





Abstract
Mitochondria control cellular metabolism, serve as hubs for signaling and organelle communication, and are important for the health and survival of cells. VPS13D encodes a cytoplasmic lipid transfer protein that regulates mitochondrial morphology, mitochondria and endoplasmic reticulum contact, and quality control of mitochondria. VPS13D mutations have been reported in patients displaying ataxic and spastic gait disorders with variable age of onset. Here, we used CRISPR/Cas9 gene editing to create VPS13D-related spinocerebellar ataxia-4 missense mutations and C-terminal deletion in VPS13D's ortholog vps-13D in Caenorhabditis elegans. Consistent with SCAR4 patient movement disorders and mitochondrial dysfunction, vps-13D mutant worms exhibit locomotion defects and abnormal mitochondrial morphology. Importantly, animals with a vps-13D deletion or a N3017I missense mutation exhibited an increase in mitochondrial unfolded protein response. The cellular and behavioral changes caused by VPS13D mutations in C. elegans advance the development of animal models that are needed to study SCAR4 pathogenesis.
Introduction
Mitochondria are dynamic organelles that play an important role in cellular bioenergetics and viability. The mitochondrial network undergoes continuous fusion and fission to maintain its health. In addition, mitochondria form dynamic contacts with other organelles, especially the endoplasmic reticulum, to participate in various biological processes. Damaged mitochondria are selectively cleared by autophagy, and alterations in mitochondrial removal by autophagy have been associated with neurological disorders, including Alzheimer's disease, Parkinson's disease, Huntington's disease, and amyotrophic lateral sclerosis (Collier et al. 2023).
Vacuolar protein sorting-associated protein 13D, encoded by the VPS13D gene, is a key regulator of mitochondrial processes, including mitochondrial clearance, mitochondrial morphology, and interorganelle contacts (Anding et al. 2018; Guillén-Samander et al. 2021; Shen, Fortier, Zhao, et al. 2021). VPS13D is a member of the conserved VPS13 protein family, which facilitates membrane contacts and transports lipids between organelles (Leonzino et al. 2021). Unlike the other 3 human VPS13 proteins (VPS13A–C), VPS13D possesses a unique ubiquitin-associated (UBA) domain that has been shown to interact with K63 ubiquitin chains and participate in mitochondrial health (Anding et al. 2018; Leonzino et al. 2021). Mutations in the VPS13A–C genes have been linked to specific neurological disorders, including chorea-acanthocytosis, Cohen’s syndrome, and early-onset Parkinson's disease. Importantly, biallelic pathogenic variants in the VPS13D gene have also been associated with spastic ataxia and spastic paraplegia (Gauthier et al. 2018; Seong et al. 2018).
VPS13D movement disorder, also referred to as autosomal recessive spinocerebellar ataxia-4 (SCAR4; Swartz et al. 2002; Seong et al. 2018), manifests as a hyperkinetic movement disorder characterized by dystonia, chorea, and/or ataxia. This disorder is often accompanied with varying degrees of developmental delay and cognitive impairment (Meijer 2019). In the majority of the reported 42 cases, individuals have a combination of a loss-of-function variant and a seemingly milder variant, which can be either a missense mutation or a milder splicing variant (Gauthier et al. 2018; Seong et al. 2018; Koh et al. 2020; Lee et al. 2020; Petry-Schmelzer et al. 2021; Wang Yu et al. 2021; Durand et al. 2022; Huang and Fan 2022; Öztop-Çakmak et al. 2022; Baker et al. 2023; Kistol et al. 2023, 2024; Nordli and Galan 2023; Pauly et al. 2023; Algahtani et al. 2024; Harada et al. 2024; Sultan et al. 2024). Both VPS13D-deficient human and Drosophila cells exhibit notable abnormalities in mitochondrial morphology and clearance (Anding et al. 2018; Seong et al. 2018). In addition, a genetic screen for altered mitochondria in Caenorhabditis elegans identified C25H3.11 (vps-13D) (Rolland and Conradt 2022). Recent studies have linked VPS13D to Leigh syndrome (Lee et al. 2020; Kistol et al. 2023) and the Parkinson's disease gene PINK1 (Shen, Fortier, Wang, et al. 2021), suggesting that this gene has relevance to a broader spectrum of neurological diseases.
Strong loss-of-function VPS13D alleles are lethal in Drosophila (Anding et al. 2018) and mice (Skarnes et al. 2011), thus presenting a challenge to model this disease in an intact animal. In this study, we used CRISPR/Cas9 gene editing techniques to create SCAR4 patient-specific mutations in the C. elegans ortholog of the human VPS13D gene: vps-13D. We analyzed the effects of these SCAR4 patient-specific mutations in vps-13D on worm fecundity, behavior, and mitochondrial morphology. We find that vps-13D deletion mutants display maternal effect sterility and locomotion defects. vps-13D deletions and the N3017I missense mutation displayed disrupted mitochondrial morphology and resulted in different degrees of activated mitochondrial unfolded protein response (UPRmt). Furthermore, we identified a functional link between vps-13D and the mitochondria fusion regulator fzo-1 in the same pathway to regulate mitochondrial dynamics and homeostasis. The combined biological changes caused by VPS13D mutations in C. elegans advance the development of a new animal model to study SCAR4 pathogenesis.
Materials and methods
Caenorhabditis elegans strains and GFP fusion construction
All C. elegans strains were maintained at 22°C on nematode growth medium (NGM) plates seeded with Escherichia coliOP50 bacteria. The wild-type strain was Bristol N2. Sterile mutant vps-13D strains were balanced with the mIn1 balancer chromosome (Edgley and Riddle 2001). The vps-13D mutant strains were crossed with the following transgenes: zcIs14 [Pmyo-3::GFP (mit)] (Benedetti et al. 2006) and zcIs13 [Phsp-6::GFP + lin-15 (+)] (Yoneda et al. 2004; Yang et al. 2022). A full list of strains used in this study is given in Supplementary Table 1. The Pvps-13D::GFP transcriptional reporter was generated using the following primers: F(5′-ACAGGATCCTCGCATACAATCACATCGTC-3′) and R(5′- ACAGGTACCTGTCCAGGAATTGTGGTATC-3′). These primers were used to amplify a 3.5-kb fragment corresponding to the upstream promoter sequence (including exon 1 and part of exon 2) of vps-13D. The PCR product was digested by BamHI and KpnI restriction enzymes and inserted into pPD95.75 vector. The Pvps-13D::GFP construct was microinjected at 50 ng/µl into temperature-sensitive lin-15(n765ts) mutant animals along with the lin-15 (+) rescuing plasmid (pL15EK) at 80 ng/µl and pBSK DNA at 80 ng/µl. Transgenics were selected at 22°C based on a non-Muv (Multivulva) phenotype and GFP fluorescence.
CRISPR/Cas9 design and gene editing
The Bristol N2 strain was used as the wild-type strain for all CRISPR/Cas9 editing of vps-13D. Site-specific crRNAs and a repair template donor ssODNs were manually selected and generated by IDT (Integrated DNA Technologies, Inc.) who also provided the tracrRNA. Injection mixtures were prepared following established protocols (Ghanta et al. 2021), and rol-6 was used as a co-CRISPR selection marker. All crRNAs, ssODNs, and PCR screening sequences are reported in Supplementary Tables 2–4. Correct substitution or deletion sequences were confirmed via Sanger sequencing. Sequencing of C-terminal deletion ΔC (zf197) revealed a 1,218-bp deletion (LGII 5670774–5671991) with a short 20-bp insertion positioned between the 2 breakpoints. The CRISPR-designed strains underwent 4 rounds of outcrossing to the N2 strain.
Brood size assay
Ten L4 animals of each genotype were picked and separated onto individual plates. Animals were transferred onto new plates every day until the cessation of egg laying. Plates were counted for the total number of eggs after removal of the parent animal.
Larval development analysis
Five adult animals from each genotype were transferred to a new plate. After 2 h, adult animals were removed, and the eggs were left to develop for over 96 h. Two vps-13D deletion mutant strains were maintained as balanced heterozygotes using the mIn1 [dpy-10 (e128) mIs14] balancer. This balancer includes an integrated pharyngeal GFP reporter expressed in a semi-dominant manner. Consequently, the offspring segregate into wild type with a dim GFP signal, Dpy with a bright GFP signal (mIn1 homozygotes), and non-GFP vps-13D deletion homozygotes. For the vps-13D deletion strains, only non-GFP homozygotes were evaluated at each developmental stage. The developmental timing was calculated as the proportion of larvae among the total number of hatched embryos that reached each specific developmental stage. The developmental stage of each individual was determined based on body size and stage-specific morphological features.
Multi-Worm Tracker assay
Ten synchronized 1-day-old or 3-day-old (24 h or 3 days after the L4 stage) worms were placed on a thin lawn E. coliOP50 on a medium (5 cm) NGM plate. The plate was then placed in the Multi-Worm Tracker (MWT) [https://sourceforge.net/projects/mwt/, (Swierczek et al. 2011)] and recorded for 10 min. The MWT package includes real-time image analysis software and behavioral parameter measurement software, Choreography. Tracking and analysis were conducted following previous studies (Huang et al. 2019; Florman and Alkema 2022; Kang et al. 2024). The average speed of the population was measured over 5 min after a 5-min acclimation period. Experiments were analyzed using custom MATLAB (MathWorks, Inc.) scripts to interface with Choreography analysis program. The MATLAB scripts used in this study are available at https://github.com/jeremyflorman/Tracker_GUI. Analysis was limited to objects that had been tracked for a minimum of 20 s and had moved a minimum of 5 body lengths. For the analysis of single worm tracks, a plate containing a single 3-day-old animal was recorded for 10 min with images captured every 2 s. The sequential images from the final 5 min postacclimation were stitched and processed in ImageJ to generate a continuous worm movie track.
Thrashing assay
Ten age-synchronized young adult animals were transferred to a fresh unseeded plate to remove residual bacteria. Individual animals were subsequently transferred to M9 buffer in a tiny plate. After 15-s acclimatization, the number of completed thrashes during 1 min was counted per animal using a hand counter.
Gene expression knockdown via RNA-mediated interference treatment
fzo-1 RNA-mediated interference (RNAi) bacterial clones, obtained from the Ahringer library (Kamath and Ahringer 2003), were selected by ampicillin (100 mg/ml) and tetracycline (12.5 mg/ml) and verified by DNA sequencing. The control L4440 or fzo-1 RNAi bacteria grown at 37°C overnight in LB with ampicillin (100 mg/ml) were concentrated (4×) and seeded on RNAi NGM plates that contain 6 mM IPTG and 100 mg/ml ampicillin. Young adult P0 animals were placed on the RNAi plates and allowed to produce offspring. L4 larvae of F1 progeny were transferred to a new RNAi plate. After 2 generations of RNAi treatment, L4 larvae of F2 animals were analyzed.
Imaging and fluorescence quantification
To assess the sterile phenotype, adult wild-type animals and vps-13D deletion mutant animals were placed on 2% agarose pads containing 60 mM sodium azide. Adult sterile animals were identified by the absence of embryos or oocytes in the uterus through DIC imaging using a Zeiss LSM 700 microscope. To explore the expression pattern of vps-13D, adult animals expressing a Pvps-13D::GFP reporter were mounted on 2% agarose pads containing 60 mM sodium azide. Images were captured using a Zeiss LSM 700 confocal microscope. To test the impact of vps-13D mutations on mitochondrial morphology, L4 animals expressing zcIs14 transgene were mounted on 2% agarose pads containing 10 mM levamisole. Images were recorded using a Zeiss LSM 700 confocal microscope. For each genotype, mitochondrial morphology in body wall muscles in the middle of the worm body was scored for 10–15 animals in total. To test the effects of vps-13D mutations on hsp-6 expression of C. elegans, 6 L4 animals expressing Phsp-6::GFP grown on E. coliOP50 were mounted together on 2% agarose pads containing 10 mM levamisole and 18–36 animals in total. The GFP images were acquired with an AxioImager Z1 microscope (Zeiss) at 10× magnification, and the maximum fluorescence intensity was quantified using ImageJ FIJI software.
Mitochondrial morphology analysis
Mitochondrial images were segmented and quantified using the Mitochondrial Segmentation Network (MitoSegNet) toolbox, a deep learning-based tool that includes the MitoS segmentation tool and the MitoA analysis tool (Fischer et al. 2020). Documentation for the MitoS and MitoA tools is available at https://github.com/mitosegnet, and the MitoSegNet segmentation model, along with the MitoA analysis and MitoS segmentation tools (GPU/CPU versions) for Linux and Windows, can be accessed at https://zenodo.org/search?page=1&size=20&q=mitosegnet. Initially, 8-bit raw images were segmented using the pretrained mitochondria-specific “basic mode” (MitoSegNet model) in the MitoS tool (CPU version). Upon completion, the program generated a prediction folder containing the segmented images. The segmented images and corresponding raw images were then subjected to quantitative measurement using the MitoA tool, which evaluates morphological features for each object and generates summary statistics for all object features in each image. The major axis lengths were determined by measuring the lengths of the line segment connecting the 2 vertices of an ellipse fitted around an object in the MitoA tool.
Statistical analysis
All statistical analysis and graph construction were performed using GraphPad Prism 8 software. The results are presented as SEM from at least 3 independent experiments. Statistical comparisons were made using ANOVA or Kruskal–Wallis H test with Dunnett's correction for multiple samples. Significance was determined when P-value < 0.05.
Results
Caenorhabditis elegans vps-13D encodes an ortholog of VPS13D
To explore the evolutionary relationships of the VPS13D proteins among various species, we first conducted a phylogenetic analysis. VPS13D proteins from different species formed a well-supported clade, indicating that they evolved from a common ancestor (Fig. 1a). The C. elegansC25H3.11 gene encodes a protein that shares the most significant similarity with VPS13D. The predicted ricin B-type lectin domain-containing protein shares 36% similarity and 22% identity with the human VPS13D protein (Supplementary Table 5). Furthermore, the highest sequence similarity between the 2 proteins was observed in the C-terminal region, which contains the VPS13 adaptor-binding (VAB) domain (Bean et al. 2018) [also known as SHR-binding domain or WD40-like region (Kumar et al. 2018)] and a Dbl homology (DH)-like domain (InterPro) (Fig. 1b). Given that C25H3.11 gene is the closest C. elegans ortholog of the human VPS13D gene, we hereafter refer to C25H3.11 as the C. elegansvps-13D gene. In humans, 14 of 30 reported families carry at least 1 missense mutation in the C-terminal region following the UBA domain, and 5 of 8 reported homozygous missense mutations in patients are also located in this region (Gauthier et al. 2018; Seong et al. 2018; Koh et al. 2020; Lee et al. 2020; Petry-Schmelzer et al. 2021; Wang Yu et al. 2021; Durand et al. 2022; Huang and Fan 2022; Öztop-Çakmak et al. 2022; Baker et al. 2023; Kistol et al. 2023, 2024; Nordli and Galan 2023; Pauly et al. 2023; Algahtani et al. 2024; Harada et al. 2024; Sultan et al. 2024). Several of these mutation sites associated with human pathogenicity are highly conserved in the C-terminal region in C. elegans VPS13D protein (Fig. 1c).
![VPS13D ortholog vps-13D is conserved in C. elegans. a) Phylogenetic tree of VPS13D proteins. VPS13D protein sequences from various species were retrieved from UniProt. The evolutionary history was inferred using the neighbor-joining method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. Evolutionary analyses were conducted in MEGA11. The species depicted from top to bottom are human (UniProt identifier: Q5THJ4), bovine (UniProt identifier: E1BIF6), mouse (UniProt identifier: B1ART2), rat (UniProt identifier: A0A8I6G572), zebrafish (UniProt identifier: A0A8M1QUR7), Drosophila (UniProt identifier: Q9VU08), C. elegans (labeled as brown diamond, UniProt identifier: A0A2K5ATR5), and yeast (UniProt identifier: Q07878). b) Human VPS13D protein topology [modified from (Guillén-Samander et al. 2021)] and predicted domains of the C. elegans VPS-13D protein with disease-related missense mutations labeled in dotted vertical lines and deletions annotated with horizontal lines. Domains and abbreviations are as follows: N, Chorein N-terminal domain; Chorein domain; Extended Chorein domain; UBA, ubiquitin-associated domain; VAB, Vps13 adaptor-binding/SHR-binding/WD40-like domain; ricin B-type lectin domain; DH, Dbl homology-like domain; and PH, pleckstrin homology domain. c) Sequence alignment of C-terminal region between C. elegans VPS-13D, human VPS13D, and fly Vps13D protein by Clustal Omega. The 3 missense mutations enrolled in this study are outlined, and the start residue of vps-13D(ΔC) deletion is labeled with asterisk. d) Representative fluorescence image of adult hermaphrodites expressing Pvps-13D::GFP under the 3,570-bp promoter (including exon 1 and part of exon 2). A, anterior; P, posterior; V, ventral side; D, dorsal side. The scale bar is 0.2 mm.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/g3journal/15/4/10.1093_g3journal_jkaf023/2/m_jkaf023f1.jpeg?Expires=1748019733&Signature=M9dwKZj6p97ISYhtRLT2XTe6q1PhGQvS1r19bjG5hLW0GuIWQcztrtnrS8BNxkajfDy3lim0HhvlrTfu5PKMoEsE1baU~pUpt4LIgdgyHRKtr6Bnac2nK~ZrGzNx02Y-OLF4vqiLQza8gX4kCIu9YYpvVFeY2IWzr1yhDjyuXy0WIPfAS9CX2cp9Pth9U5De1TVex3x0DPKUrONCTK0U7kykAgBpnHVWNVDS0i2-FTSR8RTe0gck7q0AO57H37372pxlI8I1v484EOzcy4ogktYKdhKkOtoNtGqs0XPaNVhuLepbXkxXzfL5P0k27QH77lJIfcrpU~rkPb09w9jI0A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
VPS13D ortholog vps-13D is conserved in C. elegans. a) Phylogenetic tree of VPS13D proteins. VPS13D protein sequences from various species were retrieved from UniProt. The evolutionary history was inferred using the neighbor-joining method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. Evolutionary analyses were conducted in MEGA11. The species depicted from top to bottom are human (UniProt identifier: Q5THJ4), bovine (UniProt identifier: E1BIF6), mouse (UniProt identifier: B1ART2), rat (UniProt identifier: A0A8I6G572), zebrafish (UniProt identifier: A0A8M1QUR7), Drosophila (UniProt identifier: Q9VU08), C. elegans (labeled as brown diamond, UniProt identifier: A0A2K5ATR5), and yeast (UniProt identifier: Q07878). b) Human VPS13D protein topology [modified from (Guillén-Samander et al. 2021)] and predicted domains of the C. elegans VPS-13D protein with disease-related missense mutations labeled in dotted vertical lines and deletions annotated with horizontal lines. Domains and abbreviations are as follows: N, Chorein N-terminal domain; Chorein domain; Extended Chorein domain; UBA, ubiquitin-associated domain; VAB, Vps13 adaptor-binding/SHR-binding/WD40-like domain; ricin B-type lectin domain; DH, Dbl homology-like domain; and PH, pleckstrin homology domain. c) Sequence alignment of C-terminal region between C. elegans VPS-13D, human VPS13D, and fly Vps13D protein by Clustal Omega. The 3 missense mutations enrolled in this study are outlined, and the start residue of vps-13D(ΔC) deletion is labeled with asterisk. d) Representative fluorescence image of adult hermaphrodites expressing Pvps-13D::GFP under the 3,570-bp promoter (including exon 1 and part of exon 2). A, anterior; P, posterior; V, ventral side; D, dorsal side. The scale bar is 0.2 mm.
We next focused on 3 conserved residues within the C-terminal region of VPS13D protein that are reported to be mutated in early-onset SCAR4 patients. We used CRISPR/Cas9 gene editing to create these variants in vps-13D [N2454S(zf194), N3017I(zf195), and R3144Q(zf196)] (Table 1). In addition, we generated a C-terminal deletion ΔC(zf197) that encompassed 1,218 bp, resulting in a truncation of the VPS13D protein (Fig. 1b). We also obtained a vps-13D deletion allele, Δ(ok2632), which contains a 1,740-bp deletion removing part of exons 3 and 4, resulting in a premature stop and thus most likely represents a null allele. To analyze the expression of vps-13D, we investigated its expression pattern by generating a transcriptional fusion of GFP reporter with 3.5-kb vps-13D upstream promoter fragment (including exon 1 and part of exon 2). Fluorescence from the Pvps-13D::GFP reporter was observed throughout the adult hermaphrodite, including intestine, hypodermis, muscle, glia, and neurons (Fig. 1d). Our data indicate that vps-13D is expressed in most cell types consistently with RNA expression datasets (Taylor et al. 2021).
VPS13D pathogenic missense mutations in this study.
| Caenorhabditis elegans mutations (vps-13D a.1) | VPS13D pathogenic mutations (NM_015378) | Clinical diagnosis of patients | Onset age |
|---|---|---|---|
| N2454S (zf194) | p.N3521S (c.10562A > G) | Spastic ataxia with mild intellectual disability (2 patients) | 2 years/4–5 years |
| N3017I (zf195) | p.N4107I (c. 12320A > T) | Ataxia, neuropathy, developmental delay, seizures, and optic atrophy | 1 year |
| R3144Q (zf196) | p.R4228Q (c.12683G > A) | Ataxia, muscle weakness, dystonia, mild intellectual disability | 1–2 years |
| Caenorhabditis elegans mutations (vps-13D a.1) | VPS13D pathogenic mutations (NM_015378) | Clinical diagnosis of patients | Onset age |
|---|---|---|---|
| N2454S (zf194) | p.N3521S (c.10562A > G) | Spastic ataxia with mild intellectual disability (2 patients) | 2 years/4–5 years |
| N3017I (zf195) | p.N4107I (c. 12320A > T) | Ataxia, neuropathy, developmental delay, seizures, and optic atrophy | 1 year |
| R3144Q (zf196) | p.R4228Q (c.12683G > A) | Ataxia, muscle weakness, dystonia, mild intellectual disability | 1–2 years |
VPS13D pathogenic missense mutations in this study.
| Caenorhabditis elegans mutations (vps-13D a.1) | VPS13D pathogenic mutations (NM_015378) | Clinical diagnosis of patients | Onset age |
|---|---|---|---|
| N2454S (zf194) | p.N3521S (c.10562A > G) | Spastic ataxia with mild intellectual disability (2 patients) | 2 years/4–5 years |
| N3017I (zf195) | p.N4107I (c. 12320A > T) | Ataxia, neuropathy, developmental delay, seizures, and optic atrophy | 1 year |
| R3144Q (zf196) | p.R4228Q (c.12683G > A) | Ataxia, muscle weakness, dystonia, mild intellectual disability | 1–2 years |
| Caenorhabditis elegans mutations (vps-13D a.1) | VPS13D pathogenic mutations (NM_015378) | Clinical diagnosis of patients | Onset age |
|---|---|---|---|
| N2454S (zf194) | p.N3521S (c.10562A > G) | Spastic ataxia with mild intellectual disability (2 patients) | 2 years/4–5 years |
| N3017I (zf195) | p.N4107I (c. 12320A > T) | Ataxia, neuropathy, developmental delay, seizures, and optic atrophy | 1 year |
| R3144Q (zf196) | p.R4228Q (c.12683G > A) | Ataxia, muscle weakness, dystonia, mild intellectual disability | 1–2 years |
vps-13D mutants exhibit impaired locomotion
Animals carrying either homozygous vps-13D deletion alleles [Δ(ok2632) or ΔC(zf197)] are viable. However, both homozygous vps-13D(Δ) and vps-13D(ΔC) animals display maternal effect sterility (Fig. 2a). Wild-type hermaphrodites produce oocytes that are fertilized in the spermatheca, resulting in the accumulation of developing embryos in the uterus. The germlines of either homozygous vps-13D(Δ) or vps-13D(ΔC) animals did not display visible oocytes or embryos (Fig. 2b). Due to their sterility, these 2 deletion strains were maintained as balanced heterozygotes but analyzed as homozygotes by selecting homozygous animals from a mixed population. In addition to fertility deficiencies, these 2 deletion mutants also exhibited a slower rate of development, taking 24–48 h longer than the wild type to reach the L4 stage (Fig. 2c). In contrast, 3 missense mutants vps-13D(N2454S), (N3017I), and (R3144Q) did not exhibit developmental arrest or delay and were able to produce a similar brood size compared with the wild type (Fig. 2a).
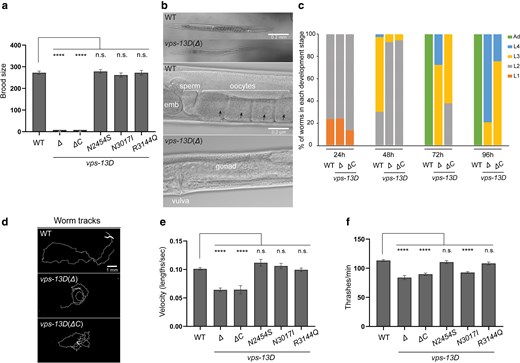
vps-13D deficiency affects worms’ fertility and locomotion. a) Total brood size (n = 30 broods) indicates the fertility ability of C. elegans to produce offspring in association with different vps-13D mutants. One-way ANOVA with Dunnett’s correction was used to compare the difference between wild-type and mutant animals. b) DIC images of whole worm and gonad in hermaphrodites of the wild-type and vps-13D (Δ) mutant strains. The scale bar in the whole worm image is 0.2 mm. The scale bar in the gonad image is 0.2 μm. Black arrows indicate oocyte nuclei. c) The percentage of worms at the different developmental stages was determined for wild-type, vps-13D(Δ), and vps-13D(ΔC) mutant animals after 96 h of growth from the embryonic stage at 22°C. d) Individual animal tracks of 3-day-old wild-type, vps-13D(Δ), and vps-13D(ΔC) mutant strains for the final 5-min recordings. The scale bar is 1 mm. e) Comparison of average velocity for 3-day-old wild-type and vps-13D animals from the final 5-min MWT recordings. The velocity has been normalized and calculated based on the worm body lengths of respective strains. The difference in average velocity between wild type and mutants was analyzed by One-way ANOVA with Dunnett's multiple comparison tests. f) Number of thrashes per minute in M9 liquid for 3-day-old wild-type and vps-13D mutant animals (n = 15–30). Differences between wild type and mutants were analyzed by one-way ANOVA with Dunnett’s correction for multiple comparisons. **** p < 0.0001; n.s., not significant.
Next, we examined whether vps-13D mutant strains possess motor defects. We conducted a population-level motility assay using an automated multiworm tracking system (MWT) (Swierczek et al. 2011). We tracked wild-type and vps-13D mutant strains for 5 min and extracted the behavioral dynamics of individual worms. One-day-old adult vps-13D mutants did not show any difference compared with the wild type (Supplementary Fig. 1). However, vps-13D deletion animals displayed a noticeable movement impairment compared with wild-type animals. The average locomotion rate of 3-day-old wild-type animals was 0.101 ± 0.005 body lengths/s, whereas either vps-13D(Δ) or vps-13D(ΔC) mutants displayed decreased locomotion rates of 0.064 ± 0.010 and 0.065 ± 0.012 body lengths/sec (Fig. 2d and e). The 3 vps-13D missense mutant strain locomotion rates were unaffected at day 3 of the adult stage (0.111 ± 0.009, 0.106 ± 0.010, and 0.099 ± 0.008 body lengths/s, respectively, Fig. 2e). Furthermore, we analyzed thrashing rates of single worm in liquid M9 medium. The average thrashing rate of 3-day-old wild-type animals was 113.2 ± 6.9 thrashes/min. Significantly, vps-13D(Δ) and vps-13D(ΔC) mutant strains showed a 20–26% decrease in thrashing rates compared with control wild-type animals (83.8 ± 11.0 and 89.8 ± 8.7 thrashes/min, respectively, Fig. 2f). Importantly, the thrashing rate of vps-13D(N3017I) mutant worms was 92.7 ± 7.1 thrashes/min, showing an 18% reduction compared with control animals (Fig. 2f), where vps-13D(N2454S) and vps-13D(R3144Q) mutants exhibited similar thrashing rates to the wild type (110.5 ± 11.0 and 108.3 ± 9.5 thrashes/min, respectively). Combined, these results indicate that the C. elegansvps-13D mutations can result in locomotion defects.
vps-13D mutants possess altered mitochondrial morphology
Dynamic changes in mitochondrial morphology are essential for mitochondrial health and homeostasis. Mitochondrial fusion and fission not only affect mitochondrial morphology but also regulate multiple mitochondrial biological processes, including mitochondrial quality control and clearance by mitophagy (Pickles et al. 2018). Previous studies indicate that VPS13D affects mitochondrial morphology and mitophagy in both human and Drosophila cells (Anding et al. 2018). To test whether vps-13D mutants exhibit a similar function to maintain mitochondrial morphology, we used a transgenic line zcIs14 {[myo-3::GFP (mit)]} that labels mitochondria in the body wall muscles (Benedetti et al. 2006) and quantified the mitochondrial morphology using MitoSegNet (Fischer et al. 2020). Although morphologies between different mutants were variable and possibly reflected allele strength, mutant strains of vps-13D displayed changes in mitochondrial morphology at the L4 larval stage compared with the wild type (Fig. 3a). Specifically, all homozygous vps-13D mutant worms possessed shorter mitochondria compared with control wild-type worms, as determined by the major axis length (Fig. 3a and b). In wild-type worms, the major axis length was 31.4 ± 11.6 pixels. By contrast, vps-13D(Δ) and vps-13D(ΔC) mutant strains displayed significantly reduced major axis lengths of 18.1 ± 3.7 and 19.0 ± 5.4 pixels, respectively (Fig. 3b). Similarly, the major axis length was also reduced in the 3 vps-13D missense mutant strains, measuring 19.7 ± 5.7, 18.0 ± 3.9, and 22.3 ± 9.5 pixels, respectively (Fig. 3b). These data are consistent with a recent report (Rolland and Conradt 2022). In addition, both vps-13D(Δ) and vps-13D(ΔC) mutant strains demonstrated a notable increase in mitochondrial area measuring 154.0 ± 32.6 and 144.7 ± 30.2 pixels, respectively, compared with 113.5 ± 19.6 pixels in wild-type worms. By contrast, vps-13D(N2454S) and vps-13D(N3017I) mutant strains display slightly smaller mitochondrial areas of 86.5 ± 20.9 and 87.6 ± 26.5 pixels, respectively, where vps-13D(R3144Q) mutant is similar to the wild type (102.3 ± 49.4 pixels) (Fig. 3a and c). These results indicate that SCAR4 mutations in the C. elegans VPS13D ortholog vps-13D lead to alterations in mitochondrial morphology, supporting VPS-13D's role in maintaining mitochondrial structure.
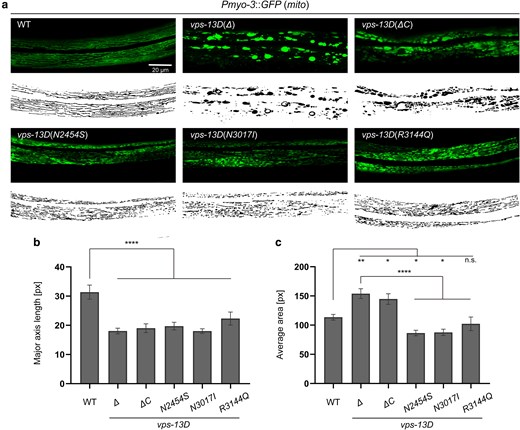
vps-13D regulates mitochondrial morphology in C. elegans. a) Mitochondrial morphology of the wild-type and vps-13D mutant animals. Original images are at the top, and MitoSegNet model segmentations are at the bottom. The scale bar is 20 μm. b) and c) Statistical analysis of mitochondrial morphology parameters. The major axis length (‘see Materials and Methods’) and average area were measured in segmented images of mitochondria from wild-type and vps-13D mutant animals (n = 15–25). One-way ANOVA with Dunnett’s correction was used to compare the difference between wild-type and mutant animals. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001; n.s., not significant.
vps-13D disruption induces mitochondrial UPRmt
Although previous assays have examined the effects of VPS13D mutations on mitochondrial morphology, their impact on mitochondrial homeostasis remains unclear (Anding et al. 2018). The mitochondrial UPRmt is a crucial cellular pathway that protects mitochondria from multiple forms of cell stress (Shpilka and Haynes 2018). To test whether vps-13D mutants affect UPRmt, we used a transgene of the transcriptional reporter Phsp-6::mtHSP70::GFP to monitor UPRmt in C. elegans (Yoneda et al. 2004; Yang et al. 2022). Interestingly, vps-13D(Δ) and vps-13D(ΔC) mutant strains exhibit a significant induction of the UPRmt with high levels of Phsp-6::mtHSP70::GFP intensity compared with wild-type animals at L4 larval age (Fig. 4a and b). While the relative fluorescence intensity of vps-13D(N2454S) and vps-13D(R3144Q) missense mutants was not significantly different from wild-type, homozygous vps-13D(N3017I) mutant worms exhibited a slight increase in UPRmt reporter expression compared with control worms (Fig. 4a and b). Combined, these results indicate that VPS13D plays a crucial role in maintaining mitochondrial homeostasis. Since the large deletion Δ(ok2632) allele and the C-terminal deletion allele ΔC (zf197) cause similar deficiencies in fertility, development, locomotion, as well as mitochondrial morphology, we cannot exclude that frameshift mutations near the C-terminus could compromise protein stability.
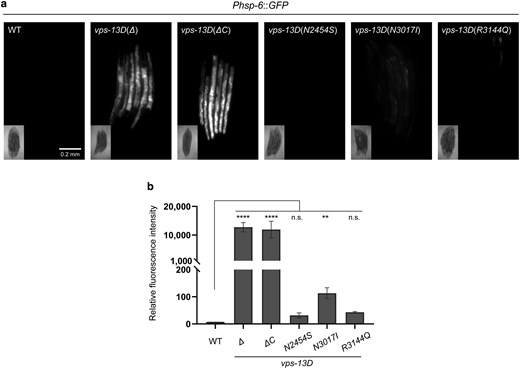
vps-13D dysfunction induces mitochondrial UPR. a) Phsp-6::GFP expression of wild-type and vps-13D mutant worms. The scale bar is 0.2 mm. b) Quantification of Phsp-6::GFP relative fluorescence intensity (n = 3–5 groups*6 animals). Kruskal–Wallis H test with Dunnett’s correction was used to compare the difference between wild-type and mutant animals. Data are presented as mean ± SEM. **P < 0.01, ****P < 0.0001; n.s., not significant.
vps-13D and fzo-1/MFN2 function in a pathway to regulate mitochondrial homeostasis
In Drosophila, the ortholog of mitofusin-2 (MFN2), Marf acts downstream of Vps13D to regulate mitochondrial fusion (Shen, Fortier, Zhao, et al. 2021). The C. elegans MFN2 ortholog FZO-1 is also involved in mitochondrial fusion (Rolland et al. 2009). Since our results indicate that vps-13D loss promotes alteration of mitochondrial morphology (Fig. 3), we examined the potential relationship between VPS-13D and FZO-1 in the regulation of mitochondrial morphology. We performed RNAi targeting fzo-1 either in vps-13D(Δ) mutant background or in control worms. fzo-1 knockdown worms exhibited a decrease in average major axis length (17.7 ± 2.5 pixels) and mitochondrial area (77.8 ± 8.5 pixels) compared with control animals, which exhibited a major axis length of 28.4 ± 6.7 pixels and a mitochondrial area of 119.2 ± 9.8 pixels (Fig. 5a–c). Knockdown of fzo-1 failed to suppress the abnormal mitochondrial morphology observed in vps-13D(Δ)-deficient animals (Fig. 5a–c). The major axis length (20.6 ± 6.5 pixels) and mitochondrial area (146.7 ± 57.4 pixels) of vps-13D(Δ); fzo-1(RNAi) worms were similar to those observed in vps-13D(Δ) control animals (19.4 ± 4.2 pixels in major axis length and 157.4 ± 40.1 pixels in area). We also examined whether fzo-1 affects mitochondrial UPRmt. Consistent with previous studies (Haeussler et al. 2020), the knockdown of fzo-1 in wild-type worms led to a significant increase in the level of the UPRmt reporter zcIs13 (Fig. 5d and e). However, fzo-1 inactivation in vps-13D(Δ) mutant worms did not result in a significant change in UPRmt compared with the vps-13D(Δ) single mutant strain (Fig. 5d and e). Collectively, these data suggest that vps-13D and fzo-1 function together to regulate mitochondria homeostasis.
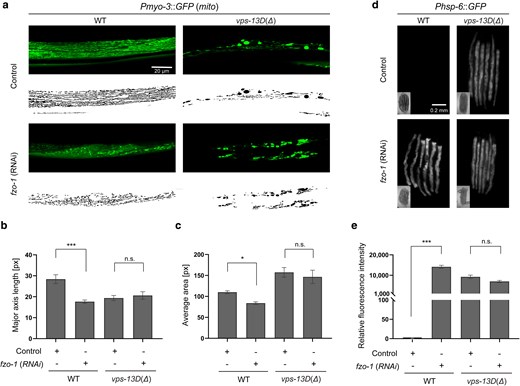
vps-13D and fzo-1 regulate mitochondrial homeostasis in similar pathways. a) Mitochondrial morphology changes of wild-type and vps-13D(Δ) animals grown on either control mock or fzo-1 (RNAi). Original images are at the top and MitoSegNet model segmentations are at the bottom. The scale bar is 20 μm. b) and c) Statistical analysis of mitochondrial morphology parameters. The major axis length (‘see Materials and Methods’) and average area were measured in segmented images of mitochondria from wild-type and vps-13D(Δ) animals grown on either control mock or fzo-1 (RNAi) (n = 10–15). d) and e) Phsp-6::GFP expression of wild-type and vps-13D(Δ) animals grown on either control mock or fzo-1 (RNAi) (n = 3–15 groups*6 animals). The scale bar is 0.2 mm. Kruskal–Wallis H test with Dunnett’s correction was used to compare the difference between wild-type and mutant animals. Data are presented as mean ± SEM. *p < 0.05, ***p < 0.001; n.s., not significant.
Discussion
We developed a new model to study VPS13D-related SCAR4 disease in the nematode C. elegans. Our results indicate that C. elegans carrying the VPS13D deletions and disease-associated missense mutations, in the worm ortholog vps-13D, are viable. However, vps-13D null deletion mutations that remove the C-terminus display maternal effect sterility. In addition, vps-13D deletions and N3017I missense mutant C. elegans exhibit impaired locomotion, thus reflecting a similar role for this gene because human SCAR4 patients exhibit movement deficiencies. Importantly, vps-13D mutant C. elegans displayed abnormal mitochondrial morphology, and vps-13D deletions and vps-13D(N3017I) mutants displayed varying levels increased mitochondrial UPR.
The key clinical features of SCAR4 patients include progressive development of hyperkinetic movement disorder (dystonia, chorea, and/or ataxia). SCAR4 patient muscle biopsies exhibited mitochondrial accumulation and mild lipidosis (Gauthier et al. 2018). Similarly, altered mitochondrial morphology and mitochondria clearance is decreased in cultured VPS13D patient-derived fibroblasts, VPS13D mutant human HeLa cells, and Drosophila (Anding et al. 2018; Seong et al. 2018; Shen, Fortier, Zhao, et al. 2021). Although mitochondrial respiratory chain function was normal in the muscle biopsy of 1 SCAR4 patient, a decrease in energy production and complex (I, III, and IV) protein levels has been detected in some patient fibroblasts (Gauthier et al. 2018; Seong et al. 2018; Durand et al. 2022). Consistent with the movement defect of SCAR4 patients, worms carrying either deletions or N3017I missense mutations in vps-13D exhibited defects in crawling and/or swimming behavior. Interestingly, the abnormal swimming ability of vps-13D mutant worms is suggestive of mitochondrial dysfunction since swimming has been shown to be more energetically demanding than crawling for C. elegans (Laranjeiro et al. 2017; Kang et al. 2024). The similarities between SCAR4 patients and vps-13D mutant worms, including locomotion defects, mitochondrial morphology, and mitochondrial homeostasis changes, suggest that C. elegans will likely be a useful model to reveal other genes in this pathway because of the strength of this genetic model.
Mitochondrial homeostasis is controlled by mitophagy machinery, including the PINK1–Parkin axis (Pickrell and Youle 2015). VPS13D functions downstream of PINK1, but bifurcates with Parkin to regulate mitochondrial clearance (Shen, Fortier, Wang, et al. 2021). Specifically, VPS13D binds to ubiquitin and affects both the PINK1 substrate ubiquitinphospho-serine 65 and localization of the mitophagy receptor Ref2p/p62 on mitochondria (Shen, Fortier, Wang, et al. 2021). Previous studies suggest mitophagy and mitochondrial UPR are both required to maintain mitochondrial health (Pellegrino and Haynes 2015). Mitophagy and mitochondrial UPR function in a complementary manner to either recycle the damaged mitochondria by autophagy or recover the less damaged mitochondria. Additionally, mitophagy inhibition also causes damaged mitochondrial DNA accumulation that triggers mitochondrial UPR. Recently, it has been shown that nonmitochondrial proteins, including VPS13D, also play a role in maintaining mitochondrial homeostasis (Rolland and Conradt 2022). Although it is unclear whether VPS13D regulates mitochondrial clearance in worms, evidence of VPS13D regulating mitophagy in different animals and human cells suggests this possibility (Anding et al. 2018; Shen, Fortier, Wang, et al. 2021). Therefore, vps-13D mutant worms may potentially accumulate damaged mitochondria to activate mitochondrial UPR.
Mitochondria and ER contact dictates interorganelle lipid transfer sites and mitochondrial membrane lipid composition (Gatta and Levine 2017). The lipid composition of mitochondrial membranes, in turn, can influence crucial processes within mitochondria (Böckler and Westermann 2014; Lahiri et al. 2015). Previous studies indicate Vmp1 and Vps13D regulate mitochondria–ER contact and mitophagy (Shen, Fortier, Zhao, et al. 2021). VPS13D binds the outer mitochondrial membrane GTPase-Miro (both Miro1 and Miro2), likely via the WD40-like/VAB domain (Guillén-Samander et al. 2021), and the conserved C-terminal regions have been shown to work synergistically with the VAB domain to facilitate the membrane targeting (Dziurdzik and Conibear 2021). Moreover, the VPS13D C-terminus is similar to the lipid transfer protein ATG2 (Öztop-Çakmak et al. 2022). Consistent with the importance of these domains, our data indicate that vps-13D C-terminus is important for mitochondrial homeostasis.
The mitochondrial protein MFN2 facilitates mitochondrial fusion, as well as mitochondria and ER contact (Filadi et al. 2018). Mutations in MFN2 are associated with Charcot–Marie–Tooth disease type 2A, in which mitochondrial morphology is abnormal in nerve tissue and fibroblasts from patients (Verhoeven et al. 2006; Amiott et al. 2008). Similarly, loss of fzo-1, the worm MFN2 homolog, resulted in progressive movement deficits, fragmented mitochondria, and mitochondrial UPR activation in C. elegans (Byrne et al. 2019; Haeussler et al. 2021). Consistent with these studies, our results indicate fzo-1 knockdown worms displayed altered mitochondrial morphology and induced mitochondrial UPR. UPRmt activation requires the key transcription factor ATFS-1, which regulates the hsp-6 reporter expression (Nargund et al. 2015; Lin et al. 2016; Shpilka et al. 2021; Xin et al. 2022). Notably, knockdown of atfs-1 significantly suppressed UPRmt in the fzo-1 mutant worms, confirming that UPRmt induction by loss of fzo-1 is ATFS-1 dependent (Chen et al. 2021). Unlike Marf/MFN2 function in the Vps13D pathway regulating mitochondrial morphology in Drosophila (Shen, Fortier, Zhao, et al. 2021), our results suggest that fzo-1 and vps-13D could function in a related pathway to affect mitochondrial UPR in C elegans. In summary, this study provides evidence supporting the conserved role of VPS13D in regulating mitochondrial morphology and function in C. elegans and provides insights into how disease mutations affect mitochondrial homeostasis and behavior.
Data availability
All data files from this study have been uploaded to Mendeley Data (DOI: 10.17632/3mwgvd2dbx.1).
Supplemental material available at G3 online.
Acknowledgments
The authors thank the Caenorhabditis Genetics Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440), for worm strains, WormBAse website, http://www.wormbase.org, release WS295, and W.K. Kang, J.T. Florman, and M. Gorczyca for technical support and helpful discussions.
Funding
This work was supported by National Institutes of Health grants R01 GM140480 (M.J.A.), R01 AG082740 and R35 GM131689 (E.H.B.).
Literature cited
Author notes
Conflicts of interest: The authors declare no conflict of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}