





Abstract
A pathogenic/likely pathogenic (P/LP) variant in SCN5A is found in 20–25% of patients with Brugada syndrome (BrS). However, the diagnostic yield and prognosis of gene panel testing in paediatric BrS is unclear. The aim of this study is to define the diagnostic yield and outcomes of SCN5A gene testing with ACMG variant classification in paediatric BrS patients compared with adults.
All consecutive patients diagnosed with BrS, between 1992 and 2022, were prospectively enrolled in the UZ Brussel BrS registry. Inclusion criteria were: (i) BrS diagnosis; (ii) genetic analysis performed with a large gene panel; and (iii) classification of gene variants following ACMG guidelines. Paediatric patients were defined as ≤16 years of age. The primary endpoint was ventricular arrhythmias (VAs). A total of 500 BrS patients were included, with 63 paediatric patients and 437 adult patients. Among children with BrS, 29 patients (46%) had a P/LP variant (P+) in SCN5A and no variants were found in 34 (54%) patients (P−). After a mean follow-up of 125.9 months, 8 children (12.7%) experienced a VA, treated with implanted cardioverter defibrillator shock. At survival analysis, P− paediatric patients had higher VA-free survival during the follow-up, compared with P+ paediatric patients. P+ status was an independent predictor of VA. There was no difference in VA-free survival between paediatric and adult BrS patients for both P− and P+.
In a large BrS cohort, the diagnostic yield for P/LP variants in the paediatric population is 46%. P+ children with BrS have a worse arrhythmic prognosis.
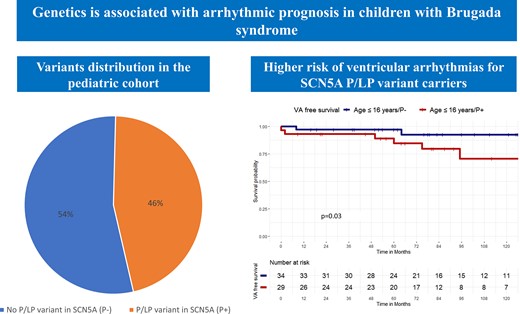
In a large cohort of Brugada syndrome (BrS) patients, undergoing genetic analysis with a large gene panel and classification of variants following ACMG guidelines, the diagnostic yield for pathogenic/likely pathogenic (P/LP) variants in the paediatric population was 46.0%.
Paediatric patients without a P/LP variant in SCN5A had higher ventricular arrhythmia (VA)-free survival during the follow-up, compared with P+ patients. SCN5A carrier status was an independent predictor of VA.
There was no difference in VA-free survival between paediatric and adult BrS patients both without and with a P/LP variant in SCN5A.
Introduction
Brugada syndrome (BrS) is an inherited primary arrhythmia syndrome associated with sudden cardiac death (SCD) in otherwise healthy subjects.1,2 In the first clinical description of BrS, three patients were children.1 Different studies reported on the clinical risk factors associated with ventricular arrhythmias (VAs) in paediatric BrS population.3
Indeed, in the study by Probst et al.,4 the risk of arrhythmic events was higher in previously symptomatic patients and in those displaying a spontaneous Type I electrocardiogram (ECG). Furthermore, Michowitz et al.5 demonstrated that the S wave in ECG Lead 1, sinus node dysfunction (SND), and atrial arrhythmias were also associated with recurrent VA in the paediatric cohort. A thorough risk stratification in children affected by BrS is of utmost clinical importance, given the high rate of device-related complications, leading to lead replacement or inappropriate shocks in this population.6–11
Genetics is emerging as a novel additional tool to predict arrhythmic prognosis in BrS.12,13 In particular, a pathogenic gene variant can be found in ≈20–25% of BrS patients.14–16 A pathogenic/likely pathogenic (P/LP) variant in the SCN5A gene has been associated with a worse electrical substrate in the epicardium of the right ventricle outflow tract.17–20 This translates into a higher risk of VA.21
Michowitz et al.5 reported a high prevalence of SCN5A among paediatric patients (58.1%). In the study by Crotti et al.,14 the yield of P/LP variants was significantly higher among BrS patients younger than 20 years of age (75%) compared with patients between 20 and 40 years of age (22%) and those older than 40 years of age (15%). However, no previous studies reported variant classification following current standardized ACMG guidelines.22 No comparison is available between the arrhythmic prognosis of children and adult BrS patients with a long-term clinical follow-up.
The aim of this study is to define the diagnostic yield for SCN5A of a gene panel with ACMG standardized variant classification in a large cohort of paediatric BrS over the last 30 years; furthermore, this study aims to find a correlation between the clinical outcomes and the genetic background of children with BrS and to make a comparison with adult BrS.
Methods
Study population
All consecutive patients diagnosed with BrS between 1992 and 2022 were prospectively enrolled in the UZ Brussel monocentric BrS registry (NCT05283759). They were included in the current study if the following inclusion criteria were fulfilled: (i) BrS diagnosed following current recommendations;23–25 (iii) genetic analysis for BrS performed with a next generation sequencing-based large gene panel; and (iii) recent reclassification of gene variants following current ACMG guidelines.22 Both probands and affected family members and both spontaneous and induced BrS Type I were included. Other diagnosis different from BrS syndrome was excluded by means of transthoracic echocardiography, magnetic resonance imaging, or computed tomography.26 Genotype-positive individuals with phenotype negativity (i.e. not diagnosed with BrS) were excluded.
The study cohort was divided into two groups as follows, based on the age at diagnosis: (i) paediatric BrS patients (children):≤16 years old and (ii) adult BrS patients (adults): >16 years old, as previously described.4 A total of 437 adult patients and 63 patients ≤16-year-old fulfilled the inclusion criteria and were analysed. Among 63 patients, 24 were described by our group in a previous report27 and of these 24 patients, 10 patients were previously included in a second study by our group.8
The study complied with the Declaration of Helsinki as revised in 2013; the ethic committee approved the study. All patients signed an informed consent that had been approved by our institutional review board.
Clinical data collection
For all patients, the risk of events at 5 years was calculated with the score system developed at our centre by Sieira et al.28 The following ECG parameters were calculated on the first available ECG: heart rate (b.p.m.), PQ interval in D2 (ms), QRS length in lead D2 (ms), QTc interval in lead D2 (ms) with Bazett correction.29 All ECGs were analysed using digital calipers by two independent physicians (L.P. and A.B.). Discrepancies >10 ms were adjudicated by a third independent physician (C.d.A.).
The following data were collected: (i) clinical history including: demographic and biometric data, syncope history, spontaneous Type I ECG, SND history, SCD history, SCD family history, and implanted cardioverter defibrillator (ICD) implantation; (ii) electrophysiological study (EPS) for VA inducibility. Electrophysiological study induction protocol was performed from the right ventricular apex site at three basic pacing cycles (600, 500, and 430 ms) with up to three ventricular premature beats down to a minimum of 200 ms or refractoriness. Electrophysiological study protocol was the same for both paediatric and adult BrS patients. Inducibility at EPS was adjudicated if a VA was induced, including ventricular fibrillation or ventricular tachycardia lasting at least 30 s. His-ventricle interval was measured with a quadripolar catheter in all patients before the induction protocol. The corrected sinus node recovery time was measured with a pacing cycle length of 600 ms.
Genetic analysis
Genetic analysis was performed in all patients with the same panel using Roche SeqCap® EZ Human Exome Probes v3.0 or Roche Nimblegen SeqCap® EZ Choice XL (113 primary cardiac arrhythmia genes and 208 cardiomyopathy genes) on a HiSeq5000 or NovaSeq6000 system. Patients were defined as (P+) if they had a P/LP variant in SCN5A following ACMG guidelines,22 using manually curated VarSome,30 Franklin (Genoox, Palo Alto, CA, USA)31 and Alamut Visual Plus (v.1.4) (SOPHiA GENETICS, Lausanne, Switzerland)-based variant classification criteria. All other patients were defined as (P−). Variants of unknown significance (VUS) in SCN5A were excluded. All reported genetic variants were reclassified in November 2022 according to the latest databases information by two independent experienced geneticists (S.V.D. and R.O.).
Follow-up
Patients were followed up in the outpatient clinic every 6 months and by remote monitoring. Patients with an ICD underwent serial device interrogations every 6 months. Patients without an ICD underwent 24 h standard 12-lead Holter-ECG every 6 months. The primary endpoint was VA occurrence, defined as documented SCD, aborted SCD, sustained ventricular tachycardia, or ventricular fibrillation or appropriate ICD intervention. The secondary endpoint was atrial fibrillation (AF) occurrence, defined following current guidelines as any documented episode of ≥30 s showing heart rhythm with no discernible P waves and irregular RR intervals.32
Statistical analysis
All variables were tested for normality with the Shapiro–Wilk test. Normally distributed variables were described as mean ± standard deviation and the groups were compared through analysis of variance, paired or unpaired t-test as appropriate, while the non-normally distributed variables were described as median (interquartile range) and compared by Mann–Whitney test or Wilcoxon signed-rank test as appropriate. The categorical variables were described as frequencies (percentages) and compared by χ2 test or Fisher’s exact test as appropriate. Cohen’s kappa statistic was used to assess interobserver agreement in ECG analysis.
Kaplan–Meier’s curves were drawn to describe the patients’ freedom from VA during the follow-up period and Log-Rank test or Pairwise Log-Rank test was used.
Cox’s proportional hazard model was performed to identify risk factors for VA. The covariates entered in the univariate and multivariate Cox model were chosen according to their clinical significance. Variables with P < 0.10 were then entered in the multivariate model and selected with a backward stepwise approach.
Survival analysis was performed with the survival33 and survminer34 packages on R software.
A P-value <0.05 was considered statistically significant. The analysis was performed using R software version 3.6.2 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Study population characteristics
A total of 500 consecutive BrS patients were included in the study. Sixty-three patients were ≤16 years of age and 437 patients were >16 years of age.
Compared with adult patients, paediatric patients were younger (11.0 years ± 2.1 vs. 42.9 years ± 13.9, P < 0.001), had higher heart rate (74.6 b.p.m. ± 16.9 vs. 71.4 b.p.m. ± 13.2, P = 0.047) and shorter PQ interval (155.3 ms ± 43.9 vs. 166.3 ms ± 31.4, P = 0.015). Good interobserver agreement was observed for ECG analysis (κ = 0.97).
An ICD was implanted in 172 patients (39.4%) [25 paediatric patients (39.7%) vs. 172 adult patients (39.4%), P = 1.00].
Complete patient characteristics are summarized in Table 1.
Clinical characteristics of paediatric and adult patients with Brugada syndrome
| Age >16 years (N = 437) | Age ≤16 years (N = 63) | Total (N = 500) | P–value | |
|---|---|---|---|---|
| Age at diagnosis (years) | 42.9 ± 13.9 | 11.0 ± 2.1 | 38.9 ± 16.9 | <0.001 |
| Gender (male) | 210 (48.1%) | 35 (55.6%) | 245 (49.0%) | 0.28 |
| Spontaneous BrS I pattern, n (%) | 76 (17.4%) | 11 (17.5%) | 87 (17.4%) | 1.00 |
| SCD family history <35 years, n (%) | 59 (13.5%) | 16 (25.4%) | 75 (15.0%) | 0.022 |
| SND, n (%) | 20 (4.6%) | 4 (6.3%) | 24 (4.8%) | 0.53 |
| History of syncope, n (%) | 143 (32.7%) | 16 (25.4%) | 159 (31.8%) | 0.31 |
| History of aborted SCD, n (%) | 22 (5.0%) | 7 (11.1%) | 29 (5.8%) | 0.07 |
| Sieira score (points) | 1.3 ± 1.7 | 1.4 ± 2.0 | 1.3 ± 1.7 | 0.67 |
| Proband, n (%) | 252 (57.7%) | 23 (36.5%) | 275 (55%) | 0.004 |
| ICD, n (%) | 172 (39.4%) | 25 (39.7%) | 197 (39.4%) | 1.00 |
| ECG HR (b.p.m.) | 71.4 ± 13.2 | 74.6 ± 16.9 | 71.8 ± 13.7 | 0.047 |
| ECG PQ (ms) | 166.3 ± 31.4 | 155.3 ± 43.9 | 165.0 ± 33.4 | 0.015 |
| ECG QRS (ms) | 100.7 ± 19.2 | 100.4 ± 22.0 | 100.7 ± 19.5 | 0.90 |
| ECG QTc (ms) | 413.7 ± 33.8 | 430.5 ± 12.4 | 420.4 ± 157.9 | 0.02 |
| EPS HV (ms) | 44.5 ± 8.1 | 43.4 ± 10.2 | 44.4 ± 8.3 | 0.49 |
| cSNRT (ms) | 321.8 ± 124.3 | 274.2 ± 116.7 | 317.1 ± 124.1 | 0.13 |
| VA inducibility at EPS, n (%) | 36 (8.2%) | 0 (0.0%) | 36 (7.2%) | 0.015 |
| SCN5A P/LP variant | 75 (17.2%) | 29 (46.0%) | 104 (20.8%) | <0.001 |
| Age >16 years (N = 437) | Age ≤16 years (N = 63) | Total (N = 500) | P–value | |
|---|---|---|---|---|
| Age at diagnosis (years) | 42.9 ± 13.9 | 11.0 ± 2.1 | 38.9 ± 16.9 | <0.001 |
| Gender (male) | 210 (48.1%) | 35 (55.6%) | 245 (49.0%) | 0.28 |
| Spontaneous BrS I pattern, n (%) | 76 (17.4%) | 11 (17.5%) | 87 (17.4%) | 1.00 |
| SCD family history <35 years, n (%) | 59 (13.5%) | 16 (25.4%) | 75 (15.0%) | 0.022 |
| SND, n (%) | 20 (4.6%) | 4 (6.3%) | 24 (4.8%) | 0.53 |
| History of syncope, n (%) | 143 (32.7%) | 16 (25.4%) | 159 (31.8%) | 0.31 |
| History of aborted SCD, n (%) | 22 (5.0%) | 7 (11.1%) | 29 (5.8%) | 0.07 |
| Sieira score (points) | 1.3 ± 1.7 | 1.4 ± 2.0 | 1.3 ± 1.7 | 0.67 |
| Proband, n (%) | 252 (57.7%) | 23 (36.5%) | 275 (55%) | 0.004 |
| ICD, n (%) | 172 (39.4%) | 25 (39.7%) | 197 (39.4%) | 1.00 |
| ECG HR (b.p.m.) | 71.4 ± 13.2 | 74.6 ± 16.9 | 71.8 ± 13.7 | 0.047 |
| ECG PQ (ms) | 166.3 ± 31.4 | 155.3 ± 43.9 | 165.0 ± 33.4 | 0.015 |
| ECG QRS (ms) | 100.7 ± 19.2 | 100.4 ± 22.0 | 100.7 ± 19.5 | 0.90 |
| ECG QTc (ms) | 413.7 ± 33.8 | 430.5 ± 12.4 | 420.4 ± 157.9 | 0.02 |
| EPS HV (ms) | 44.5 ± 8.1 | 43.4 ± 10.2 | 44.4 ± 8.3 | 0.49 |
| cSNRT (ms) | 321.8 ± 124.3 | 274.2 ± 116.7 | 317.1 ± 124.1 | 0.13 |
| VA inducibility at EPS, n (%) | 36 (8.2%) | 0 (0.0%) | 36 (7.2%) | 0.015 |
| SCN5A P/LP variant | 75 (17.2%) | 29 (46.0%) | 104 (20.8%) | <0.001 |
BrS, Brugada syndrome; cSNRT, corrected sinus node recovery time; EPS, electrophysiological study; HR, heart rate; HV, His-ventricle; P/LP, pathogenic (P)/likely pathogenic (LP) variant; RBBB, right bundle branch block; SCD, sudden cardiac death; SND, sinus node dysfunction; VA, ventricular arrhythmia.
Clinical characteristics of paediatric and adult patients with Brugada syndrome
| Age >16 years (N = 437) | Age ≤16 years (N = 63) | Total (N = 500) | P–value | |
|---|---|---|---|---|
| Age at diagnosis (years) | 42.9 ± 13.9 | 11.0 ± 2.1 | 38.9 ± 16.9 | <0.001 |
| Gender (male) | 210 (48.1%) | 35 (55.6%) | 245 (49.0%) | 0.28 |
| Spontaneous BrS I pattern, n (%) | 76 (17.4%) | 11 (17.5%) | 87 (17.4%) | 1.00 |
| SCD family history <35 years, n (%) | 59 (13.5%) | 16 (25.4%) | 75 (15.0%) | 0.022 |
| SND, n (%) | 20 (4.6%) | 4 (6.3%) | 24 (4.8%) | 0.53 |
| History of syncope, n (%) | 143 (32.7%) | 16 (25.4%) | 159 (31.8%) | 0.31 |
| History of aborted SCD, n (%) | 22 (5.0%) | 7 (11.1%) | 29 (5.8%) | 0.07 |
| Sieira score (points) | 1.3 ± 1.7 | 1.4 ± 2.0 | 1.3 ± 1.7 | 0.67 |
| Proband, n (%) | 252 (57.7%) | 23 (36.5%) | 275 (55%) | 0.004 |
| ICD, n (%) | 172 (39.4%) | 25 (39.7%) | 197 (39.4%) | 1.00 |
| ECG HR (b.p.m.) | 71.4 ± 13.2 | 74.6 ± 16.9 | 71.8 ± 13.7 | 0.047 |
| ECG PQ (ms) | 166.3 ± 31.4 | 155.3 ± 43.9 | 165.0 ± 33.4 | 0.015 |
| ECG QRS (ms) | 100.7 ± 19.2 | 100.4 ± 22.0 | 100.7 ± 19.5 | 0.90 |
| ECG QTc (ms) | 413.7 ± 33.8 | 430.5 ± 12.4 | 420.4 ± 157.9 | 0.02 |
| EPS HV (ms) | 44.5 ± 8.1 | 43.4 ± 10.2 | 44.4 ± 8.3 | 0.49 |
| cSNRT (ms) | 321.8 ± 124.3 | 274.2 ± 116.7 | 317.1 ± 124.1 | 0.13 |
| VA inducibility at EPS, n (%) | 36 (8.2%) | 0 (0.0%) | 36 (7.2%) | 0.015 |
| SCN5A P/LP variant | 75 (17.2%) | 29 (46.0%) | 104 (20.8%) | <0.001 |
| Age >16 years (N = 437) | Age ≤16 years (N = 63) | Total (N = 500) | P–value | |
|---|---|---|---|---|
| Age at diagnosis (years) | 42.9 ± 13.9 | 11.0 ± 2.1 | 38.9 ± 16.9 | <0.001 |
| Gender (male) | 210 (48.1%) | 35 (55.6%) | 245 (49.0%) | 0.28 |
| Spontaneous BrS I pattern, n (%) | 76 (17.4%) | 11 (17.5%) | 87 (17.4%) | 1.00 |
| SCD family history <35 years, n (%) | 59 (13.5%) | 16 (25.4%) | 75 (15.0%) | 0.022 |
| SND, n (%) | 20 (4.6%) | 4 (6.3%) | 24 (4.8%) | 0.53 |
| History of syncope, n (%) | 143 (32.7%) | 16 (25.4%) | 159 (31.8%) | 0.31 |
| History of aborted SCD, n (%) | 22 (5.0%) | 7 (11.1%) | 29 (5.8%) | 0.07 |
| Sieira score (points) | 1.3 ± 1.7 | 1.4 ± 2.0 | 1.3 ± 1.7 | 0.67 |
| Proband, n (%) | 252 (57.7%) | 23 (36.5%) | 275 (55%) | 0.004 |
| ICD, n (%) | 172 (39.4%) | 25 (39.7%) | 197 (39.4%) | 1.00 |
| ECG HR (b.p.m.) | 71.4 ± 13.2 | 74.6 ± 16.9 | 71.8 ± 13.7 | 0.047 |
| ECG PQ (ms) | 166.3 ± 31.4 | 155.3 ± 43.9 | 165.0 ± 33.4 | 0.015 |
| ECG QRS (ms) | 100.7 ± 19.2 | 100.4 ± 22.0 | 100.7 ± 19.5 | 0.90 |
| ECG QTc (ms) | 413.7 ± 33.8 | 430.5 ± 12.4 | 420.4 ± 157.9 | 0.02 |
| EPS HV (ms) | 44.5 ± 8.1 | 43.4 ± 10.2 | 44.4 ± 8.3 | 0.49 |
| cSNRT (ms) | 321.8 ± 124.3 | 274.2 ± 116.7 | 317.1 ± 124.1 | 0.13 |
| VA inducibility at EPS, n (%) | 36 (8.2%) | 0 (0.0%) | 36 (7.2%) | 0.015 |
| SCN5A P/LP variant | 75 (17.2%) | 29 (46.0%) | 104 (20.8%) | <0.001 |
BrS, Brugada syndrome; cSNRT, corrected sinus node recovery time; EPS, electrophysiological study; HR, heart rate; HV, His-ventricle; P/LP, pathogenic (P)/likely pathogenic (LP) variant; RBBB, right bundle branch block; SCD, sudden cardiac death; SND, sinus node dysfunction; VA, ventricular arrhythmia.
Genetic analysis
Among children with BrS, 29 patients (46.0%) had a P/LP variant (P+) in SCN5A gene. No P/LP variants could be identified in 34 (54.0%) paediatric patients (P−). There was no difference in clinical characteristics between P+ and P− paediatric patients, including spontaneous Type I ECG, history of syncope, and history of SCD (Table 2).
Clinical characteristics of paediatric patients with Brugada syndrome with and without pathogenic (p)/likely pathogenic (LP) variant
| P− (n = 34) | P+ (n = 29) | Total (n = 63) | P-value | |
|---|---|---|---|---|
| Age at diagnosis (years) | 12.1 ± 1.8 | 9.7 ± 2.6 | 11.0 ± 2.1 | 0.07 |
| Gender (male) | 22 (64.7%) | 13 (44.8%) | 35 (55.6%) | 0.13 |
| Spontaneous BrS I pattern, n (%) | 3 (8.8%) | 8 (27.6%) | 11 (17.5%) | 0.09 |
| SCD family history <35 years, n (%) | 5 (14.7%) | 11 (37.9%) | 16 (25.4%) | 0.05 |
| SND, n (%) | 3 (8.8%) | 1 (3.4%) | 4 (6.3%) | 0.62 |
| History of syncope, n (%) | 10 (29.4%) | 6 (20.7%) | 16 (25.4%) | 0.56 |
| History of aborted SCD, n (%) | 2 (5.9%) | 5 (17.2%) | 7 (11.1%) | 0.23 |
| Sieira score (points) | 1.2 ± 1.7 | 1.7 ± 2.4 | 1.4 ± 2.0 | 0.36 |
| Proband, n (%) | 13 (38.2%) | 10 (34.5%) | 23 (36.5%) | 0.79 |
| ICD, n (%) | 14 (41.2%) | 11 (37.9%) | 25 (39.7%) | 1.00 |
| ECG HR (b.p.m.) | 71.9 ± 13.8 | 77.7 ± 19.6 | 74.6 ± 16.9 | 0.18 |
| ECG PQ (ms) | 145.3 ± 43.5 | 166.2 ± 42.3 | 155.3 ± 43.9 | 0.06 |
| ECG QRS (ms) | 95.2 ± 18.4 | 106.2 ± 24.5 | 100.4 ± 22.0 | 0.05 |
| ECG QTc (ms) | 440.5 ± 15.9 | 420.4 ± 19.9 | 430.5 ± 12.4 | 0.43 |
| P− (n = 34) | P+ (n = 29) | Total (n = 63) | P-value | |
|---|---|---|---|---|
| Age at diagnosis (years) | 12.1 ± 1.8 | 9.7 ± 2.6 | 11.0 ± 2.1 | 0.07 |
| Gender (male) | 22 (64.7%) | 13 (44.8%) | 35 (55.6%) | 0.13 |
| Spontaneous BrS I pattern, n (%) | 3 (8.8%) | 8 (27.6%) | 11 (17.5%) | 0.09 |
| SCD family history <35 years, n (%) | 5 (14.7%) | 11 (37.9%) | 16 (25.4%) | 0.05 |
| SND, n (%) | 3 (8.8%) | 1 (3.4%) | 4 (6.3%) | 0.62 |
| History of syncope, n (%) | 10 (29.4%) | 6 (20.7%) | 16 (25.4%) | 0.56 |
| History of aborted SCD, n (%) | 2 (5.9%) | 5 (17.2%) | 7 (11.1%) | 0.23 |
| Sieira score (points) | 1.2 ± 1.7 | 1.7 ± 2.4 | 1.4 ± 2.0 | 0.36 |
| Proband, n (%) | 13 (38.2%) | 10 (34.5%) | 23 (36.5%) | 0.79 |
| ICD, n (%) | 14 (41.2%) | 11 (37.9%) | 25 (39.7%) | 1.00 |
| ECG HR (b.p.m.) | 71.9 ± 13.8 | 77.7 ± 19.6 | 74.6 ± 16.9 | 0.18 |
| ECG PQ (ms) | 145.3 ± 43.5 | 166.2 ± 42.3 | 155.3 ± 43.9 | 0.06 |
| ECG QRS (ms) | 95.2 ± 18.4 | 106.2 ± 24.5 | 100.4 ± 22.0 | 0.05 |
| ECG QTc (ms) | 440.5 ± 15.9 | 420.4 ± 19.9 | 430.5 ± 12.4 | 0.43 |
BrS, Brugada syndrome; HR, heart rate; HV, His-ventricle; P−, patient without any pathogenic (P)/likely pathogenic (LP) variant; P+, patient with any pathogenic (P)/likely pathogenic (LP) variant; RBBB, right bundle branch block; SCD, sudden cardiac death; SND, sinus node dysfunction; VA, ventricular arrhythmia.
Clinical characteristics of paediatric patients with Brugada syndrome with and without pathogenic (p)/likely pathogenic (LP) variant
| P− (n = 34) | P+ (n = 29) | Total (n = 63) | P-value | |
|---|---|---|---|---|
| Age at diagnosis (years) | 12.1 ± 1.8 | 9.7 ± 2.6 | 11.0 ± 2.1 | 0.07 |
| Gender (male) | 22 (64.7%) | 13 (44.8%) | 35 (55.6%) | 0.13 |
| Spontaneous BrS I pattern, n (%) | 3 (8.8%) | 8 (27.6%) | 11 (17.5%) | 0.09 |
| SCD family history <35 years, n (%) | 5 (14.7%) | 11 (37.9%) | 16 (25.4%) | 0.05 |
| SND, n (%) | 3 (8.8%) | 1 (3.4%) | 4 (6.3%) | 0.62 |
| History of syncope, n (%) | 10 (29.4%) | 6 (20.7%) | 16 (25.4%) | 0.56 |
| History of aborted SCD, n (%) | 2 (5.9%) | 5 (17.2%) | 7 (11.1%) | 0.23 |
| Sieira score (points) | 1.2 ± 1.7 | 1.7 ± 2.4 | 1.4 ± 2.0 | 0.36 |
| Proband, n (%) | 13 (38.2%) | 10 (34.5%) | 23 (36.5%) | 0.79 |
| ICD, n (%) | 14 (41.2%) | 11 (37.9%) | 25 (39.7%) | 1.00 |
| ECG HR (b.p.m.) | 71.9 ± 13.8 | 77.7 ± 19.6 | 74.6 ± 16.9 | 0.18 |
| ECG PQ (ms) | 145.3 ± 43.5 | 166.2 ± 42.3 | 155.3 ± 43.9 | 0.06 |
| ECG QRS (ms) | 95.2 ± 18.4 | 106.2 ± 24.5 | 100.4 ± 22.0 | 0.05 |
| ECG QTc (ms) | 440.5 ± 15.9 | 420.4 ± 19.9 | 430.5 ± 12.4 | 0.43 |
| P− (n = 34) | P+ (n = 29) | Total (n = 63) | P-value | |
|---|---|---|---|---|
| Age at diagnosis (years) | 12.1 ± 1.8 | 9.7 ± 2.6 | 11.0 ± 2.1 | 0.07 |
| Gender (male) | 22 (64.7%) | 13 (44.8%) | 35 (55.6%) | 0.13 |
| Spontaneous BrS I pattern, n (%) | 3 (8.8%) | 8 (27.6%) | 11 (17.5%) | 0.09 |
| SCD family history <35 years, n (%) | 5 (14.7%) | 11 (37.9%) | 16 (25.4%) | 0.05 |
| SND, n (%) | 3 (8.8%) | 1 (3.4%) | 4 (6.3%) | 0.62 |
| History of syncope, n (%) | 10 (29.4%) | 6 (20.7%) | 16 (25.4%) | 0.56 |
| History of aborted SCD, n (%) | 2 (5.9%) | 5 (17.2%) | 7 (11.1%) | 0.23 |
| Sieira score (points) | 1.2 ± 1.7 | 1.7 ± 2.4 | 1.4 ± 2.0 | 0.36 |
| Proband, n (%) | 13 (38.2%) | 10 (34.5%) | 23 (36.5%) | 0.79 |
| ICD, n (%) | 14 (41.2%) | 11 (37.9%) | 25 (39.7%) | 1.00 |
| ECG HR (b.p.m.) | 71.9 ± 13.8 | 77.7 ± 19.6 | 74.6 ± 16.9 | 0.18 |
| ECG PQ (ms) | 145.3 ± 43.5 | 166.2 ± 42.3 | 155.3 ± 43.9 | 0.06 |
| ECG QRS (ms) | 95.2 ± 18.4 | 106.2 ± 24.5 | 100.4 ± 22.0 | 0.05 |
| ECG QTc (ms) | 440.5 ± 15.9 | 420.4 ± 19.9 | 430.5 ± 12.4 | 0.43 |
BrS, Brugada syndrome; HR, heart rate; HV, His-ventricle; P−, patient without any pathogenic (P)/likely pathogenic (LP) variant; P+, patient with any pathogenic (P)/likely pathogenic (LP) variant; RBBB, right bundle branch block; SCD, sudden cardiac death; SND, sinus node dysfunction; VA, ventricular arrhythmia.
Paediatric patients had more frequently a P/LP variant, compared with adult patients [29 patients (46.0%) vs. 75 patients (17.2%), P < 0.001; Table 1). Two VUS were found in SCN5A, in two adult BrS patients, namely: c. 2398C > T and c.5812G > A. Pathogenic/likely pathogenic variants were enriched in the pore region [13 of the 28 SCN5A variants (46.4%)]. More specifically, 11 variants (39.3%) were located in the extracellular and 2 (7.1%) in the intramembranous part of the P− loop. Complete results of genetic analysis of P/LP variants in SCN5A found in the whole BrS cohort are summarized in Table 3.
Pathogenic/Likely pathogenic variants identified in SCN5A following ACMG guidelines in the whole cohort
| Sequence change | Exon# or Intron # | Variant effect | Protein change | Region | Gnomad highest population frequency | ClinVar ID | VarSome Pathogenticity predictions P/B | VarSome ACMG variant classification criteria | Original variant class | New variant classa |
|---|---|---|---|---|---|---|---|---|---|---|
| c.−53 + 1G > A | Intron 1 | Splice variant | p.? | promotor | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.183del | Exon 2 | Frameshift | p.(Lys63Serfs*34) | n-terminal-tail C | NP | NP | NP | PVS1/PM2 | 5 | 4 |
| c.361C > T | Exon 3 | Missense | p.(Arg121Trp) | n-terminal tail C | NP | 67807 | 11P/1B | PVS1/PS3/PM2/PM5/PP1/PP3/PP5 | 5 | 5 |
| c.585G > A | Exon 5 | Nonsense | p.(Trp195*) | D1 S3 | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.903G > A | Exon 7 | Nonsense | p.(Trp301*) | D1 E | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.1003T > C | Exon 9 | Missense | p.(Cys335Arg) | D1 E | NP | 1066556 | 12P/0B | PS3/PM2/PM1/PP1/PP3/PP5 | 4 | 5 |
| c.2466G > A | Exon 16 | Nonsense | p.(Trp822*) | D2 C | NP | 505895 | NP | PVS1/PM2/PP5 | 4 | 5 |
| c.2572A > C | Exon 16 | Missense + Splice variant | p.(Met858Leu) | D2 E | NP | NP | 10P/2B | PM1/PM2/PP1/PP3/PP5 | 4 | 4 |
| c.2632C > T | Exon 16 | Missense | p.(Arg878Cys) | D2 E | NP | 67744 | 12P/0B | PP5/PM1/PM2/PP1//PP3 | 5 | 4 |
| c.2658T > A | Exon 16 | Missense | p.(His886Gln) | D2 PF | NP | NP | 10P/2B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.2678G > A | Exon 16 | Missense | p.(Arg893His) | D2 PF | FIN:0.0046% | 67749 | 11P/1B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.2729C > T | Exon 16 | Missense | p.(Ser910Leu) | D2 E | NFE:0.00090% | 67753 | 11P/1B | PS3/PM2/PM1/PP1/PP3/PP5 | 5 | 5 |
| c.3673G > A | Exon 21 | Missense | p.(Glu1225Lys) | D3 E | OTH:0.017% | 67810 | 12P/0B | PS3/PM2/PP2/PP3/PP5 | 3 | 5 |
| c.3695G > A | Exon 21 | Missense | p.(Arg1232Gln) | D3 E | SAS:0.0098% | 67814 | 11P/1B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.3956G > T | Exon 22 | Missense | p.(Gly1319Val) | D3 C | AFR:0.0087% | 67838 | 12P/0B | PS3/PM2/PP3/PP5 | 4 | 5 |
| c.4083delG | Exon 23 | Frameshift | p.(Arg1362Glyfs*12) | D3 E | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.4283C > T | Exon 24 | Missense | p.(Ala1428Val) | D3 E | NP | 67876 | 11P/1B | PM2/PS3/PM1/PP3 | 5 | 5 |
| c.4300–2A > T | intron 24 | Splice variant | p.? | D3 E | NP | NP | NP | PVS1/PM1/PM2/PP1/PP3/PP5 | 4 | 5 |
| c.4343T > C | Exon 25 | Missense | p.(Ile1448Thr) | D3 S6 | NP | 67883 | 10P/2B | PM1/PM2/PP3 | 3 | 4 |
| c.4346A > G | Exon 25 | Missense | p.(Tyr1449Cys) | D3 S6 | NP | 67884 | 12P/0B | PM1/PM2/PS3/PM5/PP3/PP5 | 4 | 5 |
| c.4534C > T | Exon 26 | Missense | p.(Arg1512Trp) | ID3–4 C | OTH:0.033% | 9380 | 12P/0B | PS3/PM1/PM2/PP3/PP5 | 4 | 4 |
| c.4719C > T | Exon 27 | Synonymous + splice variant (cryptic splice site acceptor activated) | p.(Gly1573=) | D4 S2 | AMR:0.0029% | 263423 | 1P/4B | PVS1/PP3/PM2/PP5 | 4 | 4 |
| c.4813 + 3_4813 + 6dupGGGT | intron 27 | Splice variant | p.? | D4 S3 | NP | 254157 | 1P/0B | PM2/PM4/PP1/PP3/PP5 | 4 | 5 |
| c.4895G > A | Exon 28 | Missense | p.(Arg1632His) | D4 S4 | EAS:0.0054% | 67939 | 12P/0B | PS1/PS3/PM1/PM2/PM5/PP1/PP3 | 4 | 5 |
| c.4978A > G | Exon 28 | Missense | p.(Ile1660Val) | D4 S5 | NFE:0.0062% | 67947 | 11P/1B | PS1/PS3/PM1/PM2/PP3/PP5 | 5 | 4 |
| c.4981G > A | Exon 28 | Missense | p.(Gly1661Arg) | D4 S5 | NP | 201523 | 12P/0B | PS3/PM1/PM2/PP1/PP3/PP5 | 4 | 5 |
| c.5189C > A | Exon 28 | Missense | p.(Pro1730His) | D4 E | NP | NP | 11P/1B | PS3/PM1/PM2/PP1/PP3 | 3 | 5 |
| c.5356_5357del | Exon 28 | Frameshift | p.(Leu1786Glufs*2) | C-terminal tail C | NP | 254156 | NP | PVS1/PM2/PP1/PP5 | 5 | 5 |
| Sequence change | Exon# or Intron # | Variant effect | Protein change | Region | Gnomad highest population frequency | ClinVar ID | VarSome Pathogenticity predictions P/B | VarSome ACMG variant classification criteria | Original variant class | New variant classa |
|---|---|---|---|---|---|---|---|---|---|---|
| c.−53 + 1G > A | Intron 1 | Splice variant | p.? | promotor | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.183del | Exon 2 | Frameshift | p.(Lys63Serfs*34) | n-terminal-tail C | NP | NP | NP | PVS1/PM2 | 5 | 4 |
| c.361C > T | Exon 3 | Missense | p.(Arg121Trp) | n-terminal tail C | NP | 67807 | 11P/1B | PVS1/PS3/PM2/PM5/PP1/PP3/PP5 | 5 | 5 |
| c.585G > A | Exon 5 | Nonsense | p.(Trp195*) | D1 S3 | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.903G > A | Exon 7 | Nonsense | p.(Trp301*) | D1 E | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.1003T > C | Exon 9 | Missense | p.(Cys335Arg) | D1 E | NP | 1066556 | 12P/0B | PS3/PM2/PM1/PP1/PP3/PP5 | 4 | 5 |
| c.2466G > A | Exon 16 | Nonsense | p.(Trp822*) | D2 C | NP | 505895 | NP | PVS1/PM2/PP5 | 4 | 5 |
| c.2572A > C | Exon 16 | Missense + Splice variant | p.(Met858Leu) | D2 E | NP | NP | 10P/2B | PM1/PM2/PP1/PP3/PP5 | 4 | 4 |
| c.2632C > T | Exon 16 | Missense | p.(Arg878Cys) | D2 E | NP | 67744 | 12P/0B | PP5/PM1/PM2/PP1//PP3 | 5 | 4 |
| c.2658T > A | Exon 16 | Missense | p.(His886Gln) | D2 PF | NP | NP | 10P/2B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.2678G > A | Exon 16 | Missense | p.(Arg893His) | D2 PF | FIN:0.0046% | 67749 | 11P/1B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.2729C > T | Exon 16 | Missense | p.(Ser910Leu) | D2 E | NFE:0.00090% | 67753 | 11P/1B | PS3/PM2/PM1/PP1/PP3/PP5 | 5 | 5 |
| c.3673G > A | Exon 21 | Missense | p.(Glu1225Lys) | D3 E | OTH:0.017% | 67810 | 12P/0B | PS3/PM2/PP2/PP3/PP5 | 3 | 5 |
| c.3695G > A | Exon 21 | Missense | p.(Arg1232Gln) | D3 E | SAS:0.0098% | 67814 | 11P/1B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.3956G > T | Exon 22 | Missense | p.(Gly1319Val) | D3 C | AFR:0.0087% | 67838 | 12P/0B | PS3/PM2/PP3/PP5 | 4 | 5 |
| c.4083delG | Exon 23 | Frameshift | p.(Arg1362Glyfs*12) | D3 E | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.4283C > T | Exon 24 | Missense | p.(Ala1428Val) | D3 E | NP | 67876 | 11P/1B | PM2/PS3/PM1/PP3 | 5 | 5 |
| c.4300–2A > T | intron 24 | Splice variant | p.? | D3 E | NP | NP | NP | PVS1/PM1/PM2/PP1/PP3/PP5 | 4 | 5 |
| c.4343T > C | Exon 25 | Missense | p.(Ile1448Thr) | D3 S6 | NP | 67883 | 10P/2B | PM1/PM2/PP3 | 3 | 4 |
| c.4346A > G | Exon 25 | Missense | p.(Tyr1449Cys) | D3 S6 | NP | 67884 | 12P/0B | PM1/PM2/PS3/PM5/PP3/PP5 | 4 | 5 |
| c.4534C > T | Exon 26 | Missense | p.(Arg1512Trp) | ID3–4 C | OTH:0.033% | 9380 | 12P/0B | PS3/PM1/PM2/PP3/PP5 | 4 | 4 |
| c.4719C > T | Exon 27 | Synonymous + splice variant (cryptic splice site acceptor activated) | p.(Gly1573=) | D4 S2 | AMR:0.0029% | 263423 | 1P/4B | PVS1/PP3/PM2/PP5 | 4 | 4 |
| c.4813 + 3_4813 + 6dupGGGT | intron 27 | Splice variant | p.? | D4 S3 | NP | 254157 | 1P/0B | PM2/PM4/PP1/PP3/PP5 | 4 | 5 |
| c.4895G > A | Exon 28 | Missense | p.(Arg1632His) | D4 S4 | EAS:0.0054% | 67939 | 12P/0B | PS1/PS3/PM1/PM2/PM5/PP1/PP3 | 4 | 5 |
| c.4978A > G | Exon 28 | Missense | p.(Ile1660Val) | D4 S5 | NFE:0.0062% | 67947 | 11P/1B | PS1/PS3/PM1/PM2/PP3/PP5 | 5 | 4 |
| c.4981G > A | Exon 28 | Missense | p.(Gly1661Arg) | D4 S5 | NP | 201523 | 12P/0B | PS3/PM1/PM2/PP1/PP3/PP5 | 4 | 5 |
| c.5189C > A | Exon 28 | Missense | p.(Pro1730His) | D4 E | NP | NP | 11P/1B | PS3/PM1/PM2/PP1/PP3 | 3 | 5 |
| c.5356_5357del | Exon 28 | Frameshift | p.(Leu1786Glufs*2) | C-terminal tail C | NP | 254156 | NP | PVS1/PM2/PP1/PP5 | 5 | 5 |
B, benign; C, cytoplasmic; D, domain; E, extracellular; ID, interdomain linking loop; NP, not present; P, pathogenic; PF, intramembrane pore forming; S, transmembrane segment.
New variant class refers to the reclassification following ACMG guidelines in November 2022.
Pathogenic/Likely pathogenic variants identified in SCN5A following ACMG guidelines in the whole cohort
| Sequence change | Exon# or Intron # | Variant effect | Protein change | Region | Gnomad highest population frequency | ClinVar ID | VarSome Pathogenticity predictions P/B | VarSome ACMG variant classification criteria | Original variant class | New variant classa |
|---|---|---|---|---|---|---|---|---|---|---|
| c.−53 + 1G > A | Intron 1 | Splice variant | p.? | promotor | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.183del | Exon 2 | Frameshift | p.(Lys63Serfs*34) | n-terminal-tail C | NP | NP | NP | PVS1/PM2 | 5 | 4 |
| c.361C > T | Exon 3 | Missense | p.(Arg121Trp) | n-terminal tail C | NP | 67807 | 11P/1B | PVS1/PS3/PM2/PM5/PP1/PP3/PP5 | 5 | 5 |
| c.585G > A | Exon 5 | Nonsense | p.(Trp195*) | D1 S3 | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.903G > A | Exon 7 | Nonsense | p.(Trp301*) | D1 E | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.1003T > C | Exon 9 | Missense | p.(Cys335Arg) | D1 E | NP | 1066556 | 12P/0B | PS3/PM2/PM1/PP1/PP3/PP5 | 4 | 5 |
| c.2466G > A | Exon 16 | Nonsense | p.(Trp822*) | D2 C | NP | 505895 | NP | PVS1/PM2/PP5 | 4 | 5 |
| c.2572A > C | Exon 16 | Missense + Splice variant | p.(Met858Leu) | D2 E | NP | NP | 10P/2B | PM1/PM2/PP1/PP3/PP5 | 4 | 4 |
| c.2632C > T | Exon 16 | Missense | p.(Arg878Cys) | D2 E | NP | 67744 | 12P/0B | PP5/PM1/PM2/PP1//PP3 | 5 | 4 |
| c.2658T > A | Exon 16 | Missense | p.(His886Gln) | D2 PF | NP | NP | 10P/2B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.2678G > A | Exon 16 | Missense | p.(Arg893His) | D2 PF | FIN:0.0046% | 67749 | 11P/1B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.2729C > T | Exon 16 | Missense | p.(Ser910Leu) | D2 E | NFE:0.00090% | 67753 | 11P/1B | PS3/PM2/PM1/PP1/PP3/PP5 | 5 | 5 |
| c.3673G > A | Exon 21 | Missense | p.(Glu1225Lys) | D3 E | OTH:0.017% | 67810 | 12P/0B | PS3/PM2/PP2/PP3/PP5 | 3 | 5 |
| c.3695G > A | Exon 21 | Missense | p.(Arg1232Gln) | D3 E | SAS:0.0098% | 67814 | 11P/1B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.3956G > T | Exon 22 | Missense | p.(Gly1319Val) | D3 C | AFR:0.0087% | 67838 | 12P/0B | PS3/PM2/PP3/PP5 | 4 | 5 |
| c.4083delG | Exon 23 | Frameshift | p.(Arg1362Glyfs*12) | D3 E | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.4283C > T | Exon 24 | Missense | p.(Ala1428Val) | D3 E | NP | 67876 | 11P/1B | PM2/PS3/PM1/PP3 | 5 | 5 |
| c.4300–2A > T | intron 24 | Splice variant | p.? | D3 E | NP | NP | NP | PVS1/PM1/PM2/PP1/PP3/PP5 | 4 | 5 |
| c.4343T > C | Exon 25 | Missense | p.(Ile1448Thr) | D3 S6 | NP | 67883 | 10P/2B | PM1/PM2/PP3 | 3 | 4 |
| c.4346A > G | Exon 25 | Missense | p.(Tyr1449Cys) | D3 S6 | NP | 67884 | 12P/0B | PM1/PM2/PS3/PM5/PP3/PP5 | 4 | 5 |
| c.4534C > T | Exon 26 | Missense | p.(Arg1512Trp) | ID3–4 C | OTH:0.033% | 9380 | 12P/0B | PS3/PM1/PM2/PP3/PP5 | 4 | 4 |
| c.4719C > T | Exon 27 | Synonymous + splice variant (cryptic splice site acceptor activated) | p.(Gly1573=) | D4 S2 | AMR:0.0029% | 263423 | 1P/4B | PVS1/PP3/PM2/PP5 | 4 | 4 |
| c.4813 + 3_4813 + 6dupGGGT | intron 27 | Splice variant | p.? | D4 S3 | NP | 254157 | 1P/0B | PM2/PM4/PP1/PP3/PP5 | 4 | 5 |
| c.4895G > A | Exon 28 | Missense | p.(Arg1632His) | D4 S4 | EAS:0.0054% | 67939 | 12P/0B | PS1/PS3/PM1/PM2/PM5/PP1/PP3 | 4 | 5 |
| c.4978A > G | Exon 28 | Missense | p.(Ile1660Val) | D4 S5 | NFE:0.0062% | 67947 | 11P/1B | PS1/PS3/PM1/PM2/PP3/PP5 | 5 | 4 |
| c.4981G > A | Exon 28 | Missense | p.(Gly1661Arg) | D4 S5 | NP | 201523 | 12P/0B | PS3/PM1/PM2/PP1/PP3/PP5 | 4 | 5 |
| c.5189C > A | Exon 28 | Missense | p.(Pro1730His) | D4 E | NP | NP | 11P/1B | PS3/PM1/PM2/PP1/PP3 | 3 | 5 |
| c.5356_5357del | Exon 28 | Frameshift | p.(Leu1786Glufs*2) | C-terminal tail C | NP | 254156 | NP | PVS1/PM2/PP1/PP5 | 5 | 5 |
| Sequence change | Exon# or Intron # | Variant effect | Protein change | Region | Gnomad highest population frequency | ClinVar ID | VarSome Pathogenticity predictions P/B | VarSome ACMG variant classification criteria | Original variant class | New variant classa |
|---|---|---|---|---|---|---|---|---|---|---|
| c.−53 + 1G > A | Intron 1 | Splice variant | p.? | promotor | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.183del | Exon 2 | Frameshift | p.(Lys63Serfs*34) | n-terminal-tail C | NP | NP | NP | PVS1/PM2 | 5 | 4 |
| c.361C > T | Exon 3 | Missense | p.(Arg121Trp) | n-terminal tail C | NP | 67807 | 11P/1B | PVS1/PS3/PM2/PM5/PP1/PP3/PP5 | 5 | 5 |
| c.585G > A | Exon 5 | Nonsense | p.(Trp195*) | D1 S3 | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.903G > A | Exon 7 | Nonsense | p.(Trp301*) | D1 E | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.1003T > C | Exon 9 | Missense | p.(Cys335Arg) | D1 E | NP | 1066556 | 12P/0B | PS3/PM2/PM1/PP1/PP3/PP5 | 4 | 5 |
| c.2466G > A | Exon 16 | Nonsense | p.(Trp822*) | D2 C | NP | 505895 | NP | PVS1/PM2/PP5 | 4 | 5 |
| c.2572A > C | Exon 16 | Missense + Splice variant | p.(Met858Leu) | D2 E | NP | NP | 10P/2B | PM1/PM2/PP1/PP3/PP5 | 4 | 4 |
| c.2632C > T | Exon 16 | Missense | p.(Arg878Cys) | D2 E | NP | 67744 | 12P/0B | PP5/PM1/PM2/PP1//PP3 | 5 | 4 |
| c.2658T > A | Exon 16 | Missense | p.(His886Gln) | D2 PF | NP | NP | 10P/2B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.2678G > A | Exon 16 | Missense | p.(Arg893His) | D2 PF | FIN:0.0046% | 67749 | 11P/1B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.2729C > T | Exon 16 | Missense | p.(Ser910Leu) | D2 E | NFE:0.00090% | 67753 | 11P/1B | PS3/PM2/PM1/PP1/PP3/PP5 | 5 | 5 |
| c.3673G > A | Exon 21 | Missense | p.(Glu1225Lys) | D3 E | OTH:0.017% | 67810 | 12P/0B | PS3/PM2/PP2/PP3/PP5 | 3 | 5 |
| c.3695G > A | Exon 21 | Missense | p.(Arg1232Gln) | D3 E | SAS:0.0098% | 67814 | 11P/1B | PM1/PM2/PM5/PP3 | 4 | 4 |
| c.3956G > T | Exon 22 | Missense | p.(Gly1319Val) | D3 C | AFR:0.0087% | 67838 | 12P/0B | PS3/PM2/PP3/PP5 | 4 | 5 |
| c.4083delG | Exon 23 | Frameshift | p.(Arg1362Glyfs*12) | D3 E | NP | NP | NP | PVS1/PM2 | 4 | 4 |
| c.4283C > T | Exon 24 | Missense | p.(Ala1428Val) | D3 E | NP | 67876 | 11P/1B | PM2/PS3/PM1/PP3 | 5 | 5 |
| c.4300–2A > T | intron 24 | Splice variant | p.? | D3 E | NP | NP | NP | PVS1/PM1/PM2/PP1/PP3/PP5 | 4 | 5 |
| c.4343T > C | Exon 25 | Missense | p.(Ile1448Thr) | D3 S6 | NP | 67883 | 10P/2B | PM1/PM2/PP3 | 3 | 4 |
| c.4346A > G | Exon 25 | Missense | p.(Tyr1449Cys) | D3 S6 | NP | 67884 | 12P/0B | PM1/PM2/PS3/PM5/PP3/PP5 | 4 | 5 |
| c.4534C > T | Exon 26 | Missense | p.(Arg1512Trp) | ID3–4 C | OTH:0.033% | 9380 | 12P/0B | PS3/PM1/PM2/PP3/PP5 | 4 | 4 |
| c.4719C > T | Exon 27 | Synonymous + splice variant (cryptic splice site acceptor activated) | p.(Gly1573=) | D4 S2 | AMR:0.0029% | 263423 | 1P/4B | PVS1/PP3/PM2/PP5 | 4 | 4 |
| c.4813 + 3_4813 + 6dupGGGT | intron 27 | Splice variant | p.? | D4 S3 | NP | 254157 | 1P/0B | PM2/PM4/PP1/PP3/PP5 | 4 | 5 |
| c.4895G > A | Exon 28 | Missense | p.(Arg1632His) | D4 S4 | EAS:0.0054% | 67939 | 12P/0B | PS1/PS3/PM1/PM2/PM5/PP1/PP3 | 4 | 5 |
| c.4978A > G | Exon 28 | Missense | p.(Ile1660Val) | D4 S5 | NFE:0.0062% | 67947 | 11P/1B | PS1/PS3/PM1/PM2/PP3/PP5 | 5 | 4 |
| c.4981G > A | Exon 28 | Missense | p.(Gly1661Arg) | D4 S5 | NP | 201523 | 12P/0B | PS3/PM1/PM2/PP1/PP3/PP5 | 4 | 5 |
| c.5189C > A | Exon 28 | Missense | p.(Pro1730His) | D4 E | NP | NP | 11P/1B | PS3/PM1/PM2/PP1/PP3 | 3 | 5 |
| c.5356_5357del | Exon 28 | Frameshift | p.(Leu1786Glufs*2) | C-terminal tail C | NP | 254156 | NP | PVS1/PM2/PP1/PP5 | 5 | 5 |
B, benign; C, cytoplasmic; D, domain; E, extracellular; ID, interdomain linking loop; NP, not present; P, pathogenic; PF, intramembrane pore forming; S, transmembrane segment.
New variant class refers to the reclassification following ACMG guidelines in November 2022.
Follow-up
After a mean follow-up of 125.9 months ± 176.4, there were no deaths in the paediatric cohort.
Eight children with BrS (12.7%) experienced a VA, corresponding to an annual rate of 1.2%. A first VA event occurred at a mean follow-up of 44.2 months ± 36.4 and all were adjudicated as appropriate ICD shocks. Inappropriate shocks occurred in 4 patients (6.3%) in the paediatric BrS cohort. Atrial fibrillation occurred in 7 children with BrS (11.1%) at a mean follow-up of 81.6 months ± 97.9. A total of 3 children (42.8%) with AF also experienced VA.
Family members adult patients had higher VA-free survival during the follow-up, compared with probands adult patients (95.2 vs. 87.6%, Log-rank P = 0.008). Also, family members paediatric patients had higher VA-free survival during the follow-up, compared with proband paediatric patients (95.0 vs. 73.9%, Log-Rank P = 0.002).
The role of pathogenic/likely pathogenic variants in paediatric Brugada population
At survival analysis, P− paediatric patients had higher VA-free survival during the follow-up, compared with P+ paediatric patients (94.1 vs. 79.3%, Log-rank P = 0.03; Figure 1). P− adult patients had higher VA-free survival during the follow-up, compared with P+ adult patients (92.5 vs. 82.7%, Log-rank P = 0.041).
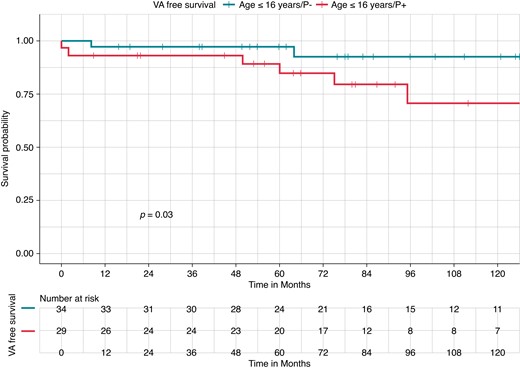
Kaplan–Meier curves of survival free from ventricular arrhythmia occurrence by P/LP variant carrier status in a paediatric Brugada syndrome population (≤16 years). Kaplan–Meier curve of survival free from VA occurrence by P/LP variant; patients without any P/LP variants (P−) (blue, upper curve) had higher VA-free survival during the follow-up, compared with those with a P/LP variant (P+) patients (red, lower curve) (94.1 vs. 79.3%, Log-rank P = 0.03). P/LP, pathogenic/likely pathogenic; VA, ventricular arrhythmia.
There was no difference in VA-free survival between children and adults with BrS (87.3 vs. 90.8%, Log-rank P = 0.47; Figure 2).
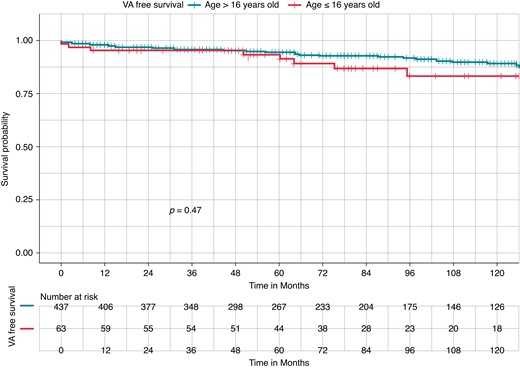
Kaplan–Meier curves of survival free from ventricular arrhythmia occurrence in Brugada syndrome by age group: paediatric cohort (≤16 years) vs. adult cohort (>16 years). Kaplan–Meier curve of survival free from ventricular arrhythmia (VA) occurrence. There was no difference in VA-free survival between paediatric Brugada syndrome (BrS) patients (red, lower curve) and adult BrS patients (blue, upper curve) (87.3 vs. 90.8%, Log-rank P = 0.47).
At survival analysis stratified for P− or P+, there was no difference in VA-free survival between paediatric and adult BrS patients both without (P−; 94.1 vs. 92.5%, pairwise Log-rank P = 0.55) and with (P+; 79.3 vs. 82.7%, pairwise Log-rank P = 0.45) a P/LP variant in SCN5A (Figure 3).
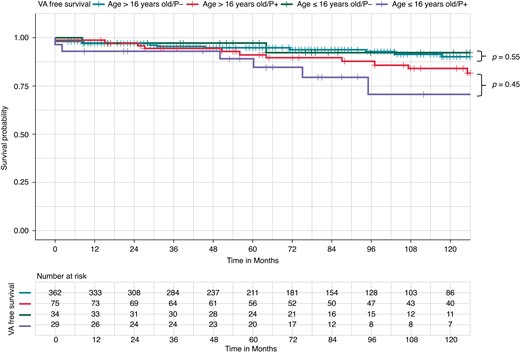
Kaplan–Meier curves of survival free from ventricular arrhythmia occurrence by pathogenic/likely pathogenic (P/LP) variant carrier status and paediatric (≤16 years) vs. adult (>16 years) Brugada syndrome. Kaplan–Meier curve of survival free from ventricular arrhythmia (VA) occurrence. There was no difference in VA-free survival between P− paediatric Brugada syndrome (BrS) patients (green curve, upper) and P− adult BrS patients (blue curve, middle upper) (94.1 vs. 92.5%, pairwise Log-rank P = 0.55). There was no difference in VA-free survival in P+ paediatric BrS patients (purple curve, lower) and P+ adult BrS patients (red curve, middle lower) (79.3 vs. 82.7%, pairwise Log-rank P = 0.45).
At Cox multivariate analysis, independent predictors of VA occurrence in the paediatric population were as follows: P/LP variant in SCN5A carrier status [hazard ratio (HR) = 3.92, 95% confidence interval (CI) 1.85–32.52, P = 0.02], spontaneous BrS I pattern (HR = 2.78, 95% CI 1.27–15.69, P = 0.02), SND (HR = 11.03, 95% CI 3.26–96.63, P = 0.003), and history of aborted SCD (HR = 20.28, 95% CI 7.9–142.40, P < 0.001).
Discussion
The main findings of this study are summarized as follows: (i) In a large cohort of BrS patients, undergoing genetic analysis with a large gene panel and classification of variants following ACMG guidelines, the diagnostic yield for P/LP variants in the paediatric population was 46.0% of which 39.3% of P/LP variants are present in the pore region; (ii) paediatric patients without a P/LP variant in SCN5A had higher VA-free survival during the follow-up, compared with P+ patients; (iii) there was no difference in VA-free survival between paediatric and adult BrS patients both without and with a P/LP variant in SCN5A; and (iv) SCN5A carrier status was an independent predictor of VA.
Diagnostic yield of genetic testing in paediatric Brugada population
In the current study, the yield of genetic testing in a large paediatric BrS cohort is 46.0%. This is higher compared with the adult BrS patients from the same cohort (17.2%) and compared with previous studies reporting a P/LP variant in 20–25% of all-comer BrS patients.14–16,35–37 However, the data presented are consistent with previous literature focusing on the paediatric population. In particular, our group previously found a total of 7 (37%) mutations in the SCN5A gene in 19 paediatric BrS patients.8 Furthermore, Righi et al.38 found a mutation in SCN5A gene in 14 of 39 patients (35.9%). Previous studies may have been hampered by inconsistent use of ACMG classification22 that brought standardization for variant classification in both research and clinical field.39 However, strict use of these guidelines might lead to a conservative interpretation, increasing VUS reporting especially for missense variants and this may increase the false-negative rate.40 The reported diagnostic yield of ≈45% for SCN5A gene in the paediatric population, despite the use of ACMG criteria, reflects a careful familial screening and a thorough co-segregation analysis.
The role of pathogenic/likely pathogenic variants in paediatric patients with Brugada syndrome
Paediatric patients with a P/LP variant in SCN5A had a worse arrhythmic prognosis compared with P− patients and SCN5A variant carrier status was an independent predictor of VA. There was no difference in VA-free survival between paediatric and adult BrS patients both without and with a P/LP variant in SCN5A.
Previous studies on all-comer BrS population demonstrated that SCN5A P/LP mutation carrier status is associated with delayed epicardial activation,18,20 worse epicardial substrate,17 and lower survival free from ventricular events.21
In paediatric BrS patients the role of SCN5A P/LP variants is controversial. Indeed, Righi et al.38 demonstrated that in BrS patients ≤12 years, SCN5A variants are associated with a higher occurrence of malignant VA. However, the low number of events did not allow to correct for potential confounders. Michowitz et al.5 found that SCN5A P/LP variant is an independent predictor of VA but only in the 13–20 years cohort, probably because of a lack of power. In the study by Andorin et al.41 SCN5A positive genotype showed a trend as a predictor of VA, although not significant (P = 0.08). In the current study, independent predictors of VA occurrence in the paediatric population were as follows: P/LP variant in SCN5A carrier status, spontaneous BrS I pattern, SND, and history of aborted SCD. These data are in agreement with Andorin et al.41 that identified spontaneous Type I ECG pattern and symptoms at diagnosis as independent predictors of VA. In particular, symptoms at diagnosis were defined as a composite of history of aborted SCD, VA or syncope. Syncope in paediatric BrS population may be associated to bradyarrhythmias and SND. The latter was an independent risk factor in our cohort, together with aborted SCD and spontaneous BrS pattern I. Our data are also consistent with Michowitz et al.5 demonstrating intraventricular conduction delay, presence of S wave in ECG Lead I, SND and atrial arrhythmias as risk factors in the paediatric group, while SCN5A mutation was associated with VA only in the adolescent group. In a previous study by our group,27 on patients ≤19 years the variables significantly associated with events and included in the final model were as follows: presentation with SCD or syncope, spontaneous Type I ECG, SND, and/or atrial tachycardia and conduction abnormality. However, only 38% of patients had a genetic test in the previous study, thus no definitive conclusion could be drawn about the role of SCN5A. This is the first study to demonstrate that a P/LP variant in SCN5A is an independent risk factor for VA in all paediatric (<16 years) BrS patients.
In the current study, there was no difference between P+ and P− paediatric patients in clinical variables known to be associated with VA and the lower survival free from ventricular events in the P+ population might be attributed to a P/LP variant in SCN5A gene.
Limitations
Limitations include referral bias due to the inclusion of patients from a tertiary centre specialized in BrS. In vitro functional characterization of variants was not performed routinely. An analysis of genotype–phenotype correlation based on a specific topographic location or variant was not performed due to the low number of specific variants available. The role of non-SCN5A genes in children with BrS remains unclear.
Conclusions
In a large paediatric BrS cohort undergoing genetic analysis with a large gene panel using ACMG standard classification of variants, a P/LP variant in SCN5A can be found in 44.7% of patients ≤16 years. Paediatric patients without a P/LP variant in SCN5A had higher VA-free survival during the follow-up, compared with P+ patients. SCN5A carrier status was an independent predictor of VA.
Funding
The genetic research was funded by the Innoviris BRIDGE strategic platform 2017 through the IGenCare (Integrated Personalised Medical Genomics Care Solution for Patients with Rare Genetic Diseases) project (BRGIMP12), by the Interdisciplinary Research Projects of the Vrje Universiteit Brussel ‘IRP8_a: IMAGica: an Integrative personalised Medical Approach for Genetic diseases, Inherited Cardiac Arrhythmias as a model’ and ‘IRP8_b: Bridging the gap between explainable AI and evidence-based medicine, a patient’s and expert’s perspective in inherited cardiac arrhythmias’ and by Wetenschappelijk Fonds Willy Gepts through the granted projects ‘Unraveling the molecular genetic pathways of Brugada syndrome by a next generation sequencing candidate gene approach’, ‘Unraveling the molecular genetic pathways of Brugada syndrome via cardiomics research’ and ‘The genetic complexity of Brugada syndrome: a multiple hit disease model’.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
Author notes
Luigi Pannone and Antonio Bisignani contributed equally and shared first authorship.
Sonia Van Dooren and Carlo de Asmundis contributed equally and shared last authorship.
Conflict of interest: A.B. is a consultant for Biotronik. A.S. received research grants from Daiichi-Sankyo and Bayer; he has received speaker fees from Menarini and Bayer. V.M. received an educational grant from the Foundation ‘Enrico and Enrica Sovena’. M.L.M. is consultant for Atricure. P.B. received compensation for teaching purposes from Biotronik. G.B.C. received compensation for teaching purposes and proctoring from Medtronic, Abbott, Biotronik, Boston Scientific, and Acutus Medical. C.d.A. receives research grants on behalf of the centre from Biotronik, Medtronic, Abbott, LivaNova, Boston Scientific, AtriCure, Philips, and Acutus; C.d.A. received compensation for teaching purposes and proctoring from Medtronic, Abbott, Biotronik, LivaNova, Boston Scientific, Atricure, Acutus Medical Daiichi Sankyo. The remaining authors have nothing to disclose.

{kind=link}
{kind=link}
{kind=link}
{kind=link}