





Abstract
Loss-of-function mutations of FIG4 are responsible for neurological disorders in human and mouse that result from reduced abundance of the signaling lipid PI(3,5)P2. In contrast, loss-of-function mutations of the phosphoinositide kinase PIP4K2C result in elevated abundance of PI(3,5)P2. These opposing effects on PI(3,5)P2 suggested that we might be able to compensate for deficiency of FIG4 by reducing expression of PIP4K2C. To test this hypothesis in a whole animal model, we generated triallelic mice with genotype Fig 4−/−, Pip4k2c+/−; these mice are null for Fig 4 and haploinsufficient for Pip4k2c. The neonatal lethality of Fig 4 null mice in the C57BL/6J strain background was rescued by reduced expression of Pip4k2c. The lysosome enlargement characteristic of Fig 4 null cells was also reduced by heterozygous loss of Pip4k2c. The data demonstrate interaction between these two genes, and suggest that inhibition of the kinase PIPK4C2 could be a target for treatment of FIG4 deficiency disorders such as Charcot-Marie-Tooth Type 4J and Yunis-Varón Syndrome.
Introduction
Recessively inherited, loss-of-function variants of the phosphoinositide phosphatase FIG4 are responsible for several rare genetic disorders. Complete loss-of-function of FIG4 results in Yunis-Varón Syndrome (OMIM 216340), a lethal multisystem disorder affecting development of the skeleton and nervous system (Campeau et al. 2013). Partial loss of FIG4 function results in the peripheral neuropathy Charcot-Marie-Tooth Type 4J (CMT4J) (OMIM 611228), most often caused by compound heterozygosity for a null allele and the partial loss-of-function variant p.Ile41Thr (Chow et al. 2007; Nicholson et al. 2011). The p.Ile41Thr variant is present at an allele frequency of 0.001 in European populations, and homozygous individuals were recently described (LaFontaine et al. 2021). Other partial loss-of-function variants of FIG4 result in polymicrogyria with epilepsy (OMIM 612619) (Baulac et al. 2014) and pediatric neurodegeneration with hypomyelination (Lenk et al. 2019a), related to the requirement for PI(3,5)P2 during oligodendrocyte maturation (Mironova et al. 2016). Deficiency of the FIG4 binding partner VAC14 results in similar neurological disorders (OMIM 617054) (Lenk et al. 2016; de Gusmao et al. 2019).
At the cell level, deficiency of FIG4 reduces the abundance of the signaling phospholipid PI(3,5)P2, leading to defective lysosome function (Chow et al. 2007; Ferguson et al. 2009; Ferguson et al. 2012; Lenk and Meisler, 2014). PI(3,5)P2 regulates the activity of lysosomal ion channels and transporters including TRPML1, TPC1, TPC2 and CLC-7 (Dong et al. 2010; She et al. 2018; Patel et al. 2022; Leray et al. 2022). Reduced PI(3,5)P2 abundance alters the regulation of lysosomal ion flux and results in osmotic swelling and hyperacidic lysosomes (Chow et al. 2007; Ferguson et al. 2009; Lenk et al. 2019b; Jin et al. 2017; Wilson et al. 2018). Interventions that increase the intracellular levels of PI(3,5)P2 could potentially be therapeutic for FIG4 deficiency disorders.
Chemical inhibition of the phosphoinositide kinase PIP4K2C is one intervention that increases intracellular levels of PI(3,5)P2 in cultured cells (Al-Ramahi et al. 2017). To determine whether this observation could be reproduced in the whole animal and compensate for loss of Fig 4, we carried out crosses between mice with mutations in the two genes. The Pip4k2c null mouse exhibits normal growth and viability (Shim et al. 2016). The Fig 4 null mouse has a lethal phenotype that includes neurodegeneration, diluted pigmentation, and tremor (Chow et al. 2007; Ferguson et al. 2012; Bissig et al. 2019). A partial loss-of-function model of Fig 4, which is seen in most patients, is not currently available in the mouse (Lenk et al. 2011). Therefore, to evaluate the effect of reduced Pip4k2c, we generated Fig 4 null mice that are heterozygous for the null allele of Pip4k2c.
FIG4 and PIP4K2C are components of the phosphoinositide metabolic pathway (Fig. 1). PI(3)P is converted to PI(3,5)P2 by a biosynthetic complex that includes the kinase PIKFYVE, the phosphatase FIG4, and the scaffold protein VAC14. The 3-phosphate is removed from PI(3,5)P2 by the myotubularins (MTMRs) to generate PI(5)P (Hnia et al. 2012). Subsequent phosphorylation by the kinase PIP4K2C generates PI(4,5)P2, an abundant phosphoinositide in cell membranes and synapses (Al-Ramahi et al. 2017).
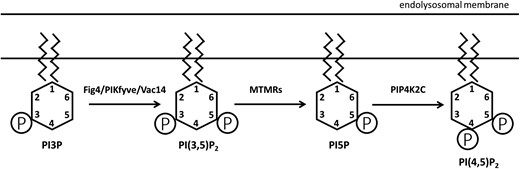
Fig 4 and Pip4k2c in phosphoinositide biosynthesis. Phosphoinositides are localized on the cytoplasmic surface of the endolysosomal compartment. In addition to the enzymes shown here, several additional enzymes catalyze the complex interconversions between the seven naturally occuring phosphoinositides (Hasegawa et al. 2017; Palamiuc et al. 2019). The abundance of the diphosphoinositide PI(3,5)P2 is reduced by inactivation of Fig 4 and increased by inactivation of Pip4k2c.
The FIG4 protein complex is localized on the cytoplasmic surface of endolysosomal vesicles (Jin et al. 2008; Botelho et al. 2008; Lees et al. 2020; Strunk et al. 2020; Rivero-Ríos & Weisman, 2022). PIP4K2C is also localized on cellular vesicles, including the endolysosome (Clarke et al. 2009). Partial loss of PIP4K2C alters the relative levels of the phosphoinositides and elevates PI(3,5)P2, resulting in enhanced autophagy and increased turnover of mutant huntingtin protein (Al-Ramahi et al. 2017).
We hypothesized that the increase in PI(3,5)P2 resulting from inactivation of Pip4k2c might compensate for the deficiency of PI(3,5)P2 in Fig 4 null mice. Consistent with this hypothesis, we observed that heterozygous loss of Pip4k2c increased the postnatal survival of Fig 4 null mice from <1 day to between 1 and 2 weeks and reduced the extensive vacuolization of Fig 4 null embryonic fibroblasts. This demonstration of genetic interaction suggests that reduction of Pip4k2c activity could be therapeutic for genetic disorders of FIG4.
Materials and methods
The spontaneous Fig 4 null mutation plt arose on a mixed genetic background (Chow et al. 2007). The congenic line C57BL/6J.Fig 4+/− was generated by more than 30 generations of backcrossing to wildtype mice of strain C57BL/6J (JAX line 017800). Pip4k2c null mice were a product of the mouse knockout mouse project and were generously provided by Dr. Lewis Cantley, Weill Cornell Medical College (Shim et al. 2016). The C57BL/6N strain background of the Pip4k2c knockout line was confirmed by genotyping with the miniMUGA panel (Sigmon et al. 2020, Yu et al. 2020)(Neogen Inc. Lincoln, NE). Genotyping of Pip4k2c mice was carried out as previously described (Shim et al. 2016). The Fig 4 mutation was genotyped by PCR with the forward primer 5′-CTTCT TTGGT GACAG GAAGA TAGA-3′ and two reverse primers. Reverse primer 5′-AGACC ACTGA AGGAT GTAGA TGTG-3′ amplifies a 200 bp fragment from the wildtype allele, and reverse primer 5′-GGAGC TAAGG CAATT TCATA CTG-3′ amplifies a 400 bp fragment from the plt mutant allele. Mouse embryonic fibroblasts (MEFs) were prepared 13 days after detection of a plug (day E13.5).
Results
Neonatal lethality of Fig 4−/− mice
Fig 4−/− null mice were generated by timed matings between congenic C57BL/6J.Fig 4+/− heterozygotes. Fig 4−/− embryos were detected at the predicted Mendelian frequency of 25% during prenatal development (15/65 total) (Table 1). At postnatal day 3, only 1 Fig 4−/− mouse was identified among 87 live births. We previously showed that Fig 4−/− homozygotes survive for 3–5 weeks on the C3HeB/FeJ or mixed strain background (Chow et al. 2007; Lenk and Meisler, 2014; Presa et al. 2015). In the current study, the congenic C57BL/6J.Fig 4+/− mice were used.
Neonatal lethality of fig 4 null mice on strain C57BL/6j. Timed matings between Fig 4+/− mice were carried out to determine the age at loss for Fig 4 null mice on the congenic C57BL/6J strain background. The probability (P-value) for the predicted Mendelian inheritance of 25% was calculated with the chi-squared test.
| Age | Fig 4−/− | Total | % Fig 4−/− | P-value |
|---|---|---|---|---|
| E14.5 | 4 | 21 | 19% | 0.7 |
| E16.5 | 4 | 15 | 27% | 1.0 |
| E17.5 | 2 | 10 | 20% | 0.6 |
| E18.5 | 5 | 19 | 26% | 1.0 |
| P3 | 1 | 87 | 1% | <0.0001 |
| Age | Fig 4−/− | Total | % Fig 4−/− | P-value |
|---|---|---|---|---|
| E14.5 | 4 | 21 | 19% | 0.7 |
| E16.5 | 4 | 15 | 27% | 1.0 |
| E17.5 | 2 | 10 | 20% | 0.6 |
| E18.5 | 5 | 19 | 26% | 1.0 |
| P3 | 1 | 87 | 1% | <0.0001 |
Neonatal lethality of fig 4 null mice on strain C57BL/6j. Timed matings between Fig 4+/− mice were carried out to determine the age at loss for Fig 4 null mice on the congenic C57BL/6J strain background. The probability (P-value) for the predicted Mendelian inheritance of 25% was calculated with the chi-squared test.
| Age | Fig 4−/− | Total | % Fig 4−/− | P-value |
|---|---|---|---|---|
| E14.5 | 4 | 21 | 19% | 0.7 |
| E16.5 | 4 | 15 | 27% | 1.0 |
| E17.5 | 2 | 10 | 20% | 0.6 |
| E18.5 | 5 | 19 | 26% | 1.0 |
| P3 | 1 | 87 | 1% | <0.0001 |
| Age | Fig 4−/− | Total | % Fig 4−/− | P-value |
|---|---|---|---|---|
| E14.5 | 4 | 21 | 19% | 0.7 |
| E16.5 | 4 | 15 | 27% | 1.0 |
| E17.5 | 2 | 10 | 20% | 0.6 |
| E18.5 | 5 | 19 | 26% | 1.0 |
| P3 | 1 | 87 | 1% | <0.0001 |
Rescue of Fig 4−/− mice by null heterozygosity for Pip4k2c+/−
Pip4k2c−/− null males were crossed with Fig 4+/− females to produce double heterozygous offspring with genotype Fig 4+/−, Pip4k2c+/− (Fig. 2). A subsequent intercross between double heterozygotes generated 304 F2 mice. Mice were genotyped on P1. The yield of Fig 4 null mice in the F2 was 0, compared with the prediction of 1/16 or 19/304. The lack of Fig 4 null mice in this F2 is consistent with the postnatal yield of only 1/87 Fig 4 null mice in the cross between Fig 4 heterozygotes shown in Table 1. The combined yield of Fig 4 null mice from the two crosses was 1 mouse compared with the combined prediction of 41 null mice (19 from the F2 and 22 from Table 1).
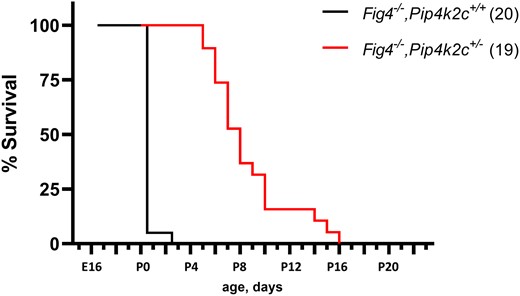
Effect of Pip4k2c on survival of fig 4 null mice. Fig 4 null mice with wildtype Pip4k2c are viable through prenatal life but do not survive after birth. (see also Table 1). Heterozygosity for the null allele of Pip4k2c rescues neonatal lethality, with mean survival of 8.2 days and maximum survival of 16 days.
The addition of the Pip4k2c null allele to the lethal Fig 4 null genotype resulted in 50% restoration of viability. The yield of F2 mice with genotype Fig 4−/−, PIP4k2c+/− was 22, compared with the prediction of 1/8 or 38/304. Comparing the two outcome of both crosses, the yield of Fig 4 null mice was increased from 1/41 to 22/38 by combination with the Pip4k2c null allele in the F2; this represents a significant increase in viability (P < 0.0001, Fisher's exact test).
Lifespan is extended in Fig 4−/−, Pip4k2c+/− mice
The average survival time of Fig 4−/−, Pip4k2c+/− mice from the F2 cross was 8.6 ± 3.2 days (n = 22), with maximum survival of 16 days (Fig. 2). In contrast, none of the Fig 4−/− mice from the F2 survived beyond P0. Heterozygosity for a null allele of Pip4k2c thus increases the postnatal viability of Fig 4 null mice (Fig. 2).
Pip4k2c+/− genotype also rescues enlarged lysosomes in mouse embryonic fibroblasts
To investigate the mechanism of rescued viability, we examined the appearance of lysosomes from mouse embryonic fibroblasts (MEFs) cultured at E13.5 from F2 mice. Most of the Fig 4 null cultured cells exhibit extensive enlargement of LAMP-positive, acidic vacuoles (Fig. 3a) (Ferguson et al. 2009; Lenk et al. 2019b). In contrast, many of the Fig 4−/−, PIP4k2c+/− fibroblasts lacked vacuoles (Fig. 3a). Quantitatively, 82% of MEFs from the Fig 4−/− mice exhibited extensive intracellular vacuolization while only 29% of MEFs from Fig 4−/−, Pip4k2c+/− mice contained enlarged vacuoles (Fig. 3b). The increased viability of the Fig 4−/−, PIP4k2c+/− mice is thus correlated with correction of the lysosome phenotype. The data demonstrate an effect of Pip4k2c haploinsufficiency on the lysosomal function of PI(3,5)P2 and provide a cellular basis for the increased viability of Fig 4−/−, Pip4k2c+/− mice.
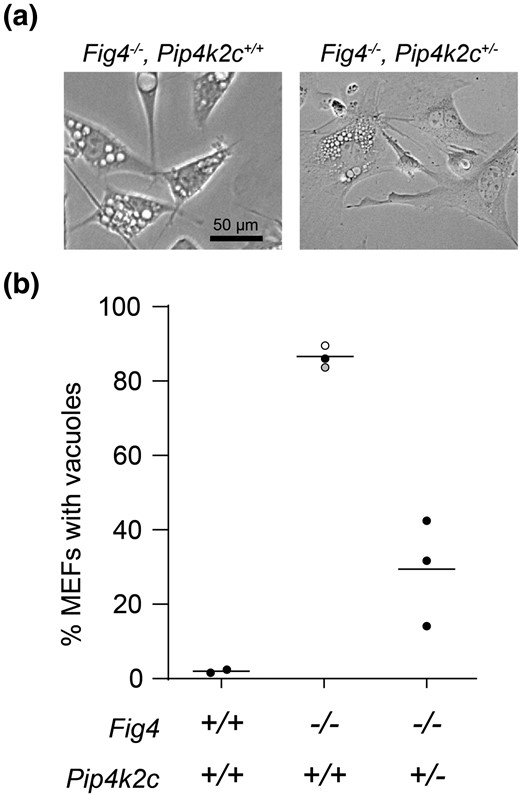
Heterozygous loss of Pip4k2c reduces vacuolization of fig 4 null MEFs. Mouse embryonic fibroblasts were isolated at day E13.5 and examined by phase contrast microscopy. (a) Each symbol in (b) represents the percent of vacuolated MEFs from one mouse, ≥ 200 cells per mouse. Solid symbols, this study; open symbol, Campeau et al. 2013; gray symbol, Lenk et al. 2016.
Discussion
The experiments described here demonstrate interaction between Fig 4 and Pip4k2c in regulation of lysosome function. The kinase PIP4K2C phosphorylates PI5P to generate PI(4,5)P2 (Fig. 1). Inhibition of PIP4K2C alters the relative proportions of these phosphoinositides (Al-Ramahi et al. 2017). The resulting elevation of PI(3,5)P2 may reflect the buildup of precursors due to the downstream block in PIP4K2C enzymatic activity. Alternatively, PIK4K2C is known to inhibit the kinase PIP5K by direct protein interaction (Wang et al. 2019). If PIP4K2C also inhibits the kinase PIKfyve, then relief of that inhibition in heterozygous null PIP4K2C cells could directly increase production of PI(3,5)P2. Regardless of the underlying mechanism, reduction of PIP4K2C provides an intervention for in vivo elevation of PI(3,5)P2.
Knockout of the phosphatase MTMR2 (Fig. 1) increases the concentration of PI(3,5)P2 in fibroblasts from Fig 4+/+ mice, but does not increase the concentration in fibroblasts from Fig−/− mice (Vaccari et al. 2014). The basis for the different effects in wildtype and mutant cells is not known. In vivo, the phenotype of Fig 4−/− mice was exacerbated by reduction of MTMR2 in the earlier work, in contrast to the improvement after reduction of the kinase PIP4K2C reported here. The different effects of reducing MTMR2 and PIP4K2C in fibroblasts and in mice may reflect their roles in metabolism of other substrates in this complex pathway.
In Fig 4 null fibroblasts, the level of PI(3,5)P2 is reduced to 50% of that of wildtype cells (Chow et al. 2007). The low level of PI(3,5)P2 results in lysosomal vacuolization and neonatal lethality. We have demonstrated that vacuolization and neonatal lethality can be corrected by 50% reduction of Pip4k2c via heterozygous knockout in Fig 4 null mice. This observation introduces a new paradigm for treatment of FIG4 deficiency.
Although striking, the beneficial effects in Fig 4−/−, Pip4k2c+/− mice are transient and maximal survival was observed at 2 weeks postnatal. This temporal limitation may be a consequence of the normal developmental increase in Pip4k2c expression that occurs between 1 and 2 weeks postnatal (Clarke et al. 2009). The developmental increase could elevate Pip4k2c level in the heterozygous null mouse beyond a critical threshold and eliminate the clearly beneficial effects seen during the first week of postnatal life. Alternatively, in vivo correction in neurons may be less efficient than observed in cultured cells.
The F2 cross that we carried out also generated double null mice with the genotype.
Fig 4−/−, Pip4k2c−/−. However, the very low yield of double null mice and their variable phenotype made it impossible to include them in the study.
The active site of the kinase Pip4k2c is a “druggable target” that has been investigated as a modifier of protein turnover and metastasis. The PIP4K2C inhibitor phenazopyridine is a widely used over-the-counter drug for treatment of urinary tract pain (Preynat-Seauve et al. 2021). The selective inhibitor NCT-504 alters the proportions of phosphoinositides in cultured cells and enhances autophagy; this inhibitor was identified in a screen for enhanced degradation of mutant huntingtin (Al-Ramahi et al. 2017). The covalent Pip4k2c inhibitor THZ-P1-2 causes defects in autophagy similar to those caused by inactivation of the gene (Sivakumaren et al. 2020). Our studies suggest that the new generation of pharmacological inhibitors of PIP4K2C could be applied to the PI(3,5)P2 deficiency disorders caused by mutations of FIG4 and VAC14.
We observed positive effects of reduced Pip4k2c in mice with a severe disorder due to complete loss of Fig 4. Since most patients have only partial loss of FIG4, the effectiveness might be greater and longer-lasting in the human disorders. Long-term rescue of the Fig 4 null mice was recently reported by gene replacement using viral delivery of the Fig 4 cDNA (Presa et al. 2021). The gene replacement did not completely restore normal expression, and combination of viral therapy with small molecule inhibitors of Pip4k2c might prove useful. Until gene replacement becomes available in the clinic, targeting of Pip4k2c offers an alternative for amelioration of the severe effects of Fig 4 deficiency.
Acknowledgements
We are grateful to Dr. L. C. Cantley for providing the Pip4k2c mutant mice.
Funding
This work was supported by the National Institutes of Health (Grant R01 GM24872) and by the CureCMT4J Foundation.
Data availability
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
Literature cited
Author notes
Conflict of interest statement The authors have no conflict of interest to report.

{kind=link}
{kind=link}
{kind=link}