bacterial (black, n=90) and arthropod (red, n=66) non-redundant subjects. (D) Sequence logo (Crooks et al. 2004) illustrating conservation of the six regions (depicted in A) across 156 antitoxins from (C).
Cognate antitoxins were found for the three other chimeric proteins harboring both the CE clan protease domain and dual PD-(D/E)XK nuclease domains. Like pLbAR_38/36, the chimeric toxins and associated antitoxins of R. gravesii, wDacB, and wStri are within modules. Thus, it appears TA modules predated the fission of chimeras that subsequently resulted in diversification of TA modules comprised of either CE clan protease domain-containing toxins or dual PD-(D/E)XK nuclease domain-containing toxins. This raises a conundrum when considering the strict codivergence of Wolbachia TA modules and the lack of CidA-CinB and CinA-CidB interactions in pull-down assays (Beckmann et al. 2017). Resolving this mystery will require characterizing how a single antitoxin interacts with a dual toxin domain-containing protein.
In total, 90 distinct antitoxins were found in bacteria, all present in rickettsial genomes (fig. 5C). Thus, the RP-inducing toxins we identified from non-rickettsial species (figs. 3A and 4A) may not function in a “modification-rescue” mechanism, or even in RP at all. Although it is possible the full complement of antitoxins was not unearthed by our analyses, refined searches based on the conserved residues of all assembled antitoxins (fig. 5D) did not yield additional proteins (data not shown). Because CI is currently the only form of RP characterized for Wolbachia TA modules, other forms of RP, such as male-killing, parthenogenesis, and feminization, may require only a toxin. This could explain why both protease and nuclease toxins, but not antitoxins, were found in four genomes of arthropod species capable of sex ratio distortion. It is also highly likely that other genetic mechanisms exist for CI as well as other forms of RP, a notion supported by the lack of perfect correlation between presence/absence of Wolbachia TA modules and particular forms of RP or mutualism (Lindsey et al. 2018).
Conversely, some of the rickettsial antitoxins are found in genomes that lack identifiable RP-inducing toxins. Again, it is possible we did not detect toxins within these genomes, or that different types of toxins work in concert with these antitoxins. The lack of TA module structures for many of these orphan antitoxins does not necessarily preclude their involvement in a “modification-rescue” mechanism. However, evidence for codivergence and coexpression of toxin and antitoxin genes within Wolbachia TA modules (Beckmann et al. 2017; Bonneau et al. 2018; Lindsey et al. 2018) indicates the tendency for associated toxins and antitoxins to be coregulated in operons. Alternatively, these orphan antitoxins may be remnants of TA module degradation, or may even have diversified in function.
Remarkably, we also identified 66 distinct antitoxins in several arthropod genomes (fig. 5C). In most cases, more than one antitoxin (or isoforms) was found, with a staggering 40 distinct antitoxins present in the American house spider (Parasteatoda tepidariorum) genome. These antitoxin genes do not appear to be contaminants, and most occur on scaffolds with numerous eukaryotic-like genes (supplementary table S1B, Supplementary Material online). None of these arthropod genomes harboring antitoxins were found to contain associated toxins. The function of these antitoxins is not clear, though a role in RP suppression (i.e., a host-mediated counterbalance to sex ratio distortion; Hornett et al. 2014; Majerus and Majerus 2010b) is an appealing concept, especially given the recent finding that CidA-like antitoxins alone can rescue wMel-induced CI in D. melanogaster (Shropshire et al. 2018). Specifically, these antitoxins may impart immunity to toxins secreted by intracellular parasites; however, such an immune arsenal would be costly to carry, plus highly impractical given the rapid divergence of TA modules. Alternatively, like arthropod-encoded orphan toxins, these arthropod-encoded antitoxins may have diversified in function.
Wolbachia do not Walk Alone: CI-Like TA Modules Abound in the Bacterial Mobilome
Although previously examined for their roles in CI induction, our analysis of the Wolbachia TA modules indicates that they 1) are part of a large family of highly mosaic genes that are dispersed across a limited number of intracellular bacteria (and in some cases their host arthropod genomes), and 2) likely comprise only a subset of a large pool of mosaic genes with roles in other modes of RP, namely parthenogenesis, feminization and male killing. The latter is supported by the presence of pLbAR_38/36 (R. felis str. LSU-Lb) and nuclease TA modules (O. tsutsugamushi) in species known to strongly correlate with parthenogenesis in their respective hosts. We also found multiple CI-like TA module components in the genome of the Cardinium endosymbiont of the sweet potato whitefly Bemisia tabaci (hereafter cBtQ1) (table 1). This was surprising, as RP phenotypes have not previously been observed in B. tabaci despite other Cardinium strains being known to induce feminization, CI or parthenogenesis in various arthropod hosts (Hunter et al. 2003; Gotoh et al. 2007; White et al. 2011). The components we found in cBtQ1 may be decaying, consistent with elevated numbers of transposable elements, recently inactivated genes and chromosomal rearrangements in the cBtQ1 genome (Santos-Garcia et al. 2014). Conversely, we did not find evidence for CI-like TA modules in the genome of the CI-inducing Cardinium strain cEper1, an endoparasite of Encarsia wasps (Penz et al. 2012), although a recent report illuminated unrelated candidate CI-inducing proteins in this species (Mann et al. 2017). Importantly, some of the cBtQ1 CI-like TA module components are encoded on a plasmid (pcBtQ1), indicating that these elements are a part of the Cardinium mobilome and should not be overlooked as factors contributing to CI and other RP phenotypes in arthropod populations harboring these endoparasites. Whether different host cytological phenotypes caused by CI-inducing Cardinium and Wolbachia strains are underpinned by similar or unrelated molecular factors remains to be determined (Gebiola et al. 2017).
Obligate Intracellular Bacteria (Excluding Wolbachia Species) Containing Multiple Factors Potentially Associated with Host Reproductive Parasitism
| Species | Hosta | DUBb | NUCc | Otherd | ATe | RPf |
|---|---|---|---|---|---|---|
| Rickettsia felis str. LSU-Lb | Liposcelis bostrychophilaB | OTU, CE, CE* | 2* | Yes | Yes | PI |
| Orientia tsutsugamushi | Leptotrombidium mites | CE* | 2 | Yes | Yes* | PI |
| Cardinium endosymbiont cBtQ1 | Bemisia tabaciW | CA | 1 | Yes | No | ? |
| “Cand. Rickettsiella isopodorum” | Terrestrial isopods | CE (2) | 2 | No | No | – |
| Diplorickettsia massiliensis | Ixodes ricinusH | CE | – | Yes | No | – |
| Rickettsia gravesii | Amblyomma limbatumS | CE, CE* | 2* | Yes | Yes | – |
| Rickettsia amblyommatis | Amblyomma ticksS | CE, CE* | 2 | No | Yes* | – |
| Rickettsia argasii | Argas dewaeH | CE* | 1 | Yes | Yes* | – |
| Rickettsia hoogstraalii | Carios capensisS | CE* | 3 | Yes | Yes* | – |
| Rickettsia bellii | Many tick speciesH,S | CE*, CE^ | – | Yes | No | – |
| Occidentia massiliensis | Ornithodoros sonraiS | – | – | Yes | Yes | – |
| Species | Hosta | DUBb | NUCc | Otherd | ATe | RPf |
|---|---|---|---|---|---|---|
| Rickettsia felis str. LSU-Lb | Liposcelis bostrychophilaB | OTU, CE, CE* | 2* | Yes | Yes | PI |
| Orientia tsutsugamushi | Leptotrombidium mites | CE* | 2 | Yes | Yes* | PI |
| Cardinium endosymbiont cBtQ1 | Bemisia tabaciW | CA | 1 | Yes | No | ? |
| “Cand. Rickettsiella isopodorum” | Terrestrial isopods | CE (2) | 2 | No | No | – |
| Diplorickettsia massiliensis | Ixodes ricinusH | CE | – | Yes | No | – |
| Rickettsia gravesii | Amblyomma limbatumS | CE, CE* | 2* | Yes | Yes | – |
| Rickettsia amblyommatis | Amblyomma ticksS | CE, CE* | 2 | No | Yes* | – |
| Rickettsia argasii | Argas dewaeH | CE* | 1 | Yes | Yes* | – |
| Rickettsia hoogstraalii | Carios capensisS | CE* | 3 | Yes | Yes* | – |
| Rickettsia bellii | Many tick speciesH,S | CE*, CE^ | – | Yes | No | – |
| Occidentia massiliensis | Ornithodoros sonraiS | – | – | Yes | Yes | – |
B, booklouse; W, silverleaf whitefly; H, hard ticks (Ixodidae); S, soft ticks (Argasidae).
Deubiquitinases, or ubiquitin-like proteases (see figs. 2 and 3): OTU, ovarian tumor-like cysteine protease; CE, clan CE protease (similar to CidB and other Type I CI-inducing toxins); CE*, clan CE protease (RickCE-like); CE^, clan CE protease (Sca15 passenger domain); CA, clan CA protease (peptidase C19R subfamily, also found in Cardinium endosymbiont cEper1 of Encarsia pergandiella, GenBank acc. no. CCM09810).
PD-(D/E)XK nuclease (see fig. 4): similar to nucleases of CinB and Type II–IV CI-inducing toxins. Asterisks indicate domains occurring within the same protein as DUB domains.
Large region with unknown function (coordinates 1174-2813 of pLbAR_38); see fig. 1 and supplementary fig. S1, Supplementary Material online.
In silico identification of pLbAR_36/CidA/CinA antitoxins (see fig. 5). Asterisks depict multiple (some possibly degrading) genes per genome.
Predicted RP phenotype: PI, parthenogenesis induction; ?, other Cardinium strains induce RP phenotypes, but not strain cBtQ1.
Obligate Intracellular Bacteria (Excluding Wolbachia Species) Containing Multiple Factors Potentially Associated with Host Reproductive Parasitism
| Species | Hosta | DUBb | NUCc | Otherd | ATe | RPf |
|---|---|---|---|---|---|---|
| Rickettsia felis str. LSU-Lb | Liposcelis bostrychophilaB | OTU, CE, CE* | 2* | Yes | Yes | PI |
| Orientia tsutsugamushi | Leptotrombidium mites | CE* | 2 | Yes | Yes* | PI |
| Cardinium endosymbiont cBtQ1 | Bemisia tabaciW | CA | 1 | Yes | No | ? |
| “Cand. Rickettsiella isopodorum” | Terrestrial isopods | CE (2) | 2 | No | No | – |
| Diplorickettsia massiliensis | Ixodes ricinusH | CE | – | Yes | No | – |
| Rickettsia gravesii | Amblyomma limbatumS | CE, CE* | 2* | Yes | Yes | – |
| Rickettsia amblyommatis | Amblyomma ticksS | CE, CE* | 2 | No | Yes* | – |
| Rickettsia argasii | Argas dewaeH | CE* | 1 | Yes | Yes* | – |
| Rickettsia hoogstraalii | Carios capensisS | CE* | 3 | Yes | Yes* | – |
| Rickettsia bellii | Many tick speciesH,S | CE*, CE^ | – | Yes | No | – |
| Occidentia massiliensis | Ornithodoros sonraiS | – | – | Yes | Yes | – |
| Species | Hosta | DUBb | NUCc | Otherd | ATe | RPf |
|---|---|---|---|---|---|---|
| Rickettsia felis str. LSU-Lb | Liposcelis bostrychophilaB | OTU, CE, CE* | 2* | Yes | Yes | PI |
| Orientia tsutsugamushi | Leptotrombidium mites | CE* | 2 | Yes | Yes* | PI |
| Cardinium endosymbiont cBtQ1 | Bemisia tabaciW | CA | 1 | Yes | No | ? |
| “Cand. Rickettsiella isopodorum” | Terrestrial isopods | CE (2) | 2 | No | No | – |
| Diplorickettsia massiliensis | Ixodes ricinusH | CE | – | Yes | No | – |
| Rickettsia gravesii | Amblyomma limbatumS | CE, CE* | 2* | Yes | Yes | – |
| Rickettsia amblyommatis | Amblyomma ticksS | CE, CE* | 2 | No | Yes* | – |
| Rickettsia argasii | Argas dewaeH | CE* | 1 | Yes | Yes* | – |
| Rickettsia hoogstraalii | Carios capensisS | CE* | 3 | Yes | Yes* | – |
| Rickettsia bellii | Many tick speciesH,S | CE*, CE^ | – | Yes | No | – |
| Occidentia massiliensis | Ornithodoros sonraiS | – | – | Yes | Yes | – |
B, booklouse; W, silverleaf whitefly; H, hard ticks (Ixodidae); S, soft ticks (Argasidae).
Deubiquitinases, or ubiquitin-like proteases (see figs. 2 and 3): OTU, ovarian tumor-like cysteine protease; CE, clan CE protease (similar to CidB and other Type I CI-inducing toxins); CE*, clan CE protease (RickCE-like); CE^, clan CE protease (Sca15 passenger domain); CA, clan CA protease (peptidase C19R subfamily, also found in Cardinium endosymbiont cEper1 of Encarsia pergandiella, GenBank acc. no. CCM09810).
PD-(D/E)XK nuclease (see fig. 4): similar to nucleases of CinB and Type II–IV CI-inducing toxins. Asterisks indicate domains occurring within the same protein as DUB domains.
Large region with unknown function (coordinates 1174-2813 of pLbAR_38); see fig. 1 and supplementary fig. S1, Supplementary Material online.
In silico identification of pLbAR_36/CidA/CinA antitoxins (see fig. 5). Asterisks depict multiple (some possibly degrading) genes per genome.
Predicted RP phenotype: PI, parthenogenesis induction; ?, other Cardinium strains induce RP phenotypes, but not strain cBtQ1.
We identified multiple CI-like TA module components in eight other bacterial species, all of which are obligate intracellular parasites of various tick hosts (table 1). To our knowledge there are no reports of RP phenotypes induced by any of these species. However, unlike the genera Diplorickettsia, Rickettsiella, and Occidentia, RP has been documented for several species of Rickettsia, all of which lack published genome sequences. Some of these species that induce male killing in buprestid and ladybird beetles (Werren et al. 1994; Lawson et al. 2001; von der Schulenburg et al. 2001; Majerus and Majerus 2010a) and parthenogenesis in eulophid wasps (Giorgini et al. 2010) are closely related to R. bellii, which carries multiple DUBs and a protein containing the pLbAR_38 central region (table 1; supplementary fig. S1B, Supplementary Material online). Other species inducing parthenogenesis in eulophid wasps or trogiid booklice are found either in Torix Group rickettsiae (Perotti et al. 2006; Gualtieri et al. 2017) or Transitional Group rickettsiae (Hagimori et al. 2006; Perotti et al. 2006; Nugnes et al. 2015), with species in the latter group closely related to R. felis str. LSU-Lb (Gillespie et al. 2015). Sequencing the genomes of these species will undoubtedly yield factors involved in RP, some of which may be similar to the protein architectures presented herein. For instance, we identified an OTU-DCP (1033 aa) within the unpublished genome of the male-killer Rickettsia parasite of the ladybird beetle Adalia bipunctata (Murray et al. 2016) (data not shown). This OTU-DCP is a strong candidate for inducing male-killing given that 1) its OTU domain is 30% identical to the pLbAR_38 OTU domain, and 2) its top Blastp hits are to the Apaf-1 domain-containing Wolbachia OTU-DCPs (protein nos. 6–9 in fig. 2A), except that the Apaf-1 domain is replaced with five tetratricopeptide repeats.
While our analyses utilized the pLbAR_38/36 domains to detect candidate RP-inducing factors in other microbes, it should be realized that reciprocal Blastp searches, as well as more sophisticated approaches guided by conserved toxin and antitoxin motifs, are likely to unearth an even greater diversity of these elements. For example, using the abovementioned OTU-DCP of the male-killer Rickettsia parasite of A. bipunctata in a Blastp search, we detected an OTU-DCP (WP_105629072: 1065 aa with a OTU domain and five ANK repeats) in the genome of Spiroplasma poulsonii, an endoparasite that induces male-killing in several Drosophila species (Harumoto et al. 2014; Masson et al. 2018). Remarkably, this S. poulsonii OTU-DCP has very recently been identified as the sought-after Spiroplasma-derived factor responsible for inducing the male-killing phenotype (Harumoto and Lemaitre 2018). Candidate RP-inducing factors from other reproductive parasites (e.g., species of Arsenophonus, certain microsporidian protists, and possibly viruses; Kageyama et al. 2012, 2017; Hurst and Frost 2015) not detected using our pLbAR_38/36-directed approach may come to light using similar approaches. Also, we predict that some Wolbachia OTU-DCPs, particularly the Apaf-1 domain-containing toxins (protein nos 6–9 in fig. 2A), will be implicated in male-killing phenotypes that seem superficially divergent from that induced by S. poulsonii (Harumoto et al. 2018).
The Origin of RP-Inducing Toxins and Antitoxins
Imperfect TA module compositions, weak correlation of such modules with specific RP phenotypes, and the presence of toxins and antitoxins (though never both) in arthropod genomes all collectively attest to the mobile nature and rapid evolutionary rates of these genes and possible neofunctionalization of their products. While little can be inferred from antitoxin architectures, the large size and domain composition of toxins (particularly the presence of furin protease cleavage sites) support eukaryotic origins (Beckmann and Fallon 2013; Bordenstein and Bordenstein 2016). It is interesting to speculate whether these toxins and antitoxins first arose in arthropods to suppress sexual reproduction by facilitating rapid distortions to sex ratios; that is, a stress response-mediated survival tactic. However, the paucity of contemporary arthropod genomes encoding such toxins and antitoxins casts doubt on this scenario. Alternatively, originating from numerous divergent eukaryotic hosts (e.g., amoebae, ciliates, primitive arthropods), DUB and nuclease domains with probable functions in mitosis likely recombined into large bacterial proteins, which served as scaffolds for construction of chimeric toxins (fig. 6, step 1). Origins for antitoxins are less clear, though one possibility is that a preexisting bacterial gene encoding a protein that binds regions of the toxin scaffold (e.g., the pLbAR_38 central region shown in supplementary fig. S1, Supplementary Material online) gave rise to modern antitoxins. Otherwise, should eukaryotic-like domains (i.e., catalase-rel and sterile-like transcription factor domains recently predicted within some Wolbachia antitoxins; Lindsey et al. 2018) be functionally characterized within antitoxins, then eukaryotic origins for these genes are probable.
![—Origins of genes underpinning reproductive parasitism. This hypothetical scenario explains how prokaryotes may have acquired eukaryotic genes that target host mitosis, and how bacteriophage mediated the return of “bacterial-tailored” reproductive parasitism (RP) genes to arthropod genomes. (1) Via transkingdom lateral gene transfer (LGT) (gray arrows), ancestral intracellular bacteria acquired eukaryotic genes encoding DNA-targeting deubiquitinase (OTU and clan CE proteases) and nuclease (PD-(D/E)XK) domains with probable functions in mitosis. These eukaryotic domains were recombined into large mosaic proteins (chimeric toxins). Another gene encoding a protein (antitoxin) able to interact with chimeric toxins was either i) acquired from eukaryotes, or ii) derived from a preexisting bacterial gene encoding a protein that recognizes regions of the toxin scaffold (e.g., the pLbAR_38 central region shown in supplementary fig. S1, Supplementary Material online). Selection favored the arrangement of genes encoding chimeric toxins and associated antitoxins into operons (shown here as plasmid-borne), establishing the ancestral TA modules that still circulate in the modern mobilome (e.g., R. felis str. LSU-Lb, R. gravesii, wDacB, and wStri). Thus, these TA modules are the missing links in the evolution of TA operons that encode toxins with specific functions in CI and other modes of RP (e.g., the male-killer [MK] toxin of Spiroplasma poulsonii) (see Inset). (2) Either by vertical descent (black arrows) or bacterial-bacterial LGT (gray arrows with internal red-dashed arrows), modern intracellular bacterial species may possess one or more complete TA modules, or just toxins or antitoxins (summarized in this study). Such species-specific TA repertoires indicate specialization within host contexts. (3) The rise of CI TA modules in WO prophage (and probably other prophage) of parasitic Wolbachia strains likely occurred by LGT from Rickettsiales Amplified Genetic Elements (RAGEs), integrative conjugative elements known heretofore from species of Rickettsiaceae (see text for details). (4) Phage now mediate the exchange of CI TA modules (e.g., Cid, Cin, and others) between contemporary Wolbachia strains (thick black dashed arrows). (5) Phage may have also facilitated the return of toxin- and antitoxin-encoding genes into arthropods that harbor Wolbachia strains, though whole or partial Wolbachia genome transfers may also account for the LGTs. Additional LGTs between non-Wolbachia species and hosts (e.g., species of Dendroctonus and Cardinium) would be facilitated by non-phage mechanisms or phage unique to these systems.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/gbe/10/9/10.1093_gbe_evy159/2/m_evy159f6.jpeg?Expires=1750203399&Signature=tS9ZKa3LPgHKZZiLKFSWEo5~5C~AGJazSTg1MLfyMY5bnhxtV3Chxr80C8vc5aAx8oNksBEC0bSpItA99IlX24X5tbOB8t5Aj3F3NWwk81n1Q7~MXA2XnRLnLP-I6PTT0EUGIDL3ooUy-UOs32AoL1vXRbmbT2MZDwiOme5myCujTEUlhYJhfpRyPccrKR8jxQ6stWqBT3z4HxIyAHZ4QEBv1k231P2pyqM3fL5DSFtlkzBXqKmWXFwCtejnR5P1ExJRcQRU3yp7lFZ~Q~XmdJAt02NiBc86ejezYg8H~Q0oyI7w6ekQJo7DyyiEkqWwxI30gg-dGZn-BQ6n~YroWw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
—Origins of genes underpinning reproductive parasitism. This hypothetical scenario explains how prokaryotes may have acquired eukaryotic genes that target host mitosis, and how bacteriophage mediated the return of “bacterial-tailored” reproductive parasitism (RP) genes to arthropod genomes. (1) Via transkingdom lateral gene transfer (LGT) (gray arrows), ancestral intracellular bacteria acquired eukaryotic genes encoding DNA-targeting deubiquitinase (OTU and clan CE proteases) and nuclease (PD-(D/E)XK) domains with probable functions in mitosis. These eukaryotic domains were recombined into large mosaic proteins (chimeric toxins). Another gene encoding a protein (antitoxin) able to interact with chimeric toxins was either i) acquired from eukaryotes, or ii) derived from a preexisting bacterial gene encoding a protein that recognizes regions of the toxin scaffold (e.g., the pLbAR_38 central region shown in supplementary fig. S1, Supplementary Material online). Selection favored the arrangement of genes encoding chimeric toxins and associated antitoxins into operons (shown here as plasmid-borne), establishing the ancestral TA modules that still circulate in the modern mobilome (e.g., R. felis str. LSU-Lb, R. gravesii, wDacB, and wStri). Thus, these TA modules are the missing links in the evolution of TA operons that encode toxins with specific functions in CI and other modes of RP (e.g., the male-killer [MK] toxin of Spiroplasma poulsonii) (see Inset). (2) Either by vertical descent (black arrows) or bacterial-bacterial LGT (gray arrows with internal red-dashed arrows), modern intracellular bacterial species may possess one or more complete TA modules, or just toxins or antitoxins (summarized in this study). Such species-specific TA repertoires indicate specialization within host contexts. (3) The rise of CI TA modules in WO prophage (and probably other prophage) of parasitic Wolbachia strains likely occurred by LGT from Rickettsiales Amplified Genetic Elements (RAGEs), integrative conjugative elements known heretofore from species of Rickettsiaceae (see text for details). (4) Phage now mediate the exchange of CI TA modules (e.g., Cid, Cin, and others) between contemporary Wolbachia strains (thick black dashed arrows). (5) Phage may have also facilitated the return of toxin- and antitoxin-encoding genes into arthropods that harbor Wolbachia strains, though whole or partial Wolbachia genome transfers may also account for the LGTs. Additional LGTs between non-Wolbachia species and hosts (e.g., species of Dendroctonus and Cardinium) would be facilitated by non-phage mechanisms or phage unique to these systems.
The arrangement of genes encoding chimeric toxins and associated antitoxins into operons established the ancestral TA modules that are best depicted by the chimeric systems still circulating in the modern mobilome (e.g., R. felis str. LSU-Lb, R. gravesii, wDacB, and wStri). These “missing links” were disseminated via vertical descent, yet more prominently LGT, and provide a clearer understanding of how streamlined TA modules evolved within host contexts to induce specific modes of RP (fig. 6, step 2 and inset). They further indicate that phage (particularly WO-phage) are not required for induction of RP, despite a proclivity for TA modules to occur within Wolbachia prophage EAMs (LePage et al. 2017). This is exemplified by the wRec parasite, which lacks functional WO phage yet possesses TA modules within remnant EAMs (Metcalf, Jo, et al. 2014). Furthermore, prior phylogenomics analyses have identified LGTs between Rickettsia and Wolbachia species, including Rickettsia plasmids and WO prophage (Gillespie et al. 2012, 2015; Baldridge et al. 2016). Thus, it is likely that WO prophage originally acquired RP-inducing TA modules from larger chimeric toxins within the rickettsial mobilome (fig. 6, step 3). This is supported by the wDacB and wStri genomes containing both chimeric TA modules and WO prophage-encoded CI-like TA modules, as well as the overall similarity across toxin arsenals from several Wolbachia (wDacB, wFol, and wStri) and Rickettsia (R. felis str. LSU-Lb, R. hoogstraalii, R. gravesii, R. amblyommatis) genomes reported here. Thus, for species/strains of Wolbachia, WO prophage genomes continually acquire variant TA modules from various sources in the mobilome (which includes WO phage), with mobilization of TA modules driven by functional WO phage (fig. 6, step 4).
The previous identification of eukaryotic genes flanking WO prophage genomes (Klasson et al. 2009; Woolfit et al. 2009; Duplouy et al. 2013), plus subsequent demonstration that phage WO can transfer such genes (Bordenstein and Bordenstein 2016), strongly indicates that phage receive eukaryotic genes and can disseminate them to other bacteria. Given the high similarity across toxins encoded by WO prophage and those we identified in the O. biroi, A. vulgare, and W. auropunctata genomes, it is likely that phage also mediate the transfer of TA module components to host arthropod genomes (fig. 6, step 5). However, such LGTs could arise from whole or partial Wolbachia genome transfers to host arthropod chromosomes, a seemingly widespread phenomenon (Hotopp et al. 2007; Dunning Hotopp 2011). Lateral gene transfer of RP-inducing TA modules from other bacteria to hosts is also evident given the similarity and genomic synteny between D. ponderosae and Cardinium plasmid-encoded toxins and flanking genes, as well as multiple origins of arthropod-encoded antitoxins based on best bidirectional Blastp scores (data not shown). Overall, the acquirement of bacterial toxins and antitoxins stands to benefit eukaryotes, either by incorporation into reproductive strategies or by imparting a defense against reproductive parasites. The latter is in line with other transkingdom transfers of bacterial genes to eukaryotes that augment the eukaryotic immune response to parasites and pathogens (Metcalf, Funkhouser-Jones, et al. 2014; Chou et al. 2015).
Conclusion
Although we continue to investigate the factors that turned a purported human pathogen, R. felis, into an obligate reproductive parasite of a non-hematophagous arthropod, our observations herein provide strong evidence linking pLbAR-encoded proteins and Wolbachia CI-inducing TA modules. What began as a one-to-one comparison escalated into 1) the identification of a broad range of highly modular toxins across diverse intracellular bacteria, 2) widespread occurrence of antitoxins that sometimes associate with these toxins, and 3) the presence in certain arthropod genomes of either toxins or antitoxins (but never both). Toxin–antitoxin self-interactions, coupled with unknown host targets, fuel incredible hyper-evolution that underpins RP, as typified by CI TA module amplification and subsequent recombination (Bonneau et al. 2018). Still, our detailed analyses of cryptic DUB and nuclease active sites within often exceedingly large toxins, as well as an unknown antitoxin architecture, provide strong evidence for a wide dispersal of RP-inducing elements across diverse intracellular bacteria and certain arthropod hosts. Identification of the pLbAR_38 central domain, which serves as a scaffolding platform for numerous eukaryotic-like domains, indicates a recapitulating theme for toxin architecture that persists evolutionarily and drives innovative strategies for commandeering host reproduction.
Through our analyses, we have identified numerous genes worthy of exploration for their roles in RP. Coupled with additional whole genome sequencing of known reproductive parasites (e.g., Rickettsia genome sequences are heavily skewed to human pathogens; Driscoll et al. 2013, 2017), focused bioinformatics efforts will continue to identify putative RP-inducing factors, potentially even unearthing RP phenotypes in some microbes for which they have yet to be observed. These collective efforts stand to immensely increase our understanding of the molecular factors triggering RP. Importantly, utilizing microbes other than Wolbachia strains for blocking disease transmission in non-dipteran, medically relevant arthropods (e.g., ticks, mites, fleas, lice) greatly expands the landscape for controlling vector transmission of important human pathogens.
Funding
This work was supported with funds from the National Institute of Health/National Institute of Allergy and Infectious Diseases grants (R01AI017828 and R01AI126853 to A.F.A., R21AI26108 to J.J.G. and M.S.R., and AI22672 to K.R.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We are grateful to John Beckmann and an anonymous reviewer for criticism that greatly improved the manuscript. We would like to thank Mark Guillotte and Stephanie Lehman for insightful discussions, and Magda Beier-Sexton for administrative support.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}