





Abstract
In the early nineties, few years before the birth of Europace, the clinical and scientific world of familial arrhythmogenic conditions was revolutionized by the identification of the first disease-causing genes. The explosion of genetic studies over a 15-year period led to the discovery of major disease-causing genes in practically all channelopathies and cardiomyopathies, bringing insight into the pathophysiological mechanisms of these conditions. The birth of next generation sequencing allowed a further step forward and other significant genes, as CALM1–3 in channelopathies and FLN C and TTN in cardiomyopathies were identified. Genotype–phenotype studies allowed the implementation of the genetic results in diagnosis, risk stratification, and therapeutic management with a different level of evidence in different arrhythmogenic conditions. The influence of common genetic variants, i.e. SNPs, on disease manifestation was proved in mid-twenties, and in the last 10 years with the advent of genome-wide association studies performed in familial arrhythmogenic diseases, the concept of polygenic risk score has been consolidated. Now, we are at the start of another amazing phase, i.e. the initiation of first gene therapy clinical trials.
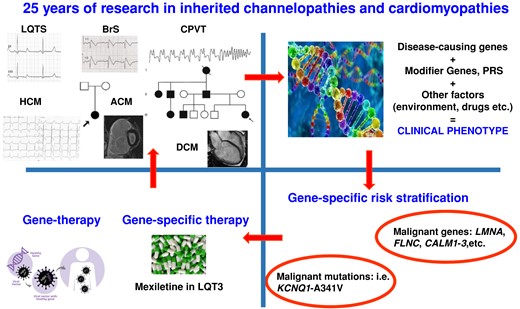
The upper panel on the left shows feature of all major channelopathies (LQTS, long QT syndrome; BrS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia) and cardiomyopathies (HCM, hypertrophic cardiomyopathy; ACM, arrhythmogenic cardiomyopathy; DCM, dilated cardiomyopathy) and two family trees. The other panels show how discovery of disease-causing genes and common variants influencing the phenotype (PRS, polygenic risk score) have an impact on clinical management. Lower panel of the left also shows the near future of gene therapy clinical trial.
This review is highlighting how genetic studies evolved during the last 25 years and how this has impacted our knowledges in the field of channelopathies and cardiomyopathies.
Gene therapy approaches evaluated so far in different channelopathies and cardiomyopathies are discussed.
Introduction
In 1999, Europace was born, the same year in which the lamin A/C gene was identified in the Emery-Dreifuss muscular dystrophy1 and a few months later in dilated cardiomyopathy (DCM) associated with conduction system disease.2 The genetic era was in its infancy. The evolution and progress in the understanding of the relationship between genetics and cardiac arrhythmias since then has been mind-boggling and worth revisiting.
In the early nineties, the clinical and scientific world of familial arrhythmogenic conditions was completely revolutionized by the identification of the first disease-causing genes for hypertrophic cardiomyopathy (HCM)3 and long QT syndrome (LQTS).4 Initially, disease-causing genes were identified through linkage analysis, a genome-wide approach that associated a genetic area within a given chromosome with the disease present in large families with clearly affected and unaffected family members.1–4 The actual gene was then identified through the screening of genes present in that region. The methods used were fascinating, such as manual DNA extraction when, all of a sudden, a white floating ball of DNA would appear. Screening was performed using single-strand conformation polymorphism (SSCP), and latterly denaturing high-pressure liquid chromatography (DHPLC) analysis, to identify samples that would deserve Sanger sequencing, a prolonged procedure requiring the use of radioactively labelled nucleotides to identify the genetic variation capable of causing the disease. The explosion of genetic studies over a 15-year period led to the discovery of major disease-causing genes in practically all channelopathies and cardiomyopathies5 and brought understanding of the pathophysiological mechanisms of the different arrhythmogenic conditions. When large families became increasingly unavailable, preventing linkage analysis, a candidate gene approach was employed, meaning that one or more biologically plausible genes were screened in affected individuals. However, our ability to make clever and careful use of such an approach was limited, and we ended up considering genes with weak evidence of pathogenicity as disease causing. At that time, we were unaware of how common it was to have ultra-rare genetic variants in our genome, and a very limited number of controls were used to state that a variant was pathogenic. However, despite the number of genes with a disputed or limited association with the different diseases identified with the candidate gene approach, there are some outstanding exceptions: CACNA1C in Timothy syndrome and KCNH2 in the short QT syndrome.5
Technological development, with SNP arrays and the birth of next generation sequencing, revolutionized genetic studies. The big step forward in genetic studies occurred with the Human Genome Project in 2003, where the first human genome was sequenced after 13 years using Sanger sequencing.6 In 2008, the 1000 Genome Project was launched, with the aim of sequencing 1000 individuals from different ethnicities, and after 2 years the first results were published.7 Next generation sequencing allowed the quick screening of big genes, impossible to be screened with older techniques, and this allowed, for instance, the identification of TTN as one of the major disease-causing genes in familial DCM. Subsequently, whole exome (the whole protein coding frame of the genome) and whole genome sequencing became available and, together with SNP arrays, allowed a return to a genome-wide approach to identify novel disease-causing genes.5 Through these approaches, CALM genes were identified8,9 and are now recognized as genes with a definite association with both LQTS and catecholaminergic polymorphic ventricular tachycardia (CPVT).5
Furthermore, the concept was proposed that common variants, or SNPs, present in the unaffected general population could influence the phenotypic manifestation of a monogenic disease acting as modifiers.10 Initially, the influence of single modifier genes with very small effect sizes was evaluated. But in 2013, with the first genome-wide association study (GWAS) performed in a familial arrhythmogenic disease, i.e. Brugada syndrome (BrS),9 the research world moved towards the concept of the polygenic risk score, the sum of the effect sizes of SNPs influencing the occurrence of a given phenotype.5,11
Advances in genetic knowledge always complemented by clinical data and genotype-phenotype correlation studies allowed the implementation of genetic analysis in diagnosis and risk stratification. Furthermore, gene-specific therapies are already a reality in many familial arrhythmogenic conditions. Now, we are ready for the next revolution, the initiation of gene therapy clinical trials. In the current review, we will go in the details of the evolution that occurred in the last 25 years in the different genetically transmitted arrhythmogenic conditions.
Long QT syndrome
No one disputes the fact that the identification of the three major LQTS genes in 1995–96 had a major impact on clinical management even though this impact has not been uniform.5 Here, we will briefly touch on the most impressive breakthroughs and progress while reminding the readers of useful sources for more detailed information.
The first came to light already in 1995 when, within months from the discovery that pathogenic variants in the cardiac sodium channel gene SCN5A were responsible for what we now call LQT3,12 Schwartz et al. proposed to use mexiletine to shorten the QT interval.13 This was the first gene-specific therapy for LQTS, and the concept has been validated by multiple studies. Currently, mexiletine is an integral part of our therapeutic armamentarium,14 and it helps to reduce excessively long QT intervals with beneficial effects on arrhythmic risk.15 Unfortunately, availability in Europe is an issue.16
In 2001, the initial studies on the genotype-phenotype correlation provided unexpected evidence for a tight link between genotype and arrhythmia triggers, i.e. the conditions associated with the life-threatening events.17 This in turn led to gene-specific management, such as the recommendations to avoid cold water swimming for LQT1 patients and sudden noises when waking up for LQT2 patients.17 Furthermore, we now know that some variants, regions in the protein18 and/or functional consequences of variants, are associated with different arrhythmic profile.19,20 Impressively, the impact of the evolution in genetic understanding has extended also to the worlds of sport21,22 and of criminal trials.23
Another major breakthrough was provided by the development of the concept of ‘modifier genes’, i.e. those genetic variants inherited by chance together with an LQTS disease-causing mutation and capable of altering, in either direction, the clinical severity directly due to the specific mutation.10 Besides the immediate and obvious impact on risk stratification, interest is focusing now on the protective modifier genes because, as their mechanism of action is being unravelled, the hopes for the development of novel therapies are increasing. A recent example is provided by the evidence that MTMR4 variants may interfere with the Nedd4L-dependent ubiquination resulting in decreased channel protein degradation and, in turn, in a reduction of the arrhythmogenic substrate for LQT1.24
Lastly, recent efforts are focusing on various aspects of gene- and mutation-specific therapies, including gene therapy, as part of precision medicine.25 These attempts were favoured by the growing use of induced pluripotent stem cells differentiated into cardiomyocytes (iPSC-CMs) and by the practical interest for drug repurposing, which relies on drugs already approved for clinical use in different diseases. Following the report that lumacaftor, a drug acting on protein trafficking used for the treatment of cystic fibrosis, can restore the trafficking of KCNH2 protein in iPSC-CMs derived from LQT2 patients,26 this drug was tested in two of the patients whose iPSC-CMs responded to lumacaftor and indeed shortened their QTc.27 A pilot clinical trial is currently ongoing. While lumacaftor may or may not turn out to be sufficiently useful for LQT2 patients with trafficking defects, these experimental and clinical studies have shown that iPSC-CMs represent a valid tool for drug-repurposing. Furthermore, iPSC-CMs are a valuable tool also to test new drugs as recently shown for serum/glucocorticoid-regulated kinase 1 inhibitors.28 Another interesting and promising development is represented by suppression/replacement gene therapy.29 While understandable excitement surrounds these developments, it is necessary to constantly keep in mind that: (i) safety of the various forms of gene therapy must be proven beyond doubt prior to its use in place of the currently employed therapies and (ii) the patients who are largely unprotected by current therapies are limited in numbers 30 and most novel therapies would be directed to the minority of patients who do not tolerate β-blockers.
For scientific fairness, it should be remembered that the most effective treatment modalities for LQTS, i.e. β-blockers25,31 and left cardiac sympathetic denervation,30 were conceived and implemented on the basis of pathophysiological considerations well before the dawn of genetic discoveries and that their safety is unquestionable.
Brugada syndrome
Despite an intense research impetus that started a few years after its first description in 1992 and perpetuated over the last 25 years, the complex genetic background underlying BrS is still considered not fully defined.32 The first genetic variant associated with BrS was identified in 1998 in the SCN5A gene, encoding the α-subunit of cardiac sodium channel Na(v)1.5.33 Although more than 20 genes have been so far associated with the condition, only rare variants in the SCN5A gene, identified only in 20% of patients with BrS, are currently considered disease-causing and clinically actionable.5,34 Gene disease clinical validity of rare variants in genes other than SCN5A has been disputed, and these variants should not be reported routinely for BrS genetic testing in a diagnostic setting.5 An European Heart Rhythm Association (EHRA) survey has recently shown that the proportion of centres with and without dedicated IAS units undertaking routinely genetic testing for BrS is 53% and 83%, respectively; thus, testing is still not ubiquitous.35
In the absence of rare SCN5A variants, BrS is considered to be largely polygenic.11 Moreover, the contribution of different genetic loci harbouring common variants associated with BrS may explain the highly variable disease expressivity of the syndrome, as shown by case–control GWAS and derived polygenic risk scores.11,36
The presence of rare SCN5A variants has been associated with a higher rate of spontaneous Brugada Type 1 electrocardiogram (ECG), symptoms, conduction abnormalities, and worse arrhythmic outcomes.37–39 Because of the risk of conduction disturbances and the more pronounced epicardial electrical abnormalities at invasive cardiac mapping observed in variant carriers, the presence of SCN5A pathogenic variants should be carefully considered during the risk assessment process and when selecting an implantable device type.39–41
Diagnostic yield of SCN5A gene testing is higher in paediatric patients with BrS, with SCN5A rare variants present in up to 46% of cases.41,42 A worse arrhythmic prognosis is observed in genotype-positive BrS paediatric patients compared with genotype-negative cases.42 Recently, circulating micro RNAs have drawn increased attention as a new potential prognostic biomarker in BrS patients.43
Na(v)1.5 sodium channel α-subunit components affected by SCN5A variants can result not only in defective gating properties but also in reduced trafficking to the cell membrane. Therefore, new mechanisms have been identified as potential therapeutic targets to increase the trafficking of NaV1.5 to plasma membranes.44 MOG1 increases plasma membrane expression of Na(v)1.5 and sodium current density, and it can rescue defective trafficking of Na(v)1.5 mutations in mouse cardiomyocytes.44 Moreover, a knock-in mouse model of BrS has been used to demonstrate that gene therapy using a small chaperone protein and targeting the protein trafficking regulator MOG1 can successfully reverse the cardiac functional abnormalities associated with BrS.44 The research roadmap on gene therapy in BrS is still open and keen to see further progress shortly.
Cardiac conduction defects and sick sinus syndrome
Cardiac conduction defects (CCDs) and sick sinus syndrome (SSS) are two pathologies frequently found in the general population. These pathologies represent the two most frequent causes for pacemaker implantation. However, idiopathic CCD in adults younger than 50 years is a very rare condition with an incidence of 0.7 per 100 000 persons/year.45
Although most cases are not of genetic origin, over the past 25 years, research has been conducted to identify the genetic basis of these conditions and to develop treatments tailored to each individual patient.
The first gene associated with CCD was SCN5A. SCN5A pathogenic variants lead to reduced sodium current, which slows intra-atrial and intra-ventricular conduction leading to a progressive alteration of the conduction parameters.46,47
TRPM4 gain-of-function mutations have been identified in families with isolated progressive cardiac conduction disease (PCCD). Conduction defects were related to an elevated TRPM4 channel density at the cell surface secondary to impaired endocytosis and deregulation of Small Ubiquitin MOdifier conjugation.47
Mutations in the NKX2.5 or TBX5 genes, usually associated with congenital cardiomyopathies, can be identified in subjects with isolated conduction disorders. This may also be the case for the LMNA or DES genes, which are usually associated with neuromuscular disorders and cardiomyopathies. It is therefore important to look for minor signs of cardiomyopathy and a potential family history of cardiomyopathy, sudden death at a young age, or neuropathy, which should lead to a genetic test.48 As pathogenic variants in SCN5A, DES, and LMNA genes have been associated with higher risk of SCD, identification of pathogenic variants in these genes may lead to implantable cardioverter defibrillator (ICD) implantation rather than a pacemaker in a patient with severe conduction defects.49
Genetic defects have also been identified in SSS. The most prevalent gene is HCN4 that should lead to isolated SSS.50HCN4 pathogenic variants are also associated with diverse phenotypes, such as sinus bradycardia, inappropriate sinus tachycardia, early-onset atrial fibrillation, atrioventricular block, left ventricular non-compaction (LVNC), idiopathic ventricular tachycardia (VT), ventricular fibrillation (VF), dilation of the aorta, and mood and anxiety disorders. Atrial standstill has been reported to occur either as a recessive disorder of SCN5A51 or by digenic inheritance of a heterozygous SCN5A pathogenic variant with a promoter variant in the connexin-40 gene.52 Variants in the ankyrin B gene, also associated with LQT4 syndrome and more recently the GNB2 gene, have also been identified.53
As for PCCD, the identification of SSS in a young subject without underlying heart disease should be investigated for a genetic cause because of the risk of associated cardiomyopathy, sometimes difficult to identify like minor forms of LVNC leading to a significant risk of sudden cardiac death (SCD).
The knowledge regarding the genetics of conduction disorders and SSS has led to a better understanding of the mechanisms involved in the creation and propagation of the cardiac electrical impulse. Even if there is currently no specific treatment in case of identification of a pathogenic variant, the improvement of knowledge in this field has allowed the first works on biological pacemakers, which could become an important therapeutic option in the future.54
Catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular tachycardia is another rare inheritable arrhythmia syndrome. As suggested by its name, polymorphic VT—which can degenerate into VF and cardiac arrest—is often evoked by moments of catecholaminergic stimulation, e.g. due to exercise or emotions, which are thus traditional triggers of arrhythmic events in these patients.55,56 Likewise, beta-blockers and cardiac sympathetic denervation to decrease adrenergic cardiac stimulation are often successful therapeutic measures.55 This is further mirrored in exercise testing being the most useful diagnostic tool to evaluate treatment efficacy in CPVT patients, to screen patients suspected of CPVT, and to phenotypically evaluate family members57,58 of CPVT patients. Importantly, unrecognized CPVT patients with unexplained syncope, arrhythmia or cardiac arrest during exercise or emotion, will have an otherwise normal evaluation and will often only be diagnosed during provocations with adrenergic stimulation like an exercise test. This also implies that reluctance to perform exercise testing in these patients may withhold a timely diagnosis and subsequent appropriate treatment.
Compared to the other rare inheritable arrhythmia syndromes discussed in this paper, the prevalence of CPVT is rather low, about 1 in every 10 000 individuals. CPVT patients often present early with arrhythmic events, in childhood, adolescence, or early adulthood, and with a higher prevalence of events compared to many other arrhythmia syndromes. The genetic underpinning of CPVT has been elucidated in sentinel work in 2001 where pathogenic variants in the cardiac ryanodine receptor 2 gene (RYR2, mostly autosomal dominant, also known as CPVT1) and the calsequestrin 2 gene (CASQ2, mostly autosomal recessive, also known as CPVT2) segregated with the phenotype in families. Disease-causing variants in these genes have an impact on calcium homeostasis in the cardiac sarcoplasmatic reticulum, where (adrenergic triggered) diastolic calcium release results in delayed afterdepolarizations which in turn trigger (bidirectional) ventricular extrasystoles and subsequently polymorphic VT and VF. Later, mutations in other genes [CALM1, CALM2, CALM3, triadin (TRDN), and TECRL] have been implicated in CPVT with, interestingly, some overlap with LQTS.9–59
As to treatment, there are several important remarks to be made in addition to the earlier mentioned effects of beta-blockers and cardiac sympathetic denervation. First, like in LQTS, nadolol or propranolol are the most effective beta-blockers.55 Second, flecainide is a very effective drug, mostly as combination therapy with beta-blockers, for patients without enough suppression of exercise induced arrhythmia or breakthrough events on beta-blockers alone.55 The effect of this combination therapy, sometimes with addition of cardiac sympathetic denervation, even challenges the indication for secondary prevention ICD therapy in patients who were treatment naive or insufficiently treated. Importantly, ICDs in these patients associate with more events and complications.60 Finally, also for CPVT, the possibilities of gene therapy are currently explored.61,62
Idiopathic ventricular fibrillation/sudden arrhythmic death syndrome
Patients surviving unexplained cardiac arrest (UCA) or idiopathic VF (IVF),63 and decedents suffering an unexplained sudden death, otherwise known as the sudden arrhythmic death syndrome (SADS),15,64–66 have benefited over the last 25 years from extensive research into their genetic basis. However, to arrive at these diagnostic labels requires deep and careful phenotyping to exclude any evident causes as per published guidelines.5,15,67 Evaluation of the cardiac arrest survivor often relies upon detailed and comprehensive clinical testing including extensive electrocardiography and imaging once myocardial infarction and non-cardiac causes have been excluded.68 Careful expert autopsy with histopathology backed up by toxicology is required to avoid misclassification of the normal heart as cardiomyopathy.69
Genetic testing in index cases surviving a cardiac arrest may then be able to confirm diagnosis of potential structural genetic disorders such as HCM, DCM, and arrhythmogenic cardiomyopathy (ACM) or arrhythmia syndromes such as LQTS, BrS, and CPVT.5 Indeed, data from the CASPER registry have supported the role of combined clinical and genetic evaluation to inform the underlying diagnosis of initial UCA.70 However, once clinical evaluation has excluded acquired and known genetic heart diseases as the cause, genetic testing may diagnose the underlying cause.5 For example, the first genetic finding associating clear causation with risk was the DPP6 haplotype, a founder genetic variant in the Dutch population providing the only diagnostic clue in families and dictating the need for ICD implantation given the associated high risk.71 Furthermore, loss of function variants in the RYR2 gene thought to be responsible for the calcium release deficiency syndrome is the main diagnostic cue in this disorder, given limited phenotypic characterization and absence of any CPVT phenotype in those who host these variants.72 Furthermore, pathogenic variants in cardiomyopathy genes have been detected in UCA and IVF cases with no evidence of structural phenotype.73,74 Over follow-up, a minority develop myopathic features, but nonetheless, it appears that concealed cardiomyopathy may be an important cause.73
Family evaluation may be another route to a genetic diagnosis in UCA.75 However, larger analyses of well-characterized IVF only supported evidence of an over-representation of early repolarization pattern and BrS amongst relatives.76,77
Given the absence of any phenotype at autopsy in SADS, post-mortem gene testing (the ‘molecular autopsy’) had therefore focused attention on arrhythmia syndrome genes with diagnostic yields of up to 35%. Subsequent studies using next generation sequencing whole exome78 and cardiac gene-specific panels initially suggested expanded yields by including ‘minor’ arrhythmia and cardiomyopathy genes. The significance of these was unclear, but by using stringent ACMG criteria for variant pathogenicity, more realistic yields of 13% have been suggested.79 These studies also identify pathogenic cardiomyopathy variants and therefore concealed cardiomyopathy as the cause of death.80 The yield of diagnosis increases to up to half of the cases if molecular autopsy is combined with family evaluation, the most common diagnosis being BrS.81–84
Pathogenic variants and successful phenotyping lead to more accurate identification of family members for prevention of sudden death85 (Figure 1). Unfortunately, an EHRA survey has identified poor uptake of autopsy, molecular autopsy, and services for families with UCA and SADS across Europe.86–88
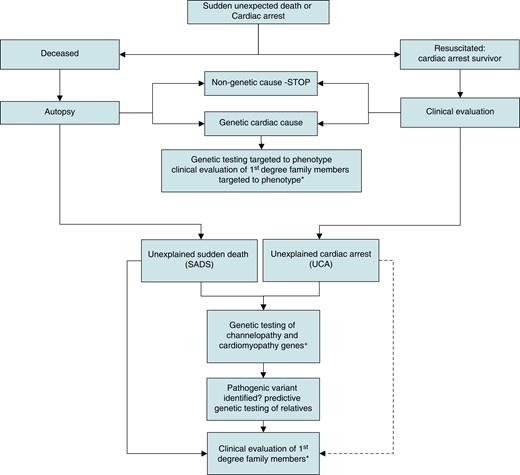
The pathway for the application of genetic and clinical evaluation following unexpected cardiac arrest or sudden death. Broken arrow indicates weaker evidence. *Predictive genetic testing will identify family members for clinical evaluation who harbour a pathogenic or likely pathogenic variant when this has been identified in a family. Otherwise, all first-degree relatives should be offered clinical evaluation. +Only genes with robust disease associations should be included. (Adapted from Behr85https://doi.org/10.1093/eurheartj/ehac172.)
Hypertrophic cardiomyopathy
hypertrophic cardiomyopathy is a common inherited cardiac disease defined by the presence of unexplained left ventricular (LV) hypertrophy.89 Although HCM has often a benign course, some patients experience severe symptoms and ventricular arrhythmias that in rare cases are fatal, leading to SCD.90
After the first description of the disease by the British pathologist Donald Teare in 1958,94 the first genetic locus associated with the condition was detected by means of a linkage analysis in 1989.91 The responsible gene was identified as MYH7. In the following decade, several other causal genes were identified, which led to a further understanding of the molecular genetic basis of HCM in large families.5
HCM is an example of monogenic disease where a single nucleotide variation is sufficient to cause a complex phenotype.92 Genetic testing identifies pathogenic or likely pathogenic variants in 30–50% of patients with HCM, and more than 1000 distinct variants have been identified. The most common are in the genes encoding for cardiac myosin binding protein C3 (MYBPC3)/myosin heavy chain 7 (MYH7) (70% of the cases) and more rarely other sarcomeric genes [TNNI3, TNNT2, α-tropomyosin gene, myosin light chain 2 (MYL2), MYL3, and actin alpha cardiac muscle 1 (ACTC1)].5 HCM is a heterogeneous disease in its clinical presentation, phenotype, and clinical course, even within the same family.5,93 Both penetrance and expressivity are likely to be influenced by epigenetic and environmental mechanisms, although the quality and extent of these interactions remain poorly understood.94 In some cases, the detection of an actionable variant is not accompanied by a clear phenotypic expression (genotype positive–phenotype negative status).95 The widespread use of genetic testing in probands and relatives has increasingly led to the identification of these individuals, who often exhibit certain ECG96 or structural abnormalities (mitral leaflet elongation, diastolic dysfunction, and myocardial crypts).89
Recent studies have led to the growing understanding that average prognosis is better in gene-negative HCM, with lower rates of SCD, heart failure, AF, and stroke. Although the mechanisms leading to gene-negative HCM remain a conundrum, recent observations have revealed that polygenic inheritance, as in the BrS, may play a significant role in this setting.97
Management of HCM includes pharmacological and non-pharmacological therapies. Symptomatic patients are offered pharmacotherapy with β-blockers or non-dihydropyridine calcium-channel blockers, and disopyramide is an option if symptoms are persistent. Non-pharmacological management mainly relies on SCD prevention through the use of ICD. Pharmacotherapies specifically targeted at interacting with the pathologic substrate and pathophysiology of HCM have been proposed, and their efficacy and safety have been recently proved through clinical trials.98 Hypercontractility appears to have a central role in the pathogenesis of HCM with the vast majority of known pathogenic variants affecting sarcomeric proteins. Mavacamten is a selective allosteric inhibitor of cardiac myosin adenosine triphosphatase which targets the underlying pathophysiology of HCM by reducing actin–myosin cross-bridge formation. A recent Phase 3 trial showed that mavacamten improves functional capacity, LV outflow tract gradient, and symptoms in patients with obstructive HCM.99
Knowledge on the mechanistic effects of variants in sarcomeric proteins has increased significantly in the last decade. This has opened up new horizons to treat cardiomyopathies, using various techniques, such as gene editing and repair using CRISPR-Cas9 techniques, antisense therapies, and RNA therapies. Pre-clinical data of gene therapy in HCM mouse models, mouse, and human engineered heart tissues have shown that gene replacement therapy via adeno-associated virus vector transfer may be an option to replace the abnormal sarcomeric protein in cardiomyocytes for severe forms of HCM.100
Future research is focused on the development of other drug agents that may be effective both in obstructive and non-obstructive patients; gene therapy holds the potential to correct or silence pathogenic variants and prevent the development of HCM.101
Arrhythmogenic right ventricular cardiomyopathy
In 1982, Marcus et al.102 published the first major description of arrhythmogenic right ventricular cardiomyopathy (ARVC). While it was soon recognized to be a familial condition most common in athletes, the genetic basis for the disease was unknown until 2000. The first breakthrough in discovering the genetic basis of ARVC was by McKoy et al.103 in 2000. These authors identified a genetic variant in plakoglobin as the genetic basis of Naxos disease.103,104 Naxos disease has similar cardiac manifestations as ARVC combined with woolly hair and palmoplantar keratosis. Two years later, Rampazzo et al.105 reported a variant in desmoplakin as a cause of ARVC. Two years after this paper, Gerull et al.106 reported that variants in plakophilin-2 are a common cause of ARVC,106 and subsequently also disease-causing variants in desmoglein-2 were described.104 Since that time, additional variants in desmosomal (desmocollin-2) and non-desmosomal proteins (phospholamban, transmembrane protein 43, desmin, lamin, cadherin 2107 and others) have been identified as causative of ARVC.110 Today, a pathogenic variant can be identified in approximately two-thirds of patients who meet the 2010 Task Force Criteria for ARVC.104,108,109
In addition to remarkable breakthroughs in understanding the genetic basis of ARVC, great progress has been made in the management of ARVC. The management of ARVC can be considered a four-legged stool. The first step is to establish an accurate diagnosis.108 Once this is done, the next step is to risk stratify the individual for sudden death risk and determine if an ICD is warranted.110 In the past few years, risk stratification algorithms have been developed to risk stratify ARVC patients for sustained ventricular arrhythmias111 and VF.112 The third leg of the stool is to prevent or minimize sustained VT events. This can be accomplished with exercise restriction, beta-blockade, antiarrhythmic drug therapy, and/or catheter ablation.113–116 The fourth leg of the stool is to slow the progression of ARVC. It is now well-recognized that exercise restriction is the most important approach to slowing progression. Beta blocks and angiotensin receptor blockers are also prescribed based on their important role in other types of heart failure.
In addition to remarkable breakthroughs in understanding the genetic basis of ARVC, considerable progress has been made in improving the diagnostic criteria for ARVC. The first diagnostic criteria were published in 1994 by McKenna et al.117 The main goal of these criteria was to define a relatively specific phenotype that could allow further characterization of this condition. These criteria were revised in 2010 by Marcus et al.108 Since then, it has become recognized that these diagnostic criteria are imperfect, especially for the left-dominant forms of the disease.118 Corrado et al.119 attempted to address these limitations by proposing the revised Padua criteria for ARVC. While these criteria stimulated further research and discussion, they have not been endorsed broadly in the EP international community. Another notable contribution to this discussion appeared in 2019 when the Heart Rhythm Society published an expert consensus document on ACM.120 Arrhythmogenic cardiomyopathy was defined as an arrhythmogenic heart muscle disorder not explained by ischaemic, hypertensive, or valvular heart disease. The document further states that ACM may present clinically as symptoms or documentation of AF, conduction disease, and/or right or LV arrhythmias. This document emphasizes the importance of genetic testing early in the workup of patients with arresting early in the workup of patients with arrhythmogenic cardiomyopathy. Most recently, a group of international experts on ARVC is leading an effort to develop new diagnostic criteria for ARVC based on genotype. It is now well recognized that the phenotypic characteristics of various variant-based subgroups of ARVC differ in their phenotypic expression, risk of sudden death, risk of heart failure, and relationship to exercise.121,122
In summary, our understanding of ARVC has progressed dramatically over more than 40 years since it was first described. It is now recognized that the way forward for diagnosis and management is best approached with a gene-first approach.
Dilated cardiomyopathy
Dilated cardiomyopathy is a severe and misunderstood disease that affect millions of young patients worldwide.123 Its complex and diverse clinical phenotype stems from dynamic interactions between multiple genetic and non-genetic factors and explains the substantial differences in DCM penetrance.5 Family screening is essential in relatives of patients with DCM, and the guidelines promote post-mortem genetic testing in the context of SCD.15,124 Despite high-throughput sequencing technologies such as gene chips and next generation sequencing, the detection rate of DCM has remained relatively constant over the last decade with a prevalence of pathogenic variants between 15% and 25% in unselected DCM patients and between 20% and 40% in familial DCM patients. More than a thousand pathogenic variants have been identified, indicating that diverse pathways cause DCM. These are often patient specific, relevant for a person or family. They may also be group specific, or the hallmark of sub-specific populations. Some monogenetic causes of DCM associated with ventricular arrhythmias have been identified (TTN, LMNA, FLNC, PLN).125 Pathogenic variants in the splicing factor RBM20 are known to be associated with early onset end-stage heart failure.126 The G213D variant in Nav1.5 in the SCN5A gene is associated with a severe form of arrhythmic DCM that may be cure by antiarrhythmic drugs.127 The role of common variants has also been reported thanks to GWAS analysis that have identified new DCM-associated SNPs.128 Polygenic risk scores have recently been shown to have predictive value for DCM prognostic.129
Over-treating or under-treating misclassified patients is still frequent because genotype interpretation is often difficult, especially with the surge of variants of uncertain significance. There is also a lack of widespread and easy-to-use stratification risk, especially at the earliest stage of the disease. The aforementioned facts explain why medical diagnostic errors in DCM patients are frequent and the prognostic poor. They emphasized the need to decipher the mechanisms of DCM. Mechanistic insights have been shown to serve for the identification of the disease at its early stage as well as drugable targets. Human induced pluripotent stem cell-derived cardiomyocytes have helped to pinpoint molecular changes such as abnormal regulation of calcium.130 Repair of a TTN truncating variant by CRISPR/Cas 9 gene editing in engineered heart muscle fully rescued contractility.131 Genetically modified zebrafish that carry a human A-band TTN have now been generated and shown to spontaneously develop DCM with age.131 Finally, accumulating evidence shows that epigenetic alterations play a role in arrhythmogenic cardiomyopathies.132
A key challenge for better and earlier detection of complications of DCM patients will be to integrate clinical, ECG, CMR, and deep genotyping data.133 Antisense and RNA therapies also represent new avenues to treat DCM patients.134 Ultimately, through large-scale multi-omic studies, increased clinical experience in CMD genetics, and improved understanding of the disease processes, optimization of DCM management will be obtained also through the implementation of genotype-tailored strategies.135
Conclusion
The last 25 years have seen enormous development in genomic technologies and their impact upon the understanding of human genetic variation, disease mechanisms, and ultimately the potential to affect patients and their care. The next steps, however, are critical. Ensuring greater access to genetic testing already mandated in guidelines15,136 and consensus statements5 requires implementation in patient pathways not only in Europe but globally. Only then will access be possible for patients to personalized medical interventions and ultimately therapies tailored to genetic aetiology. This will lay the groundwork for prevention of disease progression and major complications such as sudden death and heart failure. There lies our challenge for the next 25 years.
Acknowledgements
Crotti L., Brugada P., Postema P.G., Probst V., and Schwartz P.J. are proud members of ERN GUARD-Heart. We wish to thank Dr Giovanni Bienati for editorial support.
Funding
This study was supported by AIFA Grant TRS 2018-0001470 EUDRA CT 2020-000250-94 (L.C. and P.J.S.) and Dutch Heart Foundation grant 03-003-2021-T061 (P.P.G.).
References
Author notes
Conflict of interest: H.C. is a consultant and/or has received honoraria from Medtronic, Biosense Webster, Atricure, Abbott, and Boston Scientific.

{kind=link}
{kind=link}