





Abstract
Athletes are predisposed to atrial arrhythmias but the association between intense endurance exercise training, ventricular arrhythmias (VAs), and sudden cardiac death is less well established. Thus, it is unclear whether the ‘athlete’s heart’ promotes specific arrhythmias or whether it represents a more general pro-arrhythmogenic phenotype. Whilst direct causality has not been established, it appears possible that repeated exposure to high-intensity endurance exercise in some athletes contributes to formation of pro-arrhythmic cardiac phenotypes that underlie VAs. Theories regarding potential mechanisms for exercise-induced VAs include repeated bouts of myocardial inflammation and stretch-induced cellular remodelling. Small animal models provide some insights, but larger animal and human data are sparse.
The current clinical approach to VAs in athletes is to differentiate those with and without structural or electrical heart disease. However, if the athlete’s heart involves a degree of pro-arrhythmogenic remodelling, then this may not be such a simple dichotomy. Questions are posed by athletes with VAs in combination with extreme remodelling. Some markers, such as scar on magnetic resonance imaging, may point towards a less benign phenotype but are also quite common in ostensibly healthy athletes. Other clinical and invasive electrophysiology features may be helpful in identifying the at-risk athlete.
This review seeks to discuss the association between athletic training and VAs. We will discuss the potential mechanisms, clinical significance, and approach to the management of athletes with VAs.
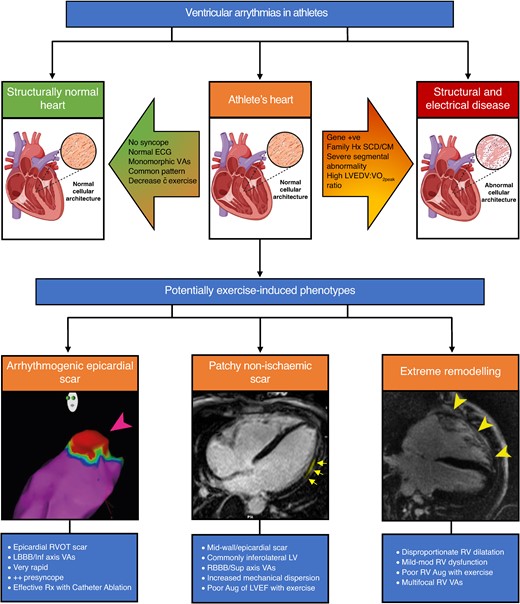
Ventricular arrhythmias in athletes. VAs can be found in athletes with structurally normal hearts, those with underlying structural/electrical heart disease or athlete’s heart. Various factors can help differentiate benign athletic remodelling from structural disease. Under the umbrella term of athlete’s heart are potentially exercise-induced phenotypes including arrhythmogenic epicardial scar, patchy non-ischaemic scar (e.g. non-ischaemic LV scar), and extreme remodelling (e.g. exercise-induced arrhythmogenic cardiomyopathy). ECG, electrocardiogram; VAs, ventricular arrhythmias; SCD, sudden cardiac death; CM, cardiomyopathy; LVEDV, left ventricular end-diastolic volume; VO2Peak, absolute peak VO2; RVOT, right ventricular outflow tract; LBBB, left bundle branch block; Rx, treated; LV, left ventricle; RBBB, right bundle branch block; Aug, augmentation; LVEF, left ventricular ejection fraction; RV, right ventricle. Image created with BioRender.com.
There are specific pro-arrhythmogenic cardiac phenotypes in athletes, some of which may be exercise-induced.
New surrogate measures of outcomes are required as clinical arrhythmia events (e.g. SCD) are unlikely to be met in the sample sizes typical of sports cardiology studies.
The addition of electroanatomic mapping to electrophysiology studies in athletes may assist in uncovering ‘hidden’ arrhythmogenic substrate.
Introduction
Whilst the cardiovascular benefits of low- to moderate-intensity exercise are well documented,1 some arrhythmias are more prevalent among athletic populations. Atrial fibrillation (AF) and flutter, for example, are more common in endurance athletes with high-intensity endurance exercise associated with an increased risk of AF,2–6 up to five-fold.7 However, this association is complex with low- and moderate-intensity exercise reducing the risk of AF8,9 and the threshold limit for the intensity and duration of physical activity that increases rather than reduces AF risk remains unknown.10 Proposed mechanisms for increased AF in endurance athletes include left atrial (LA) structural remodelling,11,12 exercise-induced inflammation and fibrosis,13–15 elevated LA pressure16,17 and systolic blood pressure during exercise,18 increased vagal tone,19 and possibly background genetic susceptibility (i.e. polygenic risk).
High-intensity endurance exercise may also be associated with increased risk of ventricular arrhythmias (VAs), although the dose-response relationship remains poorly understood. Diagnosis and management of VAs (including premature ventricular complexes, PVCs; ventricular tachycardia, VT, and ventricular fibrillation, VF) in athletes remains a conundrum for sports clinicians with a range of presentations from asymptomatic benign ectopy to sudden cardiac death (SCD). Tragically, we continue to encounter cases of unexpected SCD in young healthy athletes despite intensive pre-participation investigation. As illustrated by the case in Figure 1, investigations in endurance athletes often result in findings that would be considered abnormal in a non-athletic cohort. For example, reduced ejection fraction, reduced longitudinal strain, or delayed enhancement on cardiac magnetic resonance (CMR) are all findings that are prevalent within endurance athlete cohorts. Claessen et al.20 recently reported that one in six elite endurance athletes had a reduced ejection fraction, often in combination with small areas of scar of uncertain significance. This observation presents the conundrum of excluding athletes with variant findings from competition or allowing them to continue with careful observation. Whilst the answer to this difficult question remains contentious and approaches differ between countries, it is clear that we require a better understanding of the mechanisms and predictors of VAs in athletes.
![Twenty-three-year-old male elite cyclist with sudden cardiac death during competition. Investigations are presented for an asymptomatic elite cyclist after an initial screening 12-lead electrocardiogram (ECG), top left. T-wave inversion in V2 (a common but potentially abnormal athletic trait) prompted investigation with a transthoracic echocardiogram (TTE) on which moderate to severe chamber dilation and low-normal systolic function was identified (top right). A 24 h Holter monitor showed 370 premature ventricular complexes (PVCs) per 24 h, two couplets and one triplet [shown is a fusion beat followed by two left bundle branch block (LBBB) morphology PVCs]. Exercise stress test showed three PVCs at peak exercise [shown is one of these PVCs with right bundle branch block (RBBB) morphology]. Cardiac magnetic resonance (MRI) demonstrated athletic remodelling with dilatation of all chambers, mildly reduced LV and RV function and a small area of epicardial late gadolinium enhancement (LGE, scar) in the apical anterolateral LV wall (arrow). The athlete remained asymptomatic until suffering a cardiac arrest during competition several months after these investigations were completed. LVEDVi, left ventricular end-diastolic volume indexed to body surface area; LVESVi, left ventricular end-systolic volume indexed to body surface area; RVEDVi, right ventricular end-diastolic volume indexed to body surface area; RVESVi, right ventricular end-systolic volume indexed to body surface area; LVEF, left ventricular ejection fraction; RVEF, right ventricular ejection fraction; HR, heart rate.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/europace/26/12/10.1093_europace_euae279/8/m_euae279f1.jpeg?Expires=1747852837&Signature=CKVR2DK31BQ84Fg~9LDvxJuEaFdgswFQ0xxO5MMwG4za3QuvXNf0d8LozF8UlM9up-foZpWZBrQv-y1CMuiYB8xyM5SGoHM5wBgG4HmCNd04Fq5wBTCHcAjrlRZAe5U6u4H7KX5TjKohnk--bB9C03H8aJLK70CyYu-wNX1yL3p64iQgtNH0~F27JKEd4oBKPDwi~3cYz1IXX8VAEQnDlSJ4RDu2EstVKD2QF4K9o8YzIBQy~k3hxBBkVRiO6FoI6DuEcYd6RuTHJacE~5DclWHAuJplwAWdTgq7SMGgWGsgJKafIVvGqihH0Kv-eWNkGFu9w43LE-4EzTxdYW44mg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Twenty-three-year-old male elite cyclist with sudden cardiac death during competition. Investigations are presented for an asymptomatic elite cyclist after an initial screening 12-lead electrocardiogram (ECG), top left. T-wave inversion in V2 (a common but potentially abnormal athletic trait) prompted investigation with a transthoracic echocardiogram (TTE) on which moderate to severe chamber dilation and low-normal systolic function was identified (top right). A 24 h Holter monitor showed 370 premature ventricular complexes (PVCs) per 24 h, two couplets and one triplet [shown is a fusion beat followed by two left bundle branch block (LBBB) morphology PVCs]. Exercise stress test showed three PVCs at peak exercise [shown is one of these PVCs with right bundle branch block (RBBB) morphology]. Cardiac magnetic resonance (MRI) demonstrated athletic remodelling with dilatation of all chambers, mildly reduced LV and RV function and a small area of epicardial late gadolinium enhancement (LGE, scar) in the apical anterolateral LV wall (arrow). The athlete remained asymptomatic until suffering a cardiac arrest during competition several months after these investigations were completed. LVEDVi, left ventricular end-diastolic volume indexed to body surface area; LVESVi, left ventricular end-systolic volume indexed to body surface area; RVEDVi, right ventricular end-diastolic volume indexed to body surface area; RVESVi, right ventricular end-systolic volume indexed to body surface area; LVEF, left ventricular ejection fraction; RVEF, right ventricular ejection fraction; HR, heart rate.
For this literature review, we performed a search of four databases (PubMed, Ovid-MEDLINE, Embase, and Google Scholar) for English language peer-reviewed publications from January 1990 to August 2024. Reference lists of these articles were reviewed to identify any other relevant articles. Both clinical and experimental studies were included. Studies with unavailable full text and publication in a language other than English were excluded. Our search included the titles, abstracts and medical subject headings (MeSH) using the following search terms: athlete, endurance athlete, ventricular arrhythmia, premature ventricular complex, non-sustained ventricular tachycardia, ventricular tachycardia, sudden cardiac death, arrhythmogenic cardiomyopathy, electrophysiology study, and electroanatomic mapping. Two investigators (P.D. and A.L.G.) completed the literature search independently. Using the above mentioned terms, we initially found 380 hits. After exclusion, we ended up with a total of 250 studies of which five were systematic reviews and meta-analyses.
Our aim in this review is to critically appraise the epidemiology, the proposed mechanisms, and the current clinical approach to the management of VAs in athletes. In addition, we propose a novel characterisation of potentially exercise-induced arrhythmogenic cardiac phenotypes and provide perspective as to when an electrophysiology study (EPS), including electroanatomic mapping (EAM) and an epicardial approach, should be considered.
Epidemiology
It has been argued that SCD may be as much as three times more common in athletes during their competitive years as compared with non-athletic individuals.21 However, this has not been a universal observation and some groups have reported a similar or reduced incidence of SCD among competitive athletes.22,23 Existing data suggest that VAs are frequently detected in ostensibly healthy athletes and are generally associated with a benign prognosis.24,25 In addition, most studies have reported a similar prevalence of frequent or complex VAs between athletes and non-athletes.26–29 For example, two recent 24 h 12-lead Holter studies compared the population prevalence of VAs [defined as >10 isolated PVCs or ≥1 complex VA—PVC couplets, triplets, or non-sustained VT (NSVT)] in young and middle-aged athletes30,31 with age- and sex-matched non-athletic referents. There was no difference found in either study (respectively 10% vs. 11%, P = 0.81 in young athletes; 26% vs. 23%, P = 0.53 in middle-aged athletes). In another study by Graziano et al.,32 the prevalence and complexity of VAs did not differ between athletes and sedentary controls and were not related to the type of sport or sex of the athlete.
Female athletes are known to have a 5–10-fold lower incidence of SCD than males, despite the prevalence of predisposing cardiomyopathies and channelopathies being similar.22,33 Whilst the cause of these sex-differences in malignant arrhythmias remains unexplained, biological differences in athletic remodelling may be at least partially responsible. For example, female athletes are known to exhibit lower LV wall thickness and mass than men, even when adjusting for body size and training exposure.34,35 Notably, women are under-represented among athletes presenting with symptoms of VAs who have right ventricular (RV) abnormalities suggestive of arrhythmogenic cardiomyopathy (ACM).36–40 A majority of exercise-related SCD in young females is not associated with structural heart disease (SHD), suggesting that other underlying conditions such as inherited channelopathies could be responsible.41 Limited available evidence suggests a similar prevalence of VAs between the sexes,42,43 although all existing studies of VAs in athletes consist of predominantly male cohorts. There also appears to be racial differences in SCD with Black/African-American athletes44 and non-athletes45 at higher risk. The reasons for this are not clear. However, future research may interrogate differences in remodelling (e.g. LV wall thickness) and variants in repolarisation patterns as potential avenues of inquiry.46
Whilst there is good evidence that intensive endurance exercise is associated with significant and often profound cardiac remodelling,20,36 prospective and case–control studies in strength-trained athletes report only modest remodelling.47,48 Studies on arrhythmias in athletes, however, have found no association between VAs and the type of sport, hours of training per week, and years of sports activity.30–32 In addition, participation in endurance sports does not appear to be associated with increased risk of VAs compared to sports with comparatively lower cardiovascular demand (e.g. ‘skill’ sports like shooting).49 Studies on the acute effects of endurance exercise report electrical instability of the ventricular myocardium (increased PVCs and prolonged QTc), acute right-sided electrocardiogram (ECG) changes, acute reduction in RV function, and increase in biomarkers, suggesting myocardial injury.50–52 Recent studies of marathon athletes, however, have not demonstrated such acute effects translating into clinically significant VAs during exercise.53,54 In summary, VAs are not uncommon amongst healthy athletes. However, evaluation of those with VAs remains a priority given that it is a potential marker of VT and SCD.
Contemporary approach to VAs in athletes
The general approach to athletes with VAs is similar to non-athletes with the main goal being the differentiation between structurally normal hearts and hearts with underlying structural or electrical disease. A comprehensive clinical assessment is paramount including a detailed family history (FHx) of cardiomyopathy (CM) and/or premature SCD (men < 40 years and women < 50 years), and analysis of autopsy reports with review of relevant investigations. Electrocardiogram interpretation in athletes can be challenging due to the high prevalence of ST-T wave and other abnormalities.55 Current consensus is that any athlete with ≥2 PVCs on resting ECG [or ≥1 PVC in the case of high-endurance athletes, positive FHx of premature SCD/CM, relevant symptoms, associated ECG abnormalities, uncommon PVC morphology (discussed below), and/or short coupling interval—arbitrarily defined as interval from onset of preceding QRS to onset of PVC < 350 ms56] undergo further investigation with Holter monitoring, exercise stress testing (EST), and/or suitable imaging [i.e. transthoracic echocardiography (TTE), cardiac computed tomography, and/or contrast-enhanced CMR].55,57 Holter monitors should include an exercise session performed at usual intensity during the period of monitoring.58
Traditionally, a high PVC burden has been associated with increased probability of SHD and risk of SCD59 with burden (>500/24 h) still a minor Task Force Criteria (TFC) for ACM diagnosis.60 Data from athletic and non-athletic populations, however, suggest that a high burden can be associated with a benign prognosis (when PVC-induced CM is excluded).61,62 In a recent review, Corrado et al.58 propose a diagnostic algorithm for PVCs in athletes where PVCs are classified as ‘common’ or ‘uncommon’ based on QRS morphology, response to exercise training, complexity, coupling interval, and clinical findings including FHx. Burden is not included as a relevant prognostic marker. Common morphologies are defined as outflow tract/infundibular (left bundle branch block morphology and inferior axis) and fascicular [typical right bundle branch block (RBBB) morphology, superior/inferior axis, and QRS < 130 ms]. Athletes with ‘uncommon’ PVCs, despite unremarkable clinical assessment (including Holter monitoring, maximal EST, and TTE), are advised to undergo CMR and/or genetic testing. Many of the above recommendations are based on retrospective data evaluating the characteristics of VAs that predict underlying SHD in athletes.30,62,63
Several recent studies support the use of the above algorithm. For example, in 112 young competitive athletes referred for PVCs, 23% had a definitive diagnosis of cardiac disease correlating with ‘uncommon’ PVC morphology (P < 0.001) and VA complexity (P < 0.001). However, PVC burden was actually lower in these athletes compared to athletes without underlying disease.64 In 251 competitive athletes who underwent CMR for evaluation of VAs, presence of late gadolinium enhancement (LGE) was independently correlated with PVCs with multiple morphologies or atypical RBBB and intermediate/superior axis (i.e. uncommon) configuration (P < 0.001), NSVT at peak exercise (P = 0.002), and a lower PVC burden vs. athletes without a positive CMR (P < 0.001).65 In 205 Olympic athletes followed-up over 6 years, evaluation of PVCs identified cardiac disease requiring disqualification or serial follow-up in 12% with ‘uncommon’ vs. 1% with ‘common’ patterns.42
The current paradigm in athletes with VAs, therefore, is to focus not on burden but on ‘uncommon’ VA characteristics as a predictor of underlying pro-arrhythmic conditions.58 Athletes who have unremarkable first-line investigations, ‘common’ VA patterns and no red flag features, such as unexplained syncope or positive FHx of SCD, can be managed as having benign idiopathic VAs and are eligible for competitive sports. When underlying conditions are diagnosed, age is the main determinant of differential diagnosis. In athletes aged ≥35 years, the main cause of SCD is atherosclerotic coronary artery disease (CAD),66 though it remains unknown whether this also represents a significant cause of substrate underpinning propensity to VAs in this age group. In those aged <35 years, more common diagnoses have traditionally included primary electrical disease, inherited CMs, or anomalous origin of the coronary arteries.67 Interestingly, in a recent European study,68 the most prevalent cause of sports-related SCD in young adults aged 18–35 years was CAD, mainly through acute coronary syndrome.
Traditional pathological substrates for VAs in athletes
In addition to primary electrical disease, the main types of SHD to consider in athletes with VAs are hypertrophic cardiomyopathy (HCM), ACM, dilated cardiomyopathy (DCM), and ventricular fibrosis of unknown aetiology. Diagnosis of HCM in the athlete can be complicated by athletic hypertrophic cardiac remodelling, although wall thickness is rarely >12 mm in Caucasians and >14 mm in Black/African-American athletes.69 Cardiac magnetic resonance can aid diagnosis, with fibrosis present in ∼75% of HCM patients. Although debate persists regarding the degree to which the presence of scar aids individual risk prediction, it may be helpful as an arbiter in cases where clinical factors lead to a borderline indication and to aid in shared decision making about prophylactic implantable cardioverter defibrillator (ICD) implantation.70
Arrhythmogenic cardiomyopathy is a contemporary term used to encompass arrhythmogenic right ventricular cardiomyopathy and left-dominant forms of the disease characterised by myocardial scar and a predisposition to VAs. It is traditionally considered an inherited cardiomyopathy although phenocopies have been described in athletes that seem to be rarely associated with familial disease.37–40 It classically manifests as VAs triggered by exercise or adrenergic stress and can result in SCD as the initial presentation of disease.66,71 Diagnosis is currently based on probabilistic TFC that encompass electrophysiological, anatomical, functional, and clinical features of the disease.60 An update to the diagnostic criteria for ACM has recently been proposed, incorporating both right- and left-sided ACM phenotypes and tissue characterisation using CMR.72 Arrhythmogenic cardiomyopathy accounts for a significant proportion of SCD in young athletes with regular, high-intensity exercise and competitive sports associated with acceleration of disease and increased risk of VAs.73
Ventricular arrhythmias are common in DCM, particularly when occurring due to prior myocarditis or in association with lamin A/C mutations.74 Differentiation from athlete’s heart can be challenging as 15% of athletes participating in extreme endurance sports, such as cycling, have physiological LV dilatation that may be associated with concomitant reduction in LV ejection fraction.20,75,76 Normal/supranormal contractile reserve and absence of LGE on CMR favours athlete’s heart.20,75,77 Indexing LV end-diastolic volume (LVEDV) to absolute VO2peak has recently been shown to improve the ability to differentiate physiological and pathological LV enlargement and the LVEDV:VO2peak ratio and may prove useful in differentiating DCM from athlete’s heart.78
In athletes with a clear disease phenotype, such as ACM or HCM, genetic testing is fundamental to diagnosis, risk stratification, and family screening. In DCM phenotypes, too, discovery of pathogenic variants with a higher risk of life-threatening arrhythmias, such as lamin A/C or filamin C, mitigates restriction from all competitive or high/very-high-intensity recreational exercise, closer follow-up, and often earlier implantation of prophylactic ICD.41 Recent work is focusing on polygenic risk scores (PRS) as a possible means of identifying athletes at risk of adverse cardiac outcomes. For example, in a novel study of 281 elite endurance athletes (22 ± 8 years, 80% male) using a validated PRS previously associated with DCM,79 Claessen et al.20 found reduced ejection fraction (EF) in approximately one in six elite endurance athletes that was associated with reduced myocardial strain, increased PVC frequency, and a higher PRS (corresponding to an enrichment of background genetic variation similar to that seen in patients with DCM). Whilst none of these athletes developed heart failure or sustained arrhythmias over 4.4 years follow-up, the association between a high PRS, reduced EF, increased PVC frequency, and one case of SCD (in an athlete with all of these features) warrants further scientific scrutiny.
Emerging concepts in arrhythmogenic remodelling in athletes
Athlete’s heart is a commonly used term to describe exercise-induced structural, functional, and electrical cardiac remodelling promoted by the volume and pressure loads of exercise. The most profound changes are generally observed in endurance sports such as rowing and cycling, which encompass both dynamic and static activity over prolonged periods. However, there is often significant individual variation in cardiac remodelling between athletes with similar training volumes.36 The reasons for this remain poorly understood.
Exercise-induced ACM (EIACM), an entity first described by Heidbuchel et al.38 is hypothesised to be an extreme form of athletic remodelling with prevalent RV dilatation, RV dysfunction, and VAs in endurance athletes. A direct causal association between endurance exercise and EIACM has not been established. Furthermore, understanding of the phenotypic spectrum, prognosis, and specific management of EIACM continues to evolve. Nonetheless, the putative entity of an arrhythmogenic substrate unique to athletes is referenced in recent athlete guideline statements including the HRS Expert Consensus Statement on the management of athletes with arrhythmias.80 Despite phenotypical overlap with ‘classic’ ACM, where genetic testing reveals pathogenic variants in cardiac desmosome proteins in 40–50% of index cases,81 EIACM is typically gene-negative and there is usually no family history of disease.37,40 Patients report a history of repeated exposure to high-intensity endurance exercise. It is not clear whether there are other modifying factors such as bouts of sub-clinical myocarditis or genetic influences that are yet to be elucidated, including novel monogenic, oligogenic, or polygenic causes.
Exercise-induced ACM is thought to have phenotypic heterogeneity that can overlap with the ‘healthy’ athlete’s heart including precordial T-wave inversion, RV dilatation, and low-normal RV function.51,82–85 Lie et al.85 analysed 43 high-performance athletes (mean age 45 years, 84% male) with idiopathic VAs and found that athletes with VAs had worse RV function (measured by TTE-derived RV free wall strain) and more LGE as compared with 30 healthy athlete controls (mean age 41 years, 93% male). Those athletes with life-threatening VAs had worse LV function (LV global longitudinal strain) and greater variance in the timing of regional contraction (measured as mechanical dispersion), as compared to athletes with less severe VAs. This suggests that presence of LV dysfunction may indicate a more life-threatening phenotype whilst RV dysfunction alone, which can be more subtle and overlap with physiological athletic remodelling, may be associated with milder arrhythmias.
Recent work on EIACM has evaluated the diagnostic and prognostic value of RV free wall strain, RV and LV mechanical dispersion, VA inducibility during EPS, EAM to identify low voltage areas, LGE on CMR, and inducible RV dysfunction with exercise.38,39,85–89 However, there is a lack of consistency in these findings and differentiation of EIACM from normal athletic remodelling remains a significant challenge.90
Complicating the spectrum of athletic remodelling is a recent description by Venlet et al.39 of a rapid and highly symptomatic RV outflow tract (RVOT) tachycardia in longstanding high-level endurance athletes associated with localised anterior sub-epicardial RVOT scar. None in this group fulfilled diagnostic criteria for ACM or sarcoidosis, although it remains possible that this may also be a variant of EIACM. Epicardial EAM was performed if previous endocardial ablation had failed or an epicardial substrate suspected based on endocardial voltage and/or activation mapping. This substrate, which was hidden on CMR and only evident after endocardial unipolar or epicardial bipolar voltage mapping, highlights the superiority of EAM to CMR for detection of RV scar.91 Corrado et al.92 similarly used EAM in 27 patients (9 of whom were competitive athletes) with RVOT tachycardia and apparently normal hearts to identify low voltage regions in the RVOT and guide endomyocardial biopsy (EMB) for confirmation of ACM cases in which other disease features were absent or subtle. These studies raise the interesting discussion as to whether there should be a different threshold for progression to epicardial evaluation and treatment in athletes with VAs as compared with non-athletes.
Since the original description of EIACM,38 there has been interest in other phenotypes of athletic remodelling predisposing to VAs and SCD, particularly demonstrated by the increasing availability and quality of CMR imaging. Ventricular fibrosis of unknown aetiology, most commonly non-ischaemic LV scar (NILVS), describes a localised pattern of mid-myocardial/epicardial scar, most commonly in the inferolateral LV, which has been reported in highly-trained endurance athletes.87,88 Patients with this condition (as exemplified in Figure 1) are typically asymptomatic and have a normal ECG and TTE due to the localised nature of the fibrosis. Contrast-enhanced CMR is required to make the diagnosis. Non-ischaemic LV scar has been associated with life-threatening VAs and SCD in athletes with the majority having an atypical RBBB morphology, superior axis pattern (consistent with exit from an inferolateral LV scar).88,93,94 Despite this, prognosis appears variable with one study reporting that 22% of athletes (80% men, mean age 33 years) with NILVS experienced malignant VAs over a 3-year follow-up88 whilst the same group found no malignant VAs amongst another cohort of athletes with NILVS during a 2.5-year follow-up.95 It remains unclear why some athletes have NILVS whilst others with comparable exercise exposure do not.
Other studies have reported different exercise-induced scar patterns including isolated LGE at the insertion point of the RV with the ventricular septum. This ‘hinge-point’ pattern is not uncommonly found in endurance athletes and is thought to be related to the duration and intensity of exercise.96 Whilst hinge-LGE appears to be equally prevalent in athletes with and without arrhythmias88 and, to date, has not been associated with VAs and SCD,51,97,98 definitive data on prognosis in athletes with this phenotype are lacking.
Thus, VAs can be observed in athletes within the spectrum of exercise-induced remodelling that extends from relatively mild to quite extreme, without clear markers for when the remodelling may be considered more arrhythmogenic. In other words, various potentially exercise-induced phenotypes that can predispose to VAs may occur under the diagnostic umbrella of what is termed ‘athlete’s heart’ and it is important that sports clinicians are aware of these phenotypes, their identifying features, diagnosis, and optimal management strategies (see Graphical Abstract).
Cellular mechanisms of VAs in athletes
During exercise, concurrent activation of the sympathetic nervous system and suppression of the parasympathetic nervous system leads to changes in heart rate, atrioventricular (AV) nodal conduction, and inotropy. In cardiomyocytes, noradrenaline binds to β-adrenergic receptors and enhances the inward calcium (Ca2+) current (L-type Ca2+ channel, ICaL) leading to loading of the sarcoplasmic reticulum.99–101 Sarcoplasmic reticulum Ca2+ content is further increased by physiological adaptation of Ca2+ homeostasis underlying the increased cardiac output demand during exercise. Sarcoplasmic reticulum Ca2+ loading can result in spontaneous leakage of Ca2+ causing enhanced triggered activity. Noradrenaline also activates the slow delayed rectifier potassium channel (IKs) to shorten the QT interval and balance the QT-prolonging effect of an enhanced ICaL during exercise.99–101 Myocardial fibrosis, which is associated with underlying SHD predisposing to VAs in athletes, forms a critical substrate for conduction slowing and block, promoting reentry.102,103 Fibrosis-related uncoupling of myocardial tissue also favours increased probability of triggered arrhythmias.104,105 These mechanisms for VAs in athletes are summarised in Figure 2.
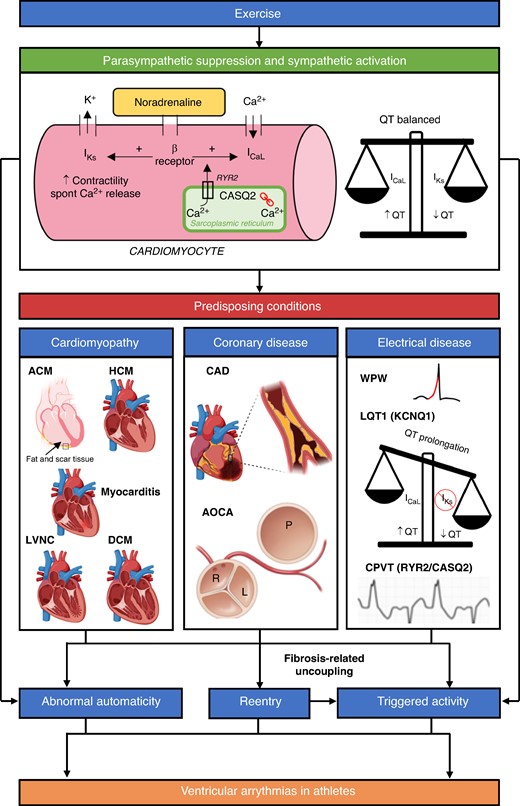
A schematic of some of the proposed mechanisms for VAs in athletes. Exercise activates the sympathetic nervous system (SNS) and suppresses the parasympathetic nervous system. Binding of noradrenaline to cardiomyocytes and physiological adaptation of Ca2+ homeostasis promotes increased triggered activity. SNS activation also leads to abnormal automaticity. Concomitant activation of IKs ensures the QT is balanced. Increase in cardiac impulse formation via the above mechanisms can lead to idiopathic VAs in structurally normal hearts. Reentry is promoted by underlying myocardial fibrosis associated with cardiomyopathies and coronary anomalies. Some idiopathic VAs (e.g. LV fascicular VT) are also caused by reentry. Reentry itself can lead to triggered activity via fibrosis-related uncoupling. The combination of abnormal cardiac impulse formulation and reentry can lead to malignant VAs and SCD in athletes with predisposing conditions. Electrical diseases also cause VAs via other mechanisms. Long QT Syndrome type 1 (LQT1) syndrome is caused by pathological variants in the KCNQ1 gene whilst catecholaminergic polymorphic ventricular tachycardia (CPVT) is caused by pathogenic variants in Ryanodine Receptor 2 (RYR2, autosomal dominant, 50%) and Calsequestrin 2 (CASQ2, autosomal recessive, 2%) genes. Please note that this schematic does not include other proposed mechanisms for VAs in athletes such as stretch-activated ion channel expression and stretch-mediated gene expression. K+, potassium; Ca2+, calcium; β Receptor, β adrenergic receptor; IKs, slow delayed rectifier potassium channel; ICaL, L-type Ca2+ channel; ACM, arrhythmogenic cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVNC, left ventricular non-compaction cardiomyopathy; DCM, dilated cardiomyopathy; CAD, coronary artery disease; AOCA, anomalous origin of the coronary arteries; WPW, Wolff–Parkinson–White syndrome. Image created with BioRender.com.
In addition to ionic changes induced by the autonomic nervous system, it is likely that mechanical stretch of the myocardium, both acutely and chronically, also contributes to triggering of arrhythmias in athletes. This is thought to be related to activation of specific mechano-sensitive ion channels as well as sodium and Ca2+ entering cardiac cells via non-selective ion channels.106 Chronic stretch on the heart also activates gene expression in cardiomyocytes and non-myocytes leading to hypertrophic remodelling via activation of factors such as angiotensin II,107,108 endothelin-1,109 and basic fibroblast growth factor.110 Hypertrophy, in turn, may contribute to electrical instability by increasing the sensitivity of mechano-electric feedback.106
Whilst existing data from small animal studies provide useful insights, understanding of additional mechanisms for VAs in human athletes remains elusive. Differences in key electrophysiological parameters, such as repolarisation properties and pattern of ion channel expression, limit conclusions that can be made from these small animal models to the human heart.111–113 Larger animal models would likely have better translational value though dedicated human studies comparing electrophysiological changes and underlying substrate in athletes with and without arrhythmias is the ‘Holy Grail’.
Recently, Polyák et al.114 analysed a canine model mimicking a 4-month elite athlete exercise regimen with trained dogs demonstrating similar remodelling to human endurance athletes. They found prolongation of cardiac repolarisation (prolonged QT in vivo and prolonged LV cardiac action potential duration in vitro) in trained dogs with the possible underlying mechanism being a reduction in magnitude of Ito current in mid-myocardial myocytes. In addition, trained dogs had significantly higher levels of LV fibrosis, increased HCN4 protein expression (HCN channels are thought to contribute to increased arrhythmogenic activity in human HCM and heart failure115), and lower intrinsic heart rate, a finding emulated in human athlete studies.116,117
Mechanisms of non-ischaemic LV scar and RVOT substrate in athletes
Mechanisms underlying patchy NILVS found in human athletes also remain under debate. Some autopsy studies, for example, suggest that NILVS is secondary to exercise-induced inflammation118 whilst others suggest that it is an LV-variant of ACM with a possible genetic aetiology.118–120 Other experts propose NILVS is a result of sub-clinical infectious myocarditis where continuation of high intensity and volume exercise results in fibrosis as shown in animal models.121–123 Other studies suggest a possible haemodynamic mechanism with cumulative and repeated exposure to exercise-induced systolic hypertension.124 The exercise-induced inflammation theory has merit given evidence of acute myocardial injury following intensive endurance exercise50,51 and association between exercise-induced cardiac dysfunction and increased expression of pro-inflammatory cytokines.125 Infectious myocarditis is, however, frequently asymptomatic126 making differentiation between this and exercise-induced inflammation extremely difficult. In addition, other factors such as toxicity of performance-enhancing drugs and genetic predisposition have not been individually assessed.
Small animal models have also demonstrated that sustained intensive endurance exercise training induces potentially adverse myocardial remodelling and arrhythmogenic substrate. For example, in a seminal study by Benito et al.,127 16 weeks of vigorous running in rats induced cardiac fibrosis, changes in ventricular function, atrial dilatation, and increased arrhythmia inducibility at EPS. Interestingly, the fibrotic changes caused by this ‘long-term’ endurance training reversed after an 8-week exercise cessation. Reduction in frequency and complexity of VAs after detraining has also been demonstrated in human athletes,128,129 though further prospective studies are required to validate these findings.
The isolated sub-epicardial substrate defined by Venlet et al.39 is intriguing. If due to exercise-induced inflammation, why is it localised to the sub-epicardial RVOT? If due to early ACM, why does the disease not appear to progress with ongoing exercise and catheter ablation is seemingly extremely effective? It is likely that this phenotype forms part of a continuum of exercise-induced RV pathology related to the known disproportionate haemodynamic effect of endurance exercise on the RV that, in some cases, does not completely recover.130,131 In addition to this haemodynamic overload, we have anecdotally observed the hypertrophic athlete’s heart directly abutting anterior and posterior structures within the limited confines of the human thorax (see Figure 3). Exercise CMR highlights the impressive mechanical rubbing between these points of contact that, interestingly, appears to mirror the location of described arrhythmogenic substrate. A similar phenomenon may also occur in the atria with one study demonstrating posterior LA fibrosis adjacent to the descending aorta, increasing with closer proximity between the two structures, suggesting a possible role for chronic, repetitive mechanical trauma inducing arrhythmogenic substrate in patients with AF.132 Whilst these observations remain purely theoretical, observed ‘hinge point’ scar in athletes may provide a precedent for how mechanical forces during exercise can result in ventricular fibrosis.
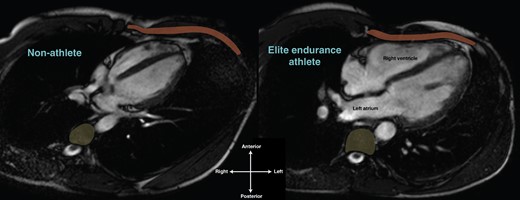
The athlete's heart within the thorax. Horizontal long axis cardiac MRI of a non-athlete (left) and elite endurance athlete’s heart (right), both 24-year-old males. Compared to the non-athlete, note the significantly greater proportion of the thorax inhabited by the athlete’s heart and the intimate relationship between the right ventricle and the chest wall (anterior, rust) and the left atrium and thoracic vertebrae (posterior, yellow). The right ventricle and left atrium of the athlete’s heart appear to be deformed in shape compared to the non-athlete.
VAs in the athlete’s heart: a contemporary approach
In athletes with structural abnormalities and complex VAs, management should incorporate medical therapy, risk stratification, possible exercise modification, and consideration of an ICD. However, for patients with less definitive structural abnormalities, differentiation between the healthy athlete's heart and more subtle phenotypes of EIACM is challenging. In these patients, additional evaluation might include evaluation of mechanical dispersion, exercise-induced RV dysfunction, and both VA inducibility and substrate identification with EAM at EPS. Ventricular arrhythmia reproducibility at repeat EST (within 12 months) has also been shown to predict underlying NILVS in athletes with VAs with an otherwise normal clinical evaluation.133 In addition, whilst the yield of genetic testing in athletes appears modest, positive findings can have important impacts on an athlete’s management and that of his or her family. Thus, genetic testing, guided by phenotypic suspicion, is recommended.
In athletes with patchy non-ischaemic scar patterns identified on CMR, the prognosis is not clear. Whilst there are some data that these scar patterns may be associated with severe sequalae,88,134 the true prevalence of NILVS and its relationship to life-threatening arrhythmias remains incompletely defined. For example, in a study of 92 seniors (mean age 69 years, 73% men), increasing levels of lifelong physical activity were not associated with NILVS despite clear demonstration of athletic cardiac remodelling and elite peak VO2 measurements in master athletes.135 In contrast, a study comparing 12 male lifelong endurance athletes (mean age 56 years) vs. 20 age-matched male veteran controls and 17 younger male endurance athletes (mean age 31 years) reported that 50% of the veteran endurance athletes had NILVS vs. none in the other two groups. The prevalence of LGE in this study was significantly associated with number of years spent training (P < 0.001), number of competitive marathons (P < 0.001), and ultra-endurance marathons completed (P < 0.007).136 A similarly high proportion of NILVS (48%; all mid-myocardial basal–lateral LV wall) was reported in a recent study of 50 asymptomatic veteran athletes.137 In this study, those with fibrosis had a greater PVC burden with higher prevalence of ventricular couplets and triplets than those without fibrosis (33% vs. 8%, P = 0.002), but the study was not adequately powered to assess for the presence of more significant arrhythmias.
In a large contemporary meta-analysis involving 1359 participants, 21% in the endurance athlete group (n = 772) had LGE detected (31% hinge point, 19% sub-endocardial, and 48% mid-myocardial/epicardial) vs. 3% in the comparison control group (n = 587). The results suggested that the prevalence of LGE was higher in the athletes group with long-term endurance exercise (OR 7.20; 95% CI 4.51–11.49, P = 0.34), with the same conclusion drawn after stratification for age.138 Other studies have a reported a similar 3–4% incidence of small, silent mid-wall NILVS in the general population.139 With such heterogenicity in reported incidence and uncertainty surrounding association with VAs and prognostic significance, it is unsurprising that there remains no consensus as to how to manage asymptomatic athletes with NILVS.
It is also unclear as to whether CMR is adequately sensitive to identify all arrhythmogenic scar. As previously discussed, Venlet et al.39 observed localised sub-epicardial arrhythmogenic substrate in athletes with sustained VT despite having no abnormal findings on TTE or CMR. Studies assessing the role of EAM ± EMB have also shown that underlying SHD can even be missed by CMR, which is relatively poor in detecting small areas of scar in the RV.140 In a cohort of 227 athletes disqualified from sports participation because of VAs, 24% of the 188 who underwent EAM (RV only in 72%, LV only in 18%, and both ventricles in 10%) had evidence of myocardial scar and/or late potentials. Amongst athletes without definitive findings after extensive non-invasive investigation (including CMR), EMB revealed a diagnosis of myocarditis in 12 and ACM in 3.119 In a smaller cohort of 13 competitive athletes with complex VAs and no underlying cardiac abnormality, again after extensive investigation including CMR, 12 of 13 had ≥1 low voltage regions at EAM with myocarditis (n = 7) or ACM (n = 5) confirmed by EAM-guided EMB. Importantly, 2 of 12 (ACM and myocarditis) had a history of unexplained syncope and another 2 of 12 (both ACM) a positive FHx of SCD.141
Thus, even in athletes with no underlying structural or electrical abnormality detected after extensive non-invasive workup including CMR, one must always consider the possibility of ‘occult’ substrate when athletes have complex VAs. In athletes with apparently structurally normal hearts, especially with red flag features such as unexplained syncope and/or a positive FHx of premature SCD or CM, consideration must be given to EPS with EAM at least of the RV (see Figure 4). Electrophysiology study in isolation provides valuable information about intrinsic conduction intervals, sinus and AV nodal function, presence of accessory pathways, and inducibility of VAs (typically reentrant) with programmed ventricular stimulation. In addition, high-dose isoprenaline infusion (up to 45 μg/min for 3 min) during EPS has been shown to induce polymorphic PVCs and sustained VT in patients with ACM and may aid in diagnosis of the disease during its early arrhythmogenic phase before significant structural changes are apparent.142,143
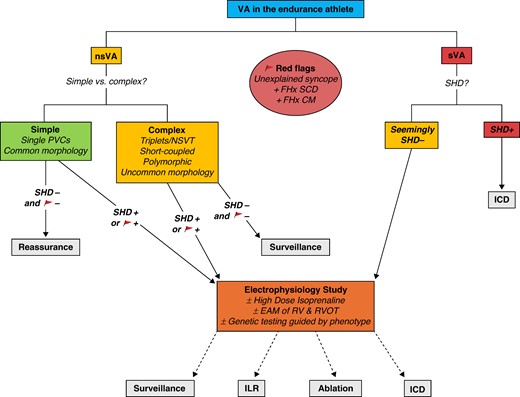
Management of VAs in athletes. nsVA, non-sustained ventricular arrhythmia—any VA lasting <30 s; sVA, sustained ventricular arrhythmia—ventricular tachycardia lasting ≥30 s and/or requiring intervention and/or ventricular fibrillation. Common premature ventricular complex (PVC) morphology: outflow tract/infundibular (LBBB morphology and inferior axis) and/or fascicular (typical RBBB morphology, superior/inferior axis, and QRS < 130 ms). Uncommon PVC morphology: all other types of PVCs. Short-coupled PVCs: PVC with interval from onset of preceding QRS to onset of PVC < 350 ms. SHD, structural heart disease; ICD, implantable cardioverter defibrillator; NSVT, non-sustained ventricular tachycardia; FHx SCD, family history of premature sudden cardiac death (male < 40 years, female < 50 years); FHx CM, family history of cardiomyopathy; EAM, electroanatomic mapping; RV, right ventricle; RVOT, right ventricular outflow tract; ILR, implantable loop recorder.
The addition of EAM to EPS allows definition of underlying arrhythmogenic substrate, most commonly by assessing tissue voltage. In general, endocardial bipolar voltage (BV) allows visualisation of sub-endocardial substrate with healthy tissue voltage defined as >1.5 mV (conventionally projected as purple) and dense scar < 0.5 mV (projected as red). Endocardial unipolar voltage (UV), which has a larger field-of-view than bipolar recordings, may better visualize ‘deeper’ intramural or epicardial substrate. Analysis of both BV and UV simultaneously in athletes allows clinicians to better understand underlying arrhythmogenic substrate. A recent expert opinion from the Italian Society of Sports Cardiology144 elaborates on the role of EAM in athletes with VAs and provides a framework for when to consider EAM-guided EMB as an additional diagnostic tool in athletes with VAs and unclear findings after comprehensive non-invasive assessment.
Epicardial mapping and ablation, which may be an effective treatment for some phenotypes,39 should be considered in patients with refractory arrhythmias, VA recurrence after endocardial ablation, VT morphology/mapping suggesting epicardial origin, and/or abnormal endocardial UV. In general, following epicardial access, formation of the EAM is the same as for an endocardial map though proceduralists must be aware of BV attenuation from epicardial fat and other structures (e.g. coronary arteries and valvular annuli). In light of these considerations, many centres will define low voltage areas as epicardial BV < 1.0–1.5 mV.
Conclusions
There is evidence of unique athletic patterns of remodelling that appear to be associated with an increased risk of VAs. Disproportionate RV remodelling, NILVS, and isolated sub-epicardial scar are arrhythmogenic phenotypes that have been discovered in athletes and require targeted evaluation and specific therapies. Understanding of these changes in humans continues to evolve and the future calls for more large animal models and human athlete studies. In the current era, cardiologists and electrophysiologists should consider arrhythmogenic phenotypes that may be specific to athletes. As in the general community, athletes may have benign VAs or may have evidence of clear pathology (e.g. CM, ischaemic injury, or sarcoidosis). However, specific to athletes, clinicians should also consider the possibility of arrhythmogenic substrates in those with complex VAs in association with more profound remodelling. Further research is required to clarify the incidence, prognosis, and mechanisms of these athletic phenotypes but current evidence suggests that careful observation is required as some athletes may develop serious arrhythmias.
Funding
P.D. is supported by a combined National Health and Medical Research Council Postgraduate Scholarship (ID: 2031119) & National Heart Foundation of Australia PhD Scholarship (ID:107659) and the Royal Australia College of Physicians Fellows Research Entry Scholarship (ID: 2023RES00039).
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
Author notes
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}