





Abstract
Coronary artery disease and heart failure carry concurrent risk for atrial fibrillation and life-threatening ventricular arrhythmias. We review evidence indicating that at therapeutic concentrations, ranolazine has potential for dual suppression of these arrhythmias. Mechanisms and clinical implications are discussed.
Introduction
Coronary artery disease and heart failure carry a dual risk for atrial fibrillation (AF) and life-threatening ventricular arrhythmias.1–4 Medical management of these conditions is challenging because of the complex and differing factors underlying arrhythmogenesis in the atria and ventricles. The difficulty of the problem is underscored by the fact that the main contemporary agents used for rhythm control in AF can result in ventricular proarrhythmia including torsade de pointes.5 Drugs with Class IC effects of blocking peak INa can cause myocardial depression, promote reentrant ventricular tachycardia, and increase mortality in patients with coronary disease. Anti-AF agents with the Class III action of blocking IKr, such as dofetilide and sotalol, can precipitate torsade de pointes. The multi-channel blocking agent amiodarone is currently widely employed, although its use can be problematic due to the potential for significant extra-cardiac toxicity, particularly in lung and thyroid tissue.6 Dronedarone, a non-iodinated benzofuran derivative of amiodarone that exhibits all four classes of antiarrhythmic action, was developed to achieve a better safety profile by eliminating the iodine moiety and substituting a methanesulfanyl group.7,8 Whereas most studies have shown that dronedarone reduces mortality and morbidity in patients with paroxysmal AF,7 it increased rates of heart failure, stroke, and death from cardiovascular causes in patients with permanent AF or moderate to severe heart failure.9 Although the exact basis for the increased risk in these patients is unclear, the potential for negative inotropy related to L-type calcium channel blockade at the currently recommended dosage has been suggested.
Ranolazine was developed as an antianginal agent and was initially thought to confer its therapeutic effects primarily through partial inhibition of fatty acid oxidation.10 These drug actions were generally observed at concentrations exceeding the plasma levels found in clinical trials (>10 μM).11 More recent evidence indicates that the anti-ischaemic effects of ranolazine relate to its inhibition of late INa, which, through an effect on reverse sodium–calcium exchange, improves diastolic compliance, thereby increasing coronary artery blood flow.12 Subsequent experimental studies have revealed a potent antiarrhythmic effect of ranolazine against both atrial and ventricular arrhythmias.12–15
Clinical evidence of ranolazine's antiarrhythmic efficacy has emerged from the MERLIN-TIMI 36 (Metabolic Efficiency with Ranolazine for Less Ischemia in Non-ST-Elevation Acute Coronary Syndrome–Thrombolysis in Myocardial Infarction 36) trial, which enrolled 6560 patients and was directed to the treatment of myocardial ischaemia in patients with non-ST elevation acute coronary syndrome.16 Ranolazine reduced the incidence of supraventricular arrhythmias with a trend towards lowering the incidence of new-onset AF across 6 days of continuous electrocardiographic recordings.17 A subsequent analysis indicated that among patients with paroxysmal AF, the overall burden tended to be lower with ranolazine than with placebo, with fewer AF-related adverse events.18 Ranolazine has also been demonstrated to be superior to amiodarone in reducing the incidence of AF following coronary artery bypass graph surgery19 and in combination therapy to improve conversion from recent onset AF over amiodarone alone.20 Murdock et al.21 recently reported in a case series that ranolazine can facilitate electrical cardioversion of AF in patients with cardioversion-resistant AF. Further analysis of the ambulatory electrocardiogram recordings from MERLIN TIMI-36 revealed that ranolazine can suppress not only supraventricular arrhythmias but also episodes of non-sustained ventricular tachycardia.22
The main goals of the present review are to summarize knowledge and discuss mechanisms of ranolazine's effects on both atrial and ventricular arrhythmogenesis.
Influence of ranolazine on atrial and ventricular electrical properties in the normal, intact heart
Programmed electrical stimulation was employed in anaesthetized canine and porcine models to evaluate the effects of intravenous ranolazine on excitability, refractoriness, dromotropy, and vulnerability to AF and ventricular fibrillation (VF).23–27
Atrial electrical properties
Experiments in large animals demonstrated ranolazine's potent atrial effects,24 consistent with findings in isolated tissue studies by Antzelevitch and colleagues.14,28,29 Specifically, significant effects on atrial refractoriness were found in each atrial site tested, namely high right atrium, right atrial appendage, and posterolateral left atrium (Table 1). These changes occurred in a non-frequency-dependent manner, in contrast to reports of IKr blockade by agents such as dofetilide and d-sotalol and to results expected with peak INa blockade and post-repolarization refractoriness. The phenomenon of non-frequency dependence may result from reverse frequency-dependent IKr inhibition coupled with frequency-dependent peak INa inhibition. The magnitude of the increase in atrial effective refractory period in atrial sites (>20% increase from baseline) was greater than the effect on the right ventricular endocardium (<10% increase from baseline). No significant changes were observed in atrial or ventricular diastolic thresholds. Ranolazine increased median atrial conduction time, a surrogate marker for conduction velocity, from high right atrium to posterolateral left atrium in a frequency-dependent manner in agreement with prior studies and consistent with inhibition of peak INa in atrial tissue.
Ranolazine's effects on atrial and ventricular effective refractory periods
| Site | Median effective refractory period (milliseconds) | Interquartile range (milliseconds) | P value |
|---|---|---|---|
| High right atrium | |||
| Control | 167 | 159–179 | 0.006 |
| Ranolazine | 202 | 183–231 | |
| Right atrial appendage | |||
| Control | 158 | 136–164 | 0.002 |
| Ranolazine | 202 | 187–211 | |
| Posterolateral left atrium | |||
| Control | 140 | 117–150 | 0.014 |
| Ranolazine | 163 | 143–193 | |
| Right ventricle | |||
| Control | 253 | 243–260 | 0.006 |
| Ranolazine | 275 | 271–281 | |
| Site | Median effective refractory period (milliseconds) | Interquartile range (milliseconds) | P value |
|---|---|---|---|
| High right atrium | |||
| Control | 167 | 159–179 | 0.006 |
| Ranolazine | 202 | 183–231 | |
| Right atrial appendage | |||
| Control | 158 | 136–164 | 0.002 |
| Ranolazine | 202 | 187–211 | |
| Posterolateral left atrium | |||
| Control | 140 | 117–150 | 0.014 |
| Ranolazine | 163 | 143–193 | |
| Right ventricle | |||
| Control | 253 | 243–260 | 0.006 |
| Ranolazine | 275 | 271–281 | |
Reprinted from ref.24 with permission from John Wiley & Sons.
Ranolazine's effects on atrial and ventricular effective refractory periods
| Site | Median effective refractory period (milliseconds) | Interquartile range (milliseconds) | P value |
|---|---|---|---|
| High right atrium | |||
| Control | 167 | 159–179 | 0.006 |
| Ranolazine | 202 | 183–231 | |
| Right atrial appendage | |||
| Control | 158 | 136–164 | 0.002 |
| Ranolazine | 202 | 187–211 | |
| Posterolateral left atrium | |||
| Control | 140 | 117–150 | 0.014 |
| Ranolazine | 163 | 143–193 | |
| Right ventricle | |||
| Control | 253 | 243–260 | 0.006 |
| Ranolazine | 275 | 271–281 | |
| Site | Median effective refractory period (milliseconds) | Interquartile range (milliseconds) | P value |
|---|---|---|---|
| High right atrium | |||
| Control | 167 | 159–179 | 0.006 |
| Ranolazine | 202 | 183–231 | |
| Right atrial appendage | |||
| Control | 158 | 136–164 | 0.002 |
| Ranolazine | 202 | 187–211 | |
| Posterolateral left atrium | |||
| Control | 140 | 117–150 | 0.014 |
| Ranolazine | 163 | 143–193 | |
| Right ventricle | |||
| Control | 253 | 243–260 | 0.006 |
| Ranolazine | 275 | 271–281 | |
Reprinted from ref.24 with permission from John Wiley & Sons.
Atrial fibrillation suppression
To test whether ranolazine alters susceptibility to AF, the muscarinic transmitter acetylcholine (ACh) was administered into the pericardial space by percutaneous transatrial access.24 This approach incorporates multiple sites of action including atrial tissue, ganglionic plexi, and pulmonary veins, all of which have been implicated clinically in atrial arrhythmogenesis. A significant reduction in AF duration (Figure 1) and tendency to suppress AF re-initiation were observed following ranolazine administration. Despite the QT prolongation associated with AF-cardioversion, torsade de pointes was not observed. These results are likely indicative of the effect of ranolazine on atrial conduction velocity due to peak INa inhibition and are similar to the actions of other potent sodium channel blockers, including flecainide and propafenone, and suggest that ranolazine affects factors that are important not only in maintaining AF but also possibly in initiating AF.
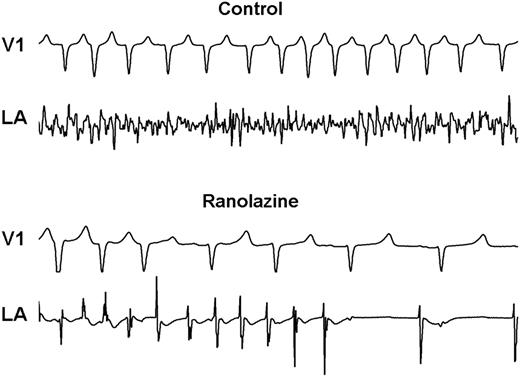
Representative example of surface (lead V1) and intracardiac electrograms (posterolateral left atrium, LA) at 12 min after atrial fibrillation (AF) initiation demonstrating early termination of AF by ranolazine. Note in the upper tracing (control) that AF is characterized by low voltage and high frequency. At the same time period, with ranolazine treatment, the arrhythmia became more organized, followed by conversion to sinus rhythm. (Reprinted from ref.24 with permission from John Wiley & Sons.).
Ranolazine reduced the dominant frequency of AF,24 an index of organization of AF and the propensity for successful defibrillation,30 in both the left and right atria. Consistent with previous reports of AF in humans and in other animal models,31 the dominant frequency of AF in the left atrium was always greater than that in the right atrial appendage, indicating that the drivers for AF in this model reside in the left atrium, where susceptibility to ACh-induced changes in rotor dynamics is enhanced.32 Ranolazine did not change this relationship, despite decreasing dominant frequency of AF in both atria.
The cellular and ionic mechanisms by which ranolazine reduces ACh-mediated AF are likely multifold: (i) depression of INa-dependent parameters, which reduces action potential upstroke, and increases in diastolic threshold of excitation and post-repolarization refractoriness; (ii) IKr inhibition, which counteracts ACh-induced shortening of action potential duration; (iii) late INa inhibition, which suppresses late phase 3 early after-depolarizations and reduces ITi, thereby suppressing delayed after-depolarizations and triggered activity.14,33 Ranolazine at the concentrations tested has minimum effect on ICa. The anti-AF capacity of ranolazine has also been shown by Lemoine et al.34 in terms of suppression of occurrence of early after-depolarizations and triggered activity in an animal model of LQT3 (SCN5A inactivation-impairing mutation).
Ventricular effects
Surface electrocardiogram analysis
Ranolazine moderately prolonged the QTc interval in the intact porcine model,23 consistent with previous reports in dogs35 but to a larger extent than is observed in humans.10,36 The drug did not alter Tpeak–Tend interval, an index of transmural dispersion of repolarization, a parameter identified in a canine wedge preparation to be attributable to differential effects on epicardial vs. M cells.14 These effects were at variance from d-sotalol, a selective IKr blocker, despite prolongation of action potential duration and the QT interval of the in-vitro-modelled electrocardiogram by both drugs.14
Electrophysiological testing
Ranolazine prolonged ventricular effective refractory period in the porcine model by a mean of 40 ms over a large range of stimulation strengths.23 Schram et al.35 had previously observed a mild but non-significant increase in effective refractory period in canines at both low and high doses of ranolazine. The significant changes in effective refractory period detected by Kumar et al.23 may be attributable to the larger sample size.
Ranolazine caused substantial increases in the magnitude of the depolarization threshold current needed to induce the repetitive extrasystoles (RE) and VF (Figure 2).23 Because the intensity of the stimulus required to provoke either RE or VF is inversely proportional to the intrinsic level of cardiac electrical heterogeneity, these measures yield an index of dispersion of refractoriness.37 The ventricular vulnerable period width has been shown to correlate with dispersion of repolarization,38 lending further support to these observations and to interpretation of the electrophysiological effects of ranolazine described. Accordingly, ranolazine's increase in the RE and VF thresholds and reduction in vulnerable period width may indicate its potential antiarrhythmic mechanism, namely, reduction in the dispersion of repolarization and refractoriness. These changes to the underlying myocardial substrate may be complementary to ranolazine's suppression of triggered activity due to early after-depolarizations.39 These findings are further supported by the report that in remodelled canine hearts with complete heart block, ranolazine suppressed drug-induced torsade de pointes, with concomitant changes in beat-to-beat variability of repolarization.40
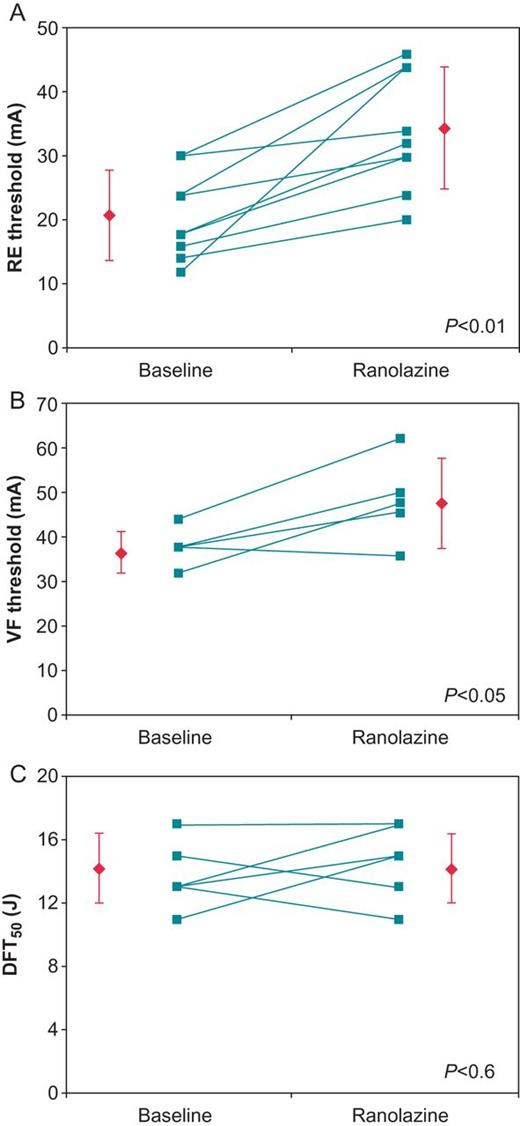
Connected dot plots of the (A) repetitive extrasystole (RE) (N= 9), (B) ventricular fibrillation (VF) (N = 5), and (C) 50% probability of defibrillation (DFT50) (N = 6) thresholds. Each line represents an individual experiment, while the diamonds represent the means ± SD. (Reprinted from ref.23 with permission from John Wiley & Sons.)
Defibrillation threshold testing
Ranolazine did not alter the 50% probability of successful ventricular defibrillation (DFT50), likely as a consequence of the countervailing influences of IKr and INa inhibition (Figure 2).23 Notably, IKr inhibition would be expected to lower DFT50, whereas INa inhibition would increase it. Ranolazine exerts minor influences on peak INa at slow to moderate heart rates, but at short cycle lengths in depolarized cells, as during VF, the drug may inhibit peak INa more effectively. Use dependence and increased binding at less negative, that is, at more depolarized, membrane potentials is a typical characteristic of sodium channel blockers and should also apply to ranolazine.41 Enhanced peak INa inhibition during VF is consistent with the finding that ranolazine reduces the dominant frequency of VF, indicative of active tissue binding during VF notwithstanding the absence of an influence on DFT50. The observation that ranolazine's antifibrillatory effect is not associated with a significant increase in ventricular defibrillation threshold indicates that the drug appears unlikely to affect the margin of safety for defibrillation. In light of the importance of this issue, further experimentation is warranted including testing defibrillation threshold during acute myocardial ischaemia.
Antifibrillatory effects of ranolazine in the ischaemic heart
Ventricular effects during severe coronary artery stenosis
Because ischaemic heart disease is associated with increased risk for ventricular arrhythmias, the effects of ranolazine during severe left anterior descending coronary artery (LAD) stenosis were assessed in the intact porcine model.25 Electrode catheters allowed assessment of the effects of severe LAD stenosis on the levels of VF threshold and T-wave alternans (TWA), a beat-to-beat fluctuation in the ST-segment and T-wave, which has been shown to reflect susceptibility to malignant ventricular arrhythmias.42

Coronary stenosis resulted in a significant increase in T-wave alternans (TWA) level, which was significantly reduced by ranolazine (N = 12). Numbers in the columns refer to the mean levels of T-wave alternans (TWA) in microvolts. (Reprinted from ref.25 with permission from Heart Rhythm Society.)
Ranolazine significantly increased VF threshold not only in the normal heart but more importantly during myocardial ischaemia, when the effects occurred independently of changes in LAD coronary blood flow, which was controlled with a hydraulic occluder. During normal coronary artery flow, blockade of IKr and consequent prolongation of action potential duration contributed to increases in VF threshold induced by both ranolazine and the IKr blocker E-4031. However, during severe myocardial ischaemia, E-4031 was profibrillatory, as it decreased VF threshold from control stenosis, whereas ranolazine increased it. Importantly, TWA tracked the ranolazine- and E-4031-induced changes in VF threshold during myocardial ischaemia, that is, ranolazine decreased but E-4031 did not alter TWA levels from the control stenosis (Figures 3 and 4).25 Studies involving assessment of lactate production by sampling coronary venous effluent or other methods involving wall motion abnormalities and measurement of regional myocardial blood flow with microspheres are required to establish an effect of ranolazine on metabolism during myocardial ischaemia.
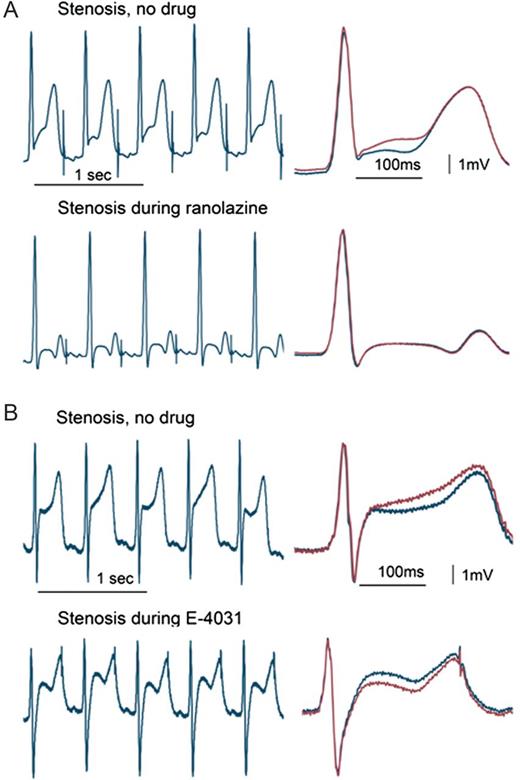
T-wave alternans (TWA) during left anterior descending coronary artery stenosis. The tracings with and without ranolazine (A) or E-4031 (B) are continuous (left panels) and with QRS-aligned superimposition (right panels). The stenosis-induced TWA was suppressed by ranolazine but not by E-4031. (Reprinted from ref.25 with permission from Heart Rhythm Society.)
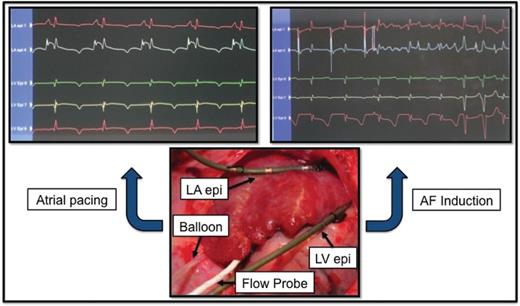
Experimental setup. Upper left panel: left atrial (LA) and left ventricular (LV) epicardial electrocardiograms obtained prior to balloon occlusion of the left circumflex (LCx) coronary artery to reduce flow by 75% during atrial pacing at 150 beats/min. Upper right panel: Induction of atrial fibrillation (AF) by a 6-mA S2 test stimulus following the last S1 pacing stimulus. Note visible T-wave heterogeneity compared to the uniform pattern observed in the upper left panel. Lower panel: Hydraulic balloon occluder positioned around the proximal LCx coronary artery upstream of the Doppler flow probe. Electrode catheters are affixed to the left atrial appendage and left ventricular epicardium within the atrial and ventricular regions supplied by the LCx. (Reprinted from ref. 47 with permission from Heart Rhythm Society.)
Ranolazine's effects on VF threshold and TWA levels indicate significant blunting of ischaemia-induced dispersion of repolarization and refractoriness.25 TWA reflects spatiotemporal heterogeneity of repolarization between neighbouring myocardial regions, as has been shown in investigations in intact large animals.43,44 and in patients with cardiomyopathy.45 The effects of ranolazine and E-4031 on VF threshold and TWA reflect their direct influences on the electrophysiological properties of the ventricular myocardium and are not attributable to changes in coronary artery blood flow as stenoses were maintained at the same level before and after drug administration. These results are consistent with findings reported by Kloner et al.,46 who showed in rats subjected to proximal left coronary artery occlusion followed by reperfusion that ranolazine at a dose consistent with inhibition of late INa exerts a potent antiarrhythmic effect and is superior to sotalol or lidocaine.
Ischaemia-induced atrial fibrillation
Ischaemic heart disease is associated not only with increased risk for malignant ventricular arrhythmias2 but also for AF.1,4 In patients with acute coronary syndromes, the incidence of AF has been reported to be 6–22% and is associated with increased short- and long-term morbidity and mortality. Nattel and coworkers27 provided experimental evidence indicating that atrial ischaemia can create a substrate for maintenance of AF by increasing local conduction slowing, leading to unidirectional block and reentry. They found that the l-type calcium channel blocker diltiazem and the beta-adrenergic receptor blocker nadolol were protective but that peak INa and IKr inhibition with flecainide and dofetilide, respectively, were not effective.26
Recently, we induced concurrent atrial and ventricular ischaemia to evaluate simultaneously the effects of antiarrhythmic agents on vulnerability to AF and ventricular tachyarrhythmias.47,48 Stenosis at the origin of the left circumflex coronary artery, which supplies all three major branches to the left atrium, namely, the proximal, intermediate, and distal arteries,49 induces significant left atrial ischaemia (Figure 5). In a preliminary report,48 we found that ranolazine at a plasma concentration in the clinical dose range completely suppressed the ischaemia-induced reduction in AF threshold. This protective effect against ischaemia-induced AF appears to be due to direct effects on atrial electrical properties independent of coronary flow, as this variable was controlled.
We also demonstrated that ranolazine and dronedarone at low dosages exhibit a synergistic, antiarrhythmic effect on vulnerability to AF and ventricular tachyarrhythmias during concurrent atrial and ventricular ischaemia.47 Specifically, when low doses of either agent were given alone, there was no protective effect, but the drug combination prevented the ischaemia-induced fall in AF threshold, reduced AF inducibility, and shortened AF duration while decreasing T-wave heterogeneity, a clinically relevant measure of susceptibility to life-threatening ventricular arrhythmias. Overall, these findings are consistent with the discovery by Burashnikov et al.50 that ranolazine and dronedarone afford synergistic antiarrhythmic actions with respect to AF susceptibility. The clinical relevance of synergistic antiarrhythmic protection by the combination of low doses of ranolazine and dronedarone is under investigation in ‘A Study to Evaluate the Effect of Ranolazine and Dronedarone When Given Alone and in Combination in Patients with Paroxysmal AF’ (HARMONY) trial (NCT 01522651). The effect of the drugs on AF burden will be studied in patients with implanted dual chamber programmable pacemakers with AF detection capabilities.
Ionic basis for ranolazine's antiarrhythmic effects
The three most likely ion channel currents responsible for the electrophysiological effects of ranolazine in the normal or ischaemic atrial and ventricular myocardium are late INa, peak INa, and IKr12 (Tables 2, 3). Analysis of excised atrial appendages from patients with AF have implicated late INa inhibition,51 while experimental studies in canine isolated perfused atrial tissue have provided evidence that ranolazine's antifibrillatory effect in the atria is attributable primarily to inhibition of peak INa.14 At therapeutic concentrations, ranolazine also inhibits the delayed rectifier potassium current (IKr),12,14 which has been extensively investigated as a target to suppress AF.52 In isolated tissue studies, evidence suggests that ranolazine's combined action on peak INa and IKr currents may enhance its efficacy in reducing susceptibility to AF, as this combination of ion channel effects contributes to atrial-selective reduction in AF vulnerability.53
Effects of ranolazine on various ion channel currents
| Ion channel current | Preparation | Potency (IC50, μM) | Comments | References |
|---|---|---|---|---|
| Peak INa | Canine ventricular myocytes | 294 | Tonic block | Undrovinas et al.56 |
| KPQ murine cardiomyocytes | 135 | Fredj et al.57 | ||
| HEK293 cells | 428 | Rajamani et al.41 | ||
| HEK293 cells | 285 | Zygmunt et al.58 | ||
| Late INa | Canine ventricular myocytes | 5.9 | Tonic block | Antzelevitch et al.53 |
| Canine ventricular myocytes KPQ | 6.5 | Undrovinas et al.56 | ||
| Murine cardiomyocytes | 15 | Fredj et al.57 | ||
| HEK293 cells | 6.9 | Wu et al.59 | ||
| Rabbit ventricular myocytes | 16.5 | Jia et al.60 | ||
| IKr | Canine ventricular myocytes | 11.5 | Antzelevitch et al.53 | |
| HEK293 cells | 14.4 | Wu et al.59 | ||
| INCX | Canine ventricular myocytes | 91 | Antzelevitch et al.53 | |
| tsA201 cells | 1.7 | Soliman et al.61 | ||
| Rabbit ventricular myocytes | >100 | Gilead, unpublished data | ||
| ICa,L | Canine ventricular myocytes | 296 | Antzelevitch et al.53 |
| Ion channel current | Preparation | Potency (IC50, μM) | Comments | References |
|---|---|---|---|---|
| Peak INa | Canine ventricular myocytes | 294 | Tonic block | Undrovinas et al.56 |
| KPQ murine cardiomyocytes | 135 | Fredj et al.57 | ||
| HEK293 cells | 428 | Rajamani et al.41 | ||
| HEK293 cells | 285 | Zygmunt et al.58 | ||
| Late INa | Canine ventricular myocytes | 5.9 | Tonic block | Antzelevitch et al.53 |
| Canine ventricular myocytes KPQ | 6.5 | Undrovinas et al.56 | ||
| Murine cardiomyocytes | 15 | Fredj et al.57 | ||
| HEK293 cells | 6.9 | Wu et al.59 | ||
| Rabbit ventricular myocytes | 16.5 | Jia et al.60 | ||
| IKr | Canine ventricular myocytes | 11.5 | Antzelevitch et al.53 | |
| HEK293 cells | 14.4 | Wu et al.59 | ||
| INCX | Canine ventricular myocytes | 91 | Antzelevitch et al.53 | |
| tsA201 cells | 1.7 | Soliman et al.61 | ||
| Rabbit ventricular myocytes | >100 | Gilead, unpublished data | ||
| ICa,L | Canine ventricular myocytes | 296 | Antzelevitch et al.53 |
Effects of ranolazine on various ion channel currents
| Ion channel current | Preparation | Potency (IC50, μM) | Comments | References |
|---|---|---|---|---|
| Peak INa | Canine ventricular myocytes | 294 | Tonic block | Undrovinas et al.56 |
| KPQ murine cardiomyocytes | 135 | Fredj et al.57 | ||
| HEK293 cells | 428 | Rajamani et al.41 | ||
| HEK293 cells | 285 | Zygmunt et al.58 | ||
| Late INa | Canine ventricular myocytes | 5.9 | Tonic block | Antzelevitch et al.53 |
| Canine ventricular myocytes KPQ | 6.5 | Undrovinas et al.56 | ||
| Murine cardiomyocytes | 15 | Fredj et al.57 | ||
| HEK293 cells | 6.9 | Wu et al.59 | ||
| Rabbit ventricular myocytes | 16.5 | Jia et al.60 | ||
| IKr | Canine ventricular myocytes | 11.5 | Antzelevitch et al.53 | |
| HEK293 cells | 14.4 | Wu et al.59 | ||
| INCX | Canine ventricular myocytes | 91 | Antzelevitch et al.53 | |
| tsA201 cells | 1.7 | Soliman et al.61 | ||
| Rabbit ventricular myocytes | >100 | Gilead, unpublished data | ||
| ICa,L | Canine ventricular myocytes | 296 | Antzelevitch et al.53 |
| Ion channel current | Preparation | Potency (IC50, μM) | Comments | References |
|---|---|---|---|---|
| Peak INa | Canine ventricular myocytes | 294 | Tonic block | Undrovinas et al.56 |
| KPQ murine cardiomyocytes | 135 | Fredj et al.57 | ||
| HEK293 cells | 428 | Rajamani et al.41 | ||
| HEK293 cells | 285 | Zygmunt et al.58 | ||
| Late INa | Canine ventricular myocytes | 5.9 | Tonic block | Antzelevitch et al.53 |
| Canine ventricular myocytes KPQ | 6.5 | Undrovinas et al.56 | ||
| Murine cardiomyocytes | 15 | Fredj et al.57 | ||
| HEK293 cells | 6.9 | Wu et al.59 | ||
| Rabbit ventricular myocytes | 16.5 | Jia et al.60 | ||
| IKr | Canine ventricular myocytes | 11.5 | Antzelevitch et al.53 | |
| HEK293 cells | 14.4 | Wu et al.59 | ||
| INCX | Canine ventricular myocytes | 91 | Antzelevitch et al.53 | |
| tsA201 cells | 1.7 | Soliman et al.61 | ||
| Rabbit ventricular myocytes | >100 | Gilead, unpublished data | ||
| ICa,L | Canine ventricular myocytes | 296 | Antzelevitch et al.53 |
Effects of ranolazine on G protein-coupled receptors (GPCR)
| GPCR | Preparation | Potency (Ki, μM) | Comments | References |
|---|---|---|---|---|
| α1 | Rat salivary gland | 8.2 | Radioligand binding | Zhao et al.62 |
| Rat liver | 19.5 | |||
| Rat ventricle | 10.5 | |||
| β1 | Rat ventricle | 8.6 | Binding | Zhao et al.62 |
| Rat heart | 1.4 | Letienne et al.63 | ||
| β2 | Guinea pig lung | 14.8 | Binding | Zhao et al.62 |
| Guinea pig lung | 0.5 | Binding | Letienne et al.63 | |
| DDT1MF-2 cells | 9.0 | Binding | Gilead, unpublished data |
| GPCR | Preparation | Potency (Ki, μM) | Comments | References |
|---|---|---|---|---|
| α1 | Rat salivary gland | 8.2 | Radioligand binding | Zhao et al.62 |
| Rat liver | 19.5 | |||
| Rat ventricle | 10.5 | |||
| β1 | Rat ventricle | 8.6 | Binding | Zhao et al.62 |
| Rat heart | 1.4 | Letienne et al.63 | ||
| β2 | Guinea pig lung | 14.8 | Binding | Zhao et al.62 |
| Guinea pig lung | 0.5 | Binding | Letienne et al.63 | |
| DDT1MF-2 cells | 9.0 | Binding | Gilead, unpublished data |
Effects of ranolazine on G protein-coupled receptors (GPCR)
| GPCR | Preparation | Potency (Ki, μM) | Comments | References |
|---|---|---|---|---|
| α1 | Rat salivary gland | 8.2 | Radioligand binding | Zhao et al.62 |
| Rat liver | 19.5 | |||
| Rat ventricle | 10.5 | |||
| β1 | Rat ventricle | 8.6 | Binding | Zhao et al.62 |
| Rat heart | 1.4 | Letienne et al.63 | ||
| β2 | Guinea pig lung | 14.8 | Binding | Zhao et al.62 |
| Guinea pig lung | 0.5 | Binding | Letienne et al.63 | |
| DDT1MF-2 cells | 9.0 | Binding | Gilead, unpublished data |
| GPCR | Preparation | Potency (Ki, μM) | Comments | References |
|---|---|---|---|---|
| α1 | Rat salivary gland | 8.2 | Radioligand binding | Zhao et al.62 |
| Rat liver | 19.5 | |||
| Rat ventricle | 10.5 | |||
| β1 | Rat ventricle | 8.6 | Binding | Zhao et al.62 |
| Rat heart | 1.4 | Letienne et al.63 | ||
| β2 | Guinea pig lung | 14.8 | Binding | Zhao et al.62 |
| Guinea pig lung | 0.5 | Binding | Letienne et al.63 | |
| DDT1MF-2 cells | 9.0 | Binding | Gilead, unpublished data |
On a cellular level, it is likely, based on results of studies in isolated single and multicellular preparations that ranolazine suppresses non-sustained ventricular tachycardia by inhibition of late INa, which results in increase in repolarization reserve and suppression of triggered activity caused by early and/or delayed after-depolarizations.14 Inhibition of late INa may also be implicated in ranolazine's capacity to reduce or abolish TWA, which results from mishandling of Ca2+ transients in the sarcoplasmic reticulum.42 Investigations in a canine heart failure model demonstrated that ranolazine reversibly blocked diastolic intracellular Ca2+ accumulation and spontaneous Ca2+ release by late INa inhibition.54 In terms of improvement in myocardial mechanical function, Lovelock et al.55 demonstrated in a murine model that diastolic dysfunction can be reversed by ranolazine, most likely resulting from a direct influence on myofilaments.
It is unlikely that ranolazine's antifibrillatory effects in both atrial and ventricular tissue during myocardial ischaemia without overt proarrhythmia are solely due to inhibition of peak INa and IKr as flecainide and E-4031, potent inhibitors of peak INa and IKr, respectively, were not protective during ischaemia, while ranolazine was antifibrillatory.25,47 Inhibition of late INa, with consequent reductions in the arrhythmogenic effects of intracellular calcium overload and improvement in repolarization reserve, is a plausible mechanism of the antifibrillatory effects of ranolazine and freedom from proarrhythmia.
Conclusions and future directions
Collectively, these experimental studies in intact large animal models indicate that at therapeutic concentrations, ranolazine exerts significant beneficial effects on electrophysiological properties of both the atria and ventricles, with greater effects on the former structure. Ranolazine's potent cardioprotective effects offer distinct advantages over other contemporary agents, as proarrhythmia was found not to be a factor in the MERLIN TIMI-36 clinical trial16,17,22 and in these large, intact animal models. Rather, the agent provides safe, dual protection against ischaemia-induced atrial and ventricular arrhythmias.25,47 Ranolazine also reduces ischaemia-induced TWA25 and T-wave heterogeneity,47 electrocardiographic phenomena indicating risk for ventricular arrhythmogenesis. As TWA can be assessed during routine exercise testing or ambulatory electrocardiogram monitoring,42 it can be employed to gauge clinical relevance of the experimental findings.
Notwithstanding this progress, there remains a need to explore further the underlying mechanisms responsible for ranolazine's antiarrhythmic action, in particular, to understand better the potential involvement of metabolic and antiischaemic influences in ranolazine's action. Also, it is important to study the effects of the agents in models in which chronic disease is simulated by infarction and electrical and structural remodelling.
Several clinical trials are underway that address these issues in populations with diverse indications, as referenced at www.clinicaltrials.gov. Collectively, the information reviewed and heightened interest in this compound with its unique mode of action are likely to contribute to important inroads in the management of supraventricular and ventricular arrhythmias.
Conflict of interest: L.B. is an employee of Gilead Sciences, Inc. None of the other authors declares a conflict of interest.
Funding
R.L.V. receives research grant support from Gilead Sciences, Inc.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}