





Abstract
This report describes the “benign” clinical course of a congenital long QT syndrome (LQTS) simulated acute coronary event in an 85 year old woman who had a history of recurrent syncope accompanied by numerous severe traumatic events from childhood. Her daughter died suddenly. LQTS was diagnosed on the basis of characteristic ECG findings, including a permanently prolonged QT interval, typical dynamic T-wave changes, and runs of torsades de pointes. A permanent DDDR pacemaker was implanted. Eighteen months after implantation there have been no further complaints of dizziness or syncope.
Introduction
Congenital long QT syndrome (LQTS) is associated with high risk of sudden death [1]. Risk stratification is based mainly on a history of syncope, torsades de pointes (TdP) or cardiac arrest [1]. LQTS is caused by mutations of cardiac ion channel genes [1,,2]. This report describes a “benign” clinical course of LQTS with potentially malignant clinical presentations.
Case report
An 85-year old woman was admitted to the emergency room after she was found unconscious at home. On admission she complained of severe dizziness, vomiting, headache and weakness. She migrated to Israel several months previously and had a history of recurrent syncope from childhood accompanied by numerous traumatic events with fractures of extremities and subdural haematoma. Her daughter, who had had no previous history of the disease, died suddenly during her sleep at the age of 50. The patient also had a history of diabetes mellitus, type II, and mild arterial hypertension. On physical examination her heart rate was 102 bpm, regular and blood pressure was 150/95 mmHg. With the exception of a prolonged QTC interval there were no pathological changes on the ECG (Fig. 1). Similar QTC prolongation was present on ECGs from previous years. Routine laboratory tests were normal. The patient was hospitalized in the internal medicine department. On the following day she complained of dizziness and chest discomfort. Significant dynamic QT and ST-T changes and several short runs of polymorphic VT were recorded on the ECG (Figs. 2 and 3). Cardiac enzyme levels were not elevated. Because an acute coronary event was suspected the patient underwent coronary angiography, which revealed 80% obstruction of the 1st marginal branch of the left circumflex artery without any changes in other vessels. Successful PTCA and stent insertion were performed. Normal heart size and normal global left ventricular function without any segmental contractility abnormalities were observed on the echo-Doppler examination. The additional five days of hospitalization were uneventful and the patient was discharged in a stable clinical condition with a diagnosis of an acute coronary event. Recommended therapy included: atenolol 12.5 mg, enalapril 10 mg and aspirin 100 mg. Twenty-four hour ambulatory ECG monitoring and myocardial perfusion imaging Tl-201 SPECT were performed one month after discharge due to complaints of dizziness and pre-syncope, but no arrhythmias or ischaemic patterns were observed and the mean heart rate was 64 bpm, range 42–74 bpm. Eight months later the patient was hospitalized with a fracture of the right pubic ramus after syncope at home. She explained that for the last several months she had had repeated episodes of dizziness and of near-syncope after emotional tension. The 12-lead ECG and monitoring strips recorded similar changes to those seen during the previous admission (Fig. 2). Congenital LQTS was diagnosed and a permanent DDDR pacemaker was implanted and programmed at a low rate of 80 bpm. Fourteen months after implantation the patient feels well, with no further complaints of dizziness or syncope. On 12-lead ECG, QT interval was 400 ms and QTC = 460 ms.
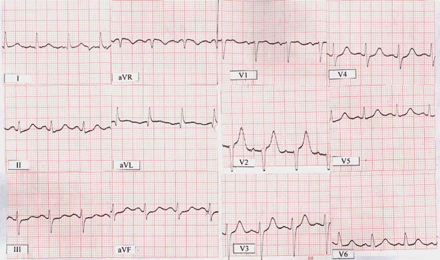
ECG on the admission shows sinus rhythm with a heart rate of 100 bpm, QT = 0.40–0.44 ms and QTC interval from 520–570 ms.
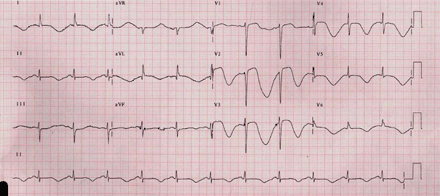
ECG recorded after complaints of dizziness and chest discomfort shows sinus rhythm, 72 bpm, QRS complex without any changes, resembling the QRS at the time of admission (Fig. 1). Significant QT and ST-T changes: extreme QT prolongation, QT = 720–760 ms and QTC interval from 789–830 ms, with concomitant giant, inverted T-waves, beat-to-beat T-wave changes – “macroscopic T wave alternans” – especially in right chest leads.
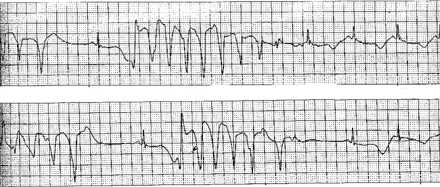
Example of monitoring ECG lead. Intermittent runs of non-sustained torsades de pointes, ventricular tachycardia and sinus rhythm with an extremely long QT interval and beat-to-beat T changes.
Discussion
This is a very unusual course (we defined it as “benign”) of congenital LQTS where the patient reached the age of 85 years despite multiple malignant clinical presentations resulting in fractures and subdural haematoma. Notwithstanding the clinical presentation, as well as the ECG between and immediately after syncope spells, showed well-known signs of LQTS, the diagnosis had been missed on numerous occasions and over many decades.
The diagnosis of LQTS was based on ECG findings, including permanently prolonged QT interval, morphology of T-waves with typical dynamic changes, runs of TdP, a long history of recurrent attacks of dizziness and syncope with multiple traumatic lesions, and a family history of sudden death. Such an ECG pattern on admission (Fig. 1) is often seen in patients with the LQT1 genotype, which is caused by KVLQT1 IKs potassium channel gene mutations [1,,2]. Genetic abnormalities (chromosome 11p15.5) that interfere with K+ conductance will result in prolongation of the QT interval due to impaired egress of K+ ions, thereby prolonging the action potential duration. The LQT1 group of patients have significantly more frequent and higher cumulative probability rate of cardiac events than other genotypes [2]. By the age of 15 years, 53% of the LQT1 subjects have had a first cardiac event, a rate significantly more frequent than the other genotypes [2]. In addition, patients from the LQT1 group are five times more likely to experience a cardiac event by the age of 40 than the LQT3 mutants; however, fatality during a cardiac event is significantly higher in patients with LQT2-3 [1,,2]. A history of exercise, emotionally triggered or shock-induced syncope has been commonly observed in patients with LQT1 [1,,2].
The patient had no identifiable reason for QT prolongation [1,,3,,4]. Suspicion that QT prolongation and arrhythmias could have been triggered by ischaemia, was ruled out after PTCA had been performed. Furthermore, a history of recurrent attacks of syncope from early childhood, retrospective analysis of ECGs and clinical presentation during the last two admissions, made the diagnosis of an acute ischaemic or neurological event as the cause of LQTS very unlikely.
Basically there are two approaches to the treatment of congenital LQTS: pharmacological and invasive. Due to a long history of syncopal attacks, accompanied by multiple traumas and relative bradycardia, treatment with β-blockers was not considered. The invasive approach includes extirpation of left [5] or right [6] stellate ganglion, ICD or permanent pacemaker implantation [7], and ablation [8]. The beneficial effects of pacing in high-risk LQTS patients are probably related to the prevention of bradycardia, pauses, and the shortening of long QT intervals, factors that are known to be arrhythmogenic. Permanent pacing was considered as an appropriate therapy in this patient due to bradycardia, long–short intervals recorded before runs of TdP and no history of aborted sudden death (a somewhat “benign” course of the disease). Our decision is supported by an uneventful eighteen-month follow-up.

{kind=link}
{kind=link}
{kind=link}