





Abstract
Sudden cardiac death (SCD) is responsible for a large proportion of non-traumatic, sudden and unexpected deaths in young individuals. Sudden cardiac death is a known manifestation of several inherited cardiac diseases. In post-mortem examinations, about two-thirds of the SCD cases show structural abnormalities at autopsy. The remaining cases stay unexplained after thorough investigations and are referred to as sudden unexplained deaths. A routine forensic investigation of the SCD victims in combination with genetic testing makes it possible to establish a likely diagnosis in some of the deaths previously characterized as unexplained. Additionally, a genetic diagnose in a SCD victim with a structural disease may not only add to the differential diagnosis, but also be of importance for pre-symptomatic family screening. In the case of SCD, the optimal establishment of the cause of death and management of the family call for standardized post-mortem procedures, genetic screening, and family screening. Studies of genetic testing in patients with primary arrhythmia disorders or cardiomyopathies and of victims of SCD presumed to be due to primary arrhythmia disorders or cardiomyopathies, were systematically identified and reviewed. The frequencies of disease-causing mutation were on average between 16 and 48% in the cardiac patient studies, compared with ∼10% in the post-mortem studies. The frequency of pathogenic mutations in heart genes in cardiac patients is up to four-fold higher than that in SCD victims in a forensic setting. Still, genetic investigation of SCD victims is important for the diagnosis and the possible investigation of relatives at risk.
Introduction
Sudden cardiac death (SCD) is defined as sudden, natural, and unexpected death due to cardiac or unknown causes that occurs, if witnessed, within 1 h of symptom onset and if unwitnessed, occurring <24 h after the deceased was last seen alive and functioning normally.1,2 Sudden cardiac death is one of the most common causes of death in the Western world with an incidence of 50–60/100 000 individuals per year.3,4 The majority occurs in the older population, often secondary to coronary heart disease.5,6 In infants, children, adolescents, and young adults, the incidence of SCD is 1–8/100 000 individuals per year7,8 and accounts for a large proportion of the total deaths in the young.7,9–11
Numerous conditions and diseases may present as SCD. If a hereditary cardiac disease associated with SCD in the young (<40 years) is present, it is in the majority of cases inherited autosomal-dominantly.12 This adds to the importance of establishing the cause of death because of the potential for screening and prophylactic treatment of family members. In studies applying thorough medico-legal investigation, including autopsy and toxicology screening, two-third of the SCD cases have been reported to have structural abnormalities of the heart.9 Additionally, approximately half of the cases (1–49 years) with structural changes can be attributed to inherited cardiac diseases including cardiomyopathies, premature ischaemic heart diseases, and aortic diseases.12,13 The remaining third of SCD cases are autopsy negative and termed sudden unexplained death (SUD) or sudden arrhythmia death syndrome (SADS), i.e. no cause of death—including normal heart and normal toxicology screening—is identified at the post-mortem examination. Many of these unexplained deaths can be attributed to primary arrhythmia disorders, i.e. inherited cardiac ion channel diseases.14–17
An optimal management strategy including prevention of future SCD in potentially at-risk relatives to SCD victims is based on a thorough and systematic medico-legal investigation.18,19 As a considerable proportion of the SCD cases are likely to have an inherited cardiac disease, molecular autopsy and genetic testing might be valuable in the forensic setting. The advances in the genetic field during the last decades have identified several genes associated with SCD. Introducing molecular autopsy as a part of the forensic investigation has the potential to enhance the diagnostic rate of SCD cases and at the same time to be a major step in the approach to screening of the family left behind. In this study, we review the reported genetic findings in young SCD victims and compare them to those reported in patients with inherited cardiac diseases.
Methods
Sources and selection criteria
Papers were systematically searched in PubMed for studies of genetic testing in diagnosed cardiac patients, SCD/SUD/SADS victims, using the search terms ‘genetic’ ‘OR’ ‘mutation’ ‘OR’ ‘screening’ ‘OR’ ‘molecular autopsy’ ‘AND’ one of the following ‘HCM’, ‘DCM’, ‘ARVC’, ‘LQTS’, ‘BrS’, ‘CPVT’, ‘SCD’, ‘SUD’, or ‘SADS’. In addition, cited articles in ‘see related articles’ in PubMed were also reviewed. Studies were included in this review if genetic analyses were performed in the commonly SCD-associated genes in a cohort and consisting of (i) patients clinically diagnosed with hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), long QT syndrome (LQTS), Brugada syndrome (BrS), or cathecholaminergic polymorphic ventricular tachycardia (CPVT); or (ii) deceased individuals with unknown cause of death or with structural abnormalities of the heart indicating the presence of an inherited cardiac disease. Below are short descriptions of the diseases included in the study.
Inherited cardiac diseases with structural abnormalities
The cardiomyopathies compose a large proportion of the inherited structural abnormalities associated with SCD. The familial cardiomyopathies are characterized by functional or structural abnormalities of the myocardium due to alterations of the anatomy, e.g. hypertrophy, loss or disarray of the muscle cells, or increased fat and scar tissue. This may lead to impaired systolic function, progressive heart failure, arrhythmias, and sudden death.20 The clinical presentation may vary from no symptom to chest discomfort, heart failure symptoms, palpitations, syncope, and SCD.21 The cardiomyopathies most frequently associated with SCD are HCM, DCM, and ARVC. All three cardiomyopathies are heterogeneous diseases with incomplete penetrance and variable expression.
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy is autosomal dominantly inherited, has a prevalence of 1:500 in the general population and is characterized by unexplained left ventricular hypertrophy. At autopsy hypertrophy, myofiber disarray and increased interstitial fibrosis are seen,21–23 but no firm criteria for the diagnosis post-mortem have been established.
The genetic causes of HCM are heterogeneous and include mutations in genes encoding sarcomeric, calcium-handling, and mitochondrial proteins.24,25 Hundreds of mutations have been identified in almost 30 genes (Supplementary material online, Table S1). Mutations in the sarcomere genes are most commonly involved and mutations in MYBPC3 and MYH7 are responsible for the majority of cases (Table 1).
Mutation rates of the most common genes associated with SCD in patients and post-mortem studies
| Gene | Protein | Mutation rate (%) | Reference | |
|---|---|---|---|---|
| Patient studies | Post-mortem studies | |||
| LQTS | ||||
| KCNQ1a | Potassium voltage-gated channel subfamily KQT member 1 | 4–92 | 2–7 | 26–37 |
| KCNH2a | Potassium voltage-gated channel subfamily H member 2 | 8–40 | 0–8 | 26–38 |
| SCN5Aa | Sodium channel protein type 5 subunit alpha | 2–33 | 0–3 | 26–37,39 |
| KCNE1 | Potassium voltage-gated channel subfamily E member 1 | 0–2 | 0–6 | 27,30–32,34–38,40 |
| KCNE2 | Potassium voltage-gated channel subfamily E member 2 | 0–1 | 0–1 | 27,30–32,34–38,40 |
| CAV3 | Caveolin-3 | 2 | – | 41 |
| CPVT | ||||
| RYR2a | Ryanodine receptor 2 | 25–60 | 9–12 | 29,42–46 |
| BrS | ||||
| SCN5Aa | Sodium channel protein type 5 subunit alpha | 14–30 | (0–3)b | 39,47–52 |
| HCM | ||||
| MYBPC3a | Myosin-binding protein C, cardiac-type | 4–39 | 2–4 | 26,53–56 |
| MYH7a | Myosin-7 (heavy chain, cardiac muscle, beta) | 2–35 | 4–5 | 26,53–65,67 |
| MYH6 | Myosin-6 (heavy chain, cardiac muscle, alpha) | 6 | 0 | 26,62 |
| TNNT2 | Troponin T, cardiac muscle | 1–6 | 0 | 26,54–65 |
| TNNI3 | Troponin I, cardiac muscle | 0–5 | 0 | 26,54–58,60–63 |
| TPM1 | Tropomyosin alpha-1 chain | 1–5 | 0 | 26,54–58,60,62 |
| MYL2 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | 1–2 | 0 | 53,55,56,58,60,62,63 |
| MYL3 | Myosin light chain 3 | 1 | 0 | 26,53,55,58,60,62 |
| ACTC1 | Actin, alpha cardiac muscle 1 | 0–1 | 0 | 26,55,58,60,62 |
| ARVC | ||||
| PKP2a | Plakophilin-2 | 11–42 | 7–16 | 53,68–77 |
| DSG2a | Desmoglein-2 | 3–20 | 0 | 53,69–77 |
| DSPa | Desmoplakin | 3–20 | 0 | 53,69–75 |
| DSC2 | Desmocollin-2 | 1–8 | 0 | 53,69–72,74–78 |
| JUP | Junction plakoglobin | 0–6 | 0 | 53,69,70,72,74,75,77 |
| TGFB3 | Transforming growth factor beta-3 | 0–3 | - | 69,70,73 |
| TMEM43 | Transmembrane protein 43 | 2 | 0 | 53,70 |
| DCM | ||||
| LMNAa | Lamin-A/C | 7–18 | 7 | 53,79–83 |
| MYH7a | Myosin-7 (heavy chain, cardiac muscle, beta) | 3–7 | (4–5)b | 79,82–84 |
| TNNT2 | Troponin T, cardiac muscle | 1–5 | 0 | 79,82–84 |
| TNNI3 | Troponin I, cardiac muscle | 1 | 0 | 79,82,83 |
| Gene | Protein | Mutation rate (%) | Reference | |
|---|---|---|---|---|
| Patient studies | Post-mortem studies | |||
| LQTS | ||||
| KCNQ1a | Potassium voltage-gated channel subfamily KQT member 1 | 4–92 | 2–7 | 26–37 |
| KCNH2a | Potassium voltage-gated channel subfamily H member 2 | 8–40 | 0–8 | 26–38 |
| SCN5Aa | Sodium channel protein type 5 subunit alpha | 2–33 | 0–3 | 26–37,39 |
| KCNE1 | Potassium voltage-gated channel subfamily E member 1 | 0–2 | 0–6 | 27,30–32,34–38,40 |
| KCNE2 | Potassium voltage-gated channel subfamily E member 2 | 0–1 | 0–1 | 27,30–32,34–38,40 |
| CAV3 | Caveolin-3 | 2 | – | 41 |
| CPVT | ||||
| RYR2a | Ryanodine receptor 2 | 25–60 | 9–12 | 29,42–46 |
| BrS | ||||
| SCN5Aa | Sodium channel protein type 5 subunit alpha | 14–30 | (0–3)b | 39,47–52 |
| HCM | ||||
| MYBPC3a | Myosin-binding protein C, cardiac-type | 4–39 | 2–4 | 26,53–56 |
| MYH7a | Myosin-7 (heavy chain, cardiac muscle, beta) | 2–35 | 4–5 | 26,53–65,67 |
| MYH6 | Myosin-6 (heavy chain, cardiac muscle, alpha) | 6 | 0 | 26,62 |
| TNNT2 | Troponin T, cardiac muscle | 1–6 | 0 | 26,54–65 |
| TNNI3 | Troponin I, cardiac muscle | 0–5 | 0 | 26,54–58,60–63 |
| TPM1 | Tropomyosin alpha-1 chain | 1–5 | 0 | 26,54–58,60,62 |
| MYL2 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | 1–2 | 0 | 53,55,56,58,60,62,63 |
| MYL3 | Myosin light chain 3 | 1 | 0 | 26,53,55,58,60,62 |
| ACTC1 | Actin, alpha cardiac muscle 1 | 0–1 | 0 | 26,55,58,60,62 |
| ARVC | ||||
| PKP2a | Plakophilin-2 | 11–42 | 7–16 | 53,68–77 |
| DSG2a | Desmoglein-2 | 3–20 | 0 | 53,69–77 |
| DSPa | Desmoplakin | 3–20 | 0 | 53,69–75 |
| DSC2 | Desmocollin-2 | 1–8 | 0 | 53,69–72,74–78 |
| JUP | Junction plakoglobin | 0–6 | 0 | 53,69,70,72,74,75,77 |
| TGFB3 | Transforming growth factor beta-3 | 0–3 | - | 69,70,73 |
| TMEM43 | Transmembrane protein 43 | 2 | 0 | 53,70 |
| DCM | ||||
| LMNAa | Lamin-A/C | 7–18 | 7 | 53,79–83 |
| MYH7a | Myosin-7 (heavy chain, cardiac muscle, beta) | 3–7 | (4–5)b | 79,82–84 |
| TNNT2 | Troponin T, cardiac muscle | 1–5 | 0 | 79,82–84 |
| TNNI3 | Troponin I, cardiac muscle | 1 | 0 | 79,82,83 |
The table shows the range of pathogenic mutations in heart genes found in studies of patients diagnosed with inherited cardiac disease and in deceased suspected of SCD.
LQTS, long QT syndrome; CPVT, cathecholaminergic polymorphic ventricular tachycardia; BrS, Brugada syndrome; HCM, hypertrophic cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; DCM, dilated cardiomyopathy.
aRecommended to be included in a restricted gene panel.
bInvestigated in a phenotypically different cohort.
Mutation rates of the most common genes associated with SCD in patients and post-mortem studies
| Gene | Protein | Mutation rate (%) | Reference | |
|---|---|---|---|---|
| Patient studies | Post-mortem studies | |||
| LQTS | ||||
| KCNQ1a | Potassium voltage-gated channel subfamily KQT member 1 | 4–92 | 2–7 | 26–37 |
| KCNH2a | Potassium voltage-gated channel subfamily H member 2 | 8–40 | 0–8 | 26–38 |
| SCN5Aa | Sodium channel protein type 5 subunit alpha | 2–33 | 0–3 | 26–37,39 |
| KCNE1 | Potassium voltage-gated channel subfamily E member 1 | 0–2 | 0–6 | 27,30–32,34–38,40 |
| KCNE2 | Potassium voltage-gated channel subfamily E member 2 | 0–1 | 0–1 | 27,30–32,34–38,40 |
| CAV3 | Caveolin-3 | 2 | – | 41 |
| CPVT | ||||
| RYR2a | Ryanodine receptor 2 | 25–60 | 9–12 | 29,42–46 |
| BrS | ||||
| SCN5Aa | Sodium channel protein type 5 subunit alpha | 14–30 | (0–3)b | 39,47–52 |
| HCM | ||||
| MYBPC3a | Myosin-binding protein C, cardiac-type | 4–39 | 2–4 | 26,53–56 |
| MYH7a | Myosin-7 (heavy chain, cardiac muscle, beta) | 2–35 | 4–5 | 26,53–65,67 |
| MYH6 | Myosin-6 (heavy chain, cardiac muscle, alpha) | 6 | 0 | 26,62 |
| TNNT2 | Troponin T, cardiac muscle | 1–6 | 0 | 26,54–65 |
| TNNI3 | Troponin I, cardiac muscle | 0–5 | 0 | 26,54–58,60–63 |
| TPM1 | Tropomyosin alpha-1 chain | 1–5 | 0 | 26,54–58,60,62 |
| MYL2 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | 1–2 | 0 | 53,55,56,58,60,62,63 |
| MYL3 | Myosin light chain 3 | 1 | 0 | 26,53,55,58,60,62 |
| ACTC1 | Actin, alpha cardiac muscle 1 | 0–1 | 0 | 26,55,58,60,62 |
| ARVC | ||||
| PKP2a | Plakophilin-2 | 11–42 | 7–16 | 53,68–77 |
| DSG2a | Desmoglein-2 | 3–20 | 0 | 53,69–77 |
| DSPa | Desmoplakin | 3–20 | 0 | 53,69–75 |
| DSC2 | Desmocollin-2 | 1–8 | 0 | 53,69–72,74–78 |
| JUP | Junction plakoglobin | 0–6 | 0 | 53,69,70,72,74,75,77 |
| TGFB3 | Transforming growth factor beta-3 | 0–3 | - | 69,70,73 |
| TMEM43 | Transmembrane protein 43 | 2 | 0 | 53,70 |
| DCM | ||||
| LMNAa | Lamin-A/C | 7–18 | 7 | 53,79–83 |
| MYH7a | Myosin-7 (heavy chain, cardiac muscle, beta) | 3–7 | (4–5)b | 79,82–84 |
| TNNT2 | Troponin T, cardiac muscle | 1–5 | 0 | 79,82–84 |
| TNNI3 | Troponin I, cardiac muscle | 1 | 0 | 79,82,83 |
| Gene | Protein | Mutation rate (%) | Reference | |
|---|---|---|---|---|
| Patient studies | Post-mortem studies | |||
| LQTS | ||||
| KCNQ1a | Potassium voltage-gated channel subfamily KQT member 1 | 4–92 | 2–7 | 26–37 |
| KCNH2a | Potassium voltage-gated channel subfamily H member 2 | 8–40 | 0–8 | 26–38 |
| SCN5Aa | Sodium channel protein type 5 subunit alpha | 2–33 | 0–3 | 26–37,39 |
| KCNE1 | Potassium voltage-gated channel subfamily E member 1 | 0–2 | 0–6 | 27,30–32,34–38,40 |
| KCNE2 | Potassium voltage-gated channel subfamily E member 2 | 0–1 | 0–1 | 27,30–32,34–38,40 |
| CAV3 | Caveolin-3 | 2 | – | 41 |
| CPVT | ||||
| RYR2a | Ryanodine receptor 2 | 25–60 | 9–12 | 29,42–46 |
| BrS | ||||
| SCN5Aa | Sodium channel protein type 5 subunit alpha | 14–30 | (0–3)b | 39,47–52 |
| HCM | ||||
| MYBPC3a | Myosin-binding protein C, cardiac-type | 4–39 | 2–4 | 26,53–56 |
| MYH7a | Myosin-7 (heavy chain, cardiac muscle, beta) | 2–35 | 4–5 | 26,53–65,67 |
| MYH6 | Myosin-6 (heavy chain, cardiac muscle, alpha) | 6 | 0 | 26,62 |
| TNNT2 | Troponin T, cardiac muscle | 1–6 | 0 | 26,54–65 |
| TNNI3 | Troponin I, cardiac muscle | 0–5 | 0 | 26,54–58,60–63 |
| TPM1 | Tropomyosin alpha-1 chain | 1–5 | 0 | 26,54–58,60,62 |
| MYL2 | Myosin regulatory light chain 2, ventricular/cardiac muscle isoform | 1–2 | 0 | 53,55,56,58,60,62,63 |
| MYL3 | Myosin light chain 3 | 1 | 0 | 26,53,55,58,60,62 |
| ACTC1 | Actin, alpha cardiac muscle 1 | 0–1 | 0 | 26,55,58,60,62 |
| ARVC | ||||
| PKP2a | Plakophilin-2 | 11–42 | 7–16 | 53,68–77 |
| DSG2a | Desmoglein-2 | 3–20 | 0 | 53,69–77 |
| DSPa | Desmoplakin | 3–20 | 0 | 53,69–75 |
| DSC2 | Desmocollin-2 | 1–8 | 0 | 53,69–72,74–78 |
| JUP | Junction plakoglobin | 0–6 | 0 | 53,69,70,72,74,75,77 |
| TGFB3 | Transforming growth factor beta-3 | 0–3 | - | 69,70,73 |
| TMEM43 | Transmembrane protein 43 | 2 | 0 | 53,70 |
| DCM | ||||
| LMNAa | Lamin-A/C | 7–18 | 7 | 53,79–83 |
| MYH7a | Myosin-7 (heavy chain, cardiac muscle, beta) | 3–7 | (4–5)b | 79,82–84 |
| TNNT2 | Troponin T, cardiac muscle | 1–5 | 0 | 79,82–84 |
| TNNI3 | Troponin I, cardiac muscle | 1 | 0 | 79,82,83 |
The table shows the range of pathogenic mutations in heart genes found in studies of patients diagnosed with inherited cardiac disease and in deceased suspected of SCD.
LQTS, long QT syndrome; CPVT, cathecholaminergic polymorphic ventricular tachycardia; BrS, Brugada syndrome; HCM, hypertrophic cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; DCM, dilated cardiomyopathy.
aRecommended to be included in a restricted gene panel.
bInvestigated in a phenotypically different cohort.
Dilated cardiomyopathy
Dilated cardiomyopathy is clinically characterized by dilation of all four heart chambers and reduced myocardial contractility. The histology is non-specific and may include lymphatic infiltration and fibrosis.23 Dilated cardiomyopathy may present in a sporadic or a familial form. The familial form is often clinically defined by including two or more affected relatives affected at a young age.85,86 Familial DCM is in the vast majority of cased autosomal dominantly inherited. The prevalence of DCM is ∼1 : 2500 individuals.87 A genetic aetiology can be identified in up to 50% of cases of familial DCM.88,89
To date, more than 30 different genes have been associated with the disease (Supplementary material online, Table S1). The genes encode a variety of proteins in the sarcomere, cytoskeleton, sarcolemma, ion channels, and nucleus membrane. Thus, the prevalence of mutations in the individual genes is low, the most frequent ones being mutations in LMNA and MYH7 (Table 1).
Arrhythmogenic right ventricular cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy is characterized by myocardial wall thinning of the right ventricle, atrophy, and fibro-fatty infiltrations of the myocardium and is in the vast majority of cases autosomal dominantly inherited. Left ventricular involvement may also be present. Arrhythmogenic right ventricular cardiomyopathy has a prevalence of 1 : 2000–5000.23,90 According to the International Task Force criteria, revised in 2010, the diagnosis of ARVC should be based on a combination of structural, histological, electrocardiogram (ECG), arrhythmia, and familial features of the disease.91,92 For ARVC, post-mortem histological criteria for determining the diagnosis have been established.92
The mutations associated with the development of ARVC are mainly located in genes encoding for cell adhesion proteins. PKP2, DSG2, and DSP are the most frequently involved genes.93 Mutations in 12 genes (Supplementary material online, Table S1) are known to be involved in the development of ARVC and these mutations are responsible for about half of the cases (Table 1).
Inherited cardiac diseases without structural abnormalities
About one-third of sudden deaths in young persons are unexplained after a thorough post-mortem investigation with no structural cardiac abnormalities found at autopsy and a negative toxicology screen. A high proportion of these cases may be associated with inherited cardiac channelopathies.7,9,15 In cardiac channelopathies, gene mutations alter the function of ion channels in the myocyte leading to cardiac rhythm disturbances, ECG abnormalities, and an increased risk of sudden death without the presence of any morphological abnormality in the heart. The affected individuals may present with dyspnoea, palpitations, dizziness, syncope, or SCD. A considerable proportion never experience symptoms before SCD.40 The most common ion channel disorders associated with SCD include LQTS, BrS, and CPVT.
Long QT syndrome
Long QT syndrome is an inherited disease affecting 1 : 2500–5000 individuals. Long QT syndrome is characterized by delayed depolarization of the myocardium seen in the ECG as QT prolongation and a morphologically normal heart without any macro- or microscopic findings.23
There are hundreds of known mutations in more than 15 genes found to be associated with LQTS, mainly affecting potassium but also sodium and calcium ion channels, either by loss- or gain-of-function (Supplementary material online, Table S1). Three genes, KCNQ1, KCNH2, and SCN5A are responsible for more than two thirds of the genotype positive LQTS cases (Table 1).
Brugada syndrome
Brugada syndrome is an inherited channelopathy with an estimated incidence of up to 66 cases per 10 000 individuals.94 It is characterized by a specific ECG pattern showing right bundle branch block and a characteristic elevation of the ST segment and can in many patients be unmasked pharmacologically by blocking the myocardial sodium channels. Similar to the other channelopathies, autopsy findings in Brugada cases are unremarkable.23,95
Brugada syndrome is inherited autosomal dominantly and has an incomplete penetrance and variable expressivity. Mutations in BrS mainly affect the cardiac sodium channel NaV1.5 (Supplementary material online, Table S1). Approximately 20% of the BrS are associated with mutations in the SCN5A gene (Table 1).
Cathecholaminergic polymorphic ventricular tachycardia
Cathecholaminergic polymorphic ventricular tachycardia is an arrhythmogenic disorder characterized by a normal resting ECG and no cardiac abnormality.96 However, emotional or physical stress can trigger ventricular tachycardia, syncope, and sudden death.42 The prevalence is not yet established.
There are several known genotypic variants of the disease (Supplementary material online, Table S1), the most common being in CPVT1, inherited in an autosomal, dominant manner, affecting the calcium channels in the sarcoplasmic reticulum in the cardiac cells. Mutations affecting the ryanodine receptor 2 (RYR2) gene are responsible for up to 60% of the CPVT cases (Table 1).
Other inherited cardiac diseases associated with sudden cardiac death
Several other inherited cardiac diseases are also associated with SCD, e.g. familial hypercholesterolemia, short QT syndrome, left ventricular non-compaction, Anderson-Fabry disease, sick sinus syndrome, familial atrial fibrillation, paroxysmal supraventricular tachycardia (Wolff–Parkinson–White), progressive cardiac conduction disorder (Lenegree–Lev disease), paroxysmal familial ventricular fibrillation as well as syndromic and non-syndromic aortic diseases. These diseases will not be further addressed in this review.
Phenotypic classification—cardiac patients and sudden cardiac death victims
The frequency of mutations identified depends on the accuracy and precision of the phenotype. This is a challenge for several reasons, including that the inherited cardiac diseases associated with SCD show incomplete, age-dependent penetrance and variable expressivity. However, by careful clinical assessment combined with modern diagnostic tools including advanced imaging, physiological or pharmacological tests, biochemistry, etc., combined with presenting symptoms and patient history, a specific diagnosis may be established in most cases.
In contrast, when performing the post-mortem investigation of the young SCD victim, several of these diagnostic tools are obviously not applicable. For this reason, the phenotype diagnosed in the cardiac clinic and the phenotype diagnosed by post-mortem investigations may not have the same accuracy. Thus, negative or only unspecific pathological findings at autopsy may be seen in the SCD victims caused by a channelopathy. At autopsy, cardiomyopathies may show a diverse variety of macroscopic and microscopic changes with overlapping characteristics between subtypes.97–99 Additional challenges for the post-mortem diagnosis may include various degrees of decomposition of the tissues of the deceased, clinical records may not be available and in unwitnessed cases, it may be impossible to ascertain the circumstances of death, including ante-mortem symptoms.
Cardiac patients
In the patient studies reviewed, the frequencies of mutations were on average between 16 and 48% (Figure 1).
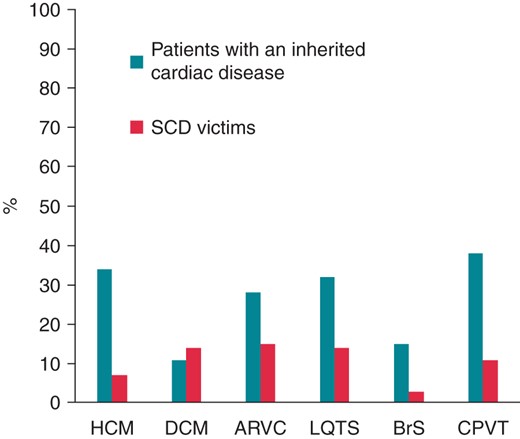
Average mutation rates in heart genes in cardiac patients and SCD victims. The figure shows the mean percentage of likely pathogenic mutations found in the reviewed studies related to HCM,26,53–67 DCM,26,53,79–84,100 ARVC,26,53,69–78 LQTS,26–28,30–40,101 BrS,26–28,40,47–49,102 and CPVT.40,42–46,103 The percentages indicate the average frequency rates of mutations in the genes associated with the different diseases. The columns ‘Patients with an inherited cardiac disease’ show results from the clinical studies and the columns ‘SCD victims’ show results from the post-mortem studies. HCM, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; LQTS, long QT syndrome; BrS, Brugada syndrome; CPVT, cathecholaminergic polymorphic ventricular tachycardia.
Sudden cardiac death victims
Post-mortem genetic testing of LQTS, BrS, and CPVT associated genes in young deceased persons with a negative autopsy classified as SUD have been reported,26–29,38,43,68 but only a few investigations of the genetic findings of SCD victims with structural abnormalities of the heart—pointing towards HCM, DCM, and ARVC—have been performed.26,53,68 Overall, the frequency of disease-causing mutations found in the post-mortem studies was ∼10% and only one-third to one-fourth of the frequencies found in the cardiac patients (Table 1 and Figure 1). These differences most likely reflect the variation in the phenotypic diagnostic specificity between the post-mortem and clinical setting. In addition, there are fewer post-mortem studies and the cohorts are smaller compared with the clinical studies, which may add to some of the differences in the reported mutation frequencies. Also, there is a bias in terms of the selection of patients vs. SCD victims for genetic testing. In the clinical setting, selection of patients for genetic screening is based on a suspicion of an inherited disease due to the presence of a phenotype based on presentation, symptoms, course of disease, clinical and paraclinical examinations, and family history. In comparison, selection of SCD victims for genetic screening is in most cases only based on the ‘SCD diagnose’, age and the fact that a number of other causes of death have been ruled out (by toxicology screens, exclusion of myocarditis, coronary occlusion, cerebral haemorrhage, etc.). Other important parameters—not least functional parameters, e.g. obtained by echocardiography—used in the cardiology department to identify the exact phenotype, are to a large extent missing. Therefore, the ‘SCD diagnose’ may represent a broad range of diagnoses. On this basis, we would a priori expect a lower frequency of pathogenic mutations in SCD victims when compared with those reported from cardiology departments.
The role of findings in the relatives to sudden cardiac death victims
The overall purpose of implementing genetic analysis as part of the routine forensic investigation should be to increase the diagnostic rate in SCD cases and to improve family screening for inherited cardiac diseases.104 As indicated, the case selection for genetic testing is essential for the outcome and should not only depend on the specific findings at the autopsy, but findings in the family may also play a major role for establishing the diagnosis. Papadakis et al.105 made a family evaluation of two groups a cohort of SADS individuals and a cohort of deceased with post-mortem structural findings of unknown significance. They found a very similar pattern in the diagnosis of the relatives in the two groups, namely that ∼50% of the relatives had an inherited cardiac disease, LQTS, and BrS being the most frequent ones. This illustrates that an accurate interpretation of autopsy findings is crucial and clear guidelines can benefit the yield of genetic testing in cases of SCD.
Molecular autopsy in forensic medicine
Molecular autopsy is important for several reasons. First, genetic investigation of SCD victims may improve the diagnostic rate and the identification of the genetic aetiology in the SCD victim allows pre-symptomatic genetic testing of relatives, i.e. a differentiation of relatives at risk and relatives not at risk before symptoms and/or clinical manifestations present. Several studies have shown that familial screening is of value for the diagnosis and prophylactic treatment of relatives to SCD victims.106–108 As suggested by Tan et al.109 genetic investigation in SCD victims should, if possible, be performed after a thorough cardiological examination of the first-degree relatives. Clinical findings may very likely be present in one or more living relatives, suggesting a diagnosis of the deceased. Phenotypic-guided genetic testing of the deceased can hereafter be performed and continued in the family if positive findings are present. However, this is not always possible and in some cases, genetic investigation in the deceased may be justified prior to clinical examinations of the relatives. Thus, e.g. in cases of SCD related to highly specific triggers (swimming, auditory stimuli, physical, or psychological stress), genetic testing for LQTS and CPVT, i.e. for mutations in RYR2, KCNQ1, KCNH2, and SCN5A, is relevant and has been reported to have led to a high frequency of positive findings in this setting.110 Also, in SCD cases fulfilling histology criteria for ARVC, screening for mutations in PKP2, DSG2, and DSP seems relevant. In cases with certain unexplained structural abnormalities of the myocardium, genetic testing of the most frequently affected cardiomyopathy-associated genes seems justified, e.g. MYBPC3, MYH7, TNNT2, and TNNI3 in suspected cases of HCM.
Secondly, a precise and complete death registry is important for the health care system; as research, evidence-based medicine and prophylactic screening are based on our knowledge of public health. However, registration of genetic test results in death registries depends on the local legislation.
Thirdly, in order to reduce the frequency of SCD, we need to expand the research in the genetics and pathogenesis of SCD. The genetic methods used in the reviewed studies were mainly first-generation sequencing. As next generation sequencing (NGS) is becoming a routinely integrated part of genetic investigations, it is tempting to speculate how this can improve the diagnostic rate in SCD cases. However, even if the efficiency and speed of NGS are evident, unselective screening of a large number of genes without a clear phenotype may be difficult in the diagnostic setting, as the bioinformatics will be very challenging.111,112 Correspondingly, rarely affected genes should probably not be a part of a routine investigation, as many mutations are private and very difficult to classify in terms of pathogenicity without functional and co-segregation family studies. The reviewed literature indicates that the majority of pathogenic mutations, corresponding to phenotype and pathology, are found in mainly 10 genes (KCNQ1, KCNH2, SCN5A, RYR2, MYH7, MYBPC3, PKP2, DSG2, DSP, and LMNA) as seen in Table 1. A restricted screening applying NGS of these genes would be cost-efficient. Thus, whole exome sequencing or broad screening including rarely affected genes, should still be a matter of scientific investigations. In general, genetic results should be interpreted with great care before a genotype–phenotype correlation can be established and the genotype is used for pre-symptomatic screening of relatives.
The introduction of routine molecular autopsy as part of the post-mortem investigation in sudden death should be based on clear recommendations including choice of tissue for investigations, indications for genetic testing, which genes to be investigated in relation to micro- and macroscopic findings, the interpretation of findings, screening, and follow-up of the family members, etc. Collaboration between forensic pathologists, clinical genetics, and cardiologists is essential in terms of identifying the familial predisposition, offer genetic counselling, clinical cardiological investigation, and prophylactic treatment to at-risk relatives of SCD victims.
Conclusion
Post-mortem genetic testing in young SCD victims identifies a likely genetic aetiology in about 10% of the cases. This frequency of identified mutations is only one-third to one-fourth of that found in patients with clinically diagnosed inherited cardiac disease. The difference relates to a number of factors including reduced accuracy in phenotypic characterization by present post-mortem diagnostic tools when compared with that of clinical characterization. The outcome of post-mortem examinations could be significantly enhanced by offering diagnostic investigations of relatives in specialized units with an interdisciplinary cooperation between forensic pathologists, cardiologists, and geneticists. Still, positive genetic findings are crucial for pre-symptomatic screening of the relatives and strategies to increase the genetic outcome are urgently needed.
Supplementary material
Supplementary material is available at Europace online.
Funding
This work was supported by Ellen and Aage Andersen's Foundation.
Conflict of interest: none declared.

{kind=link}