





Introduction
Hypertrophic cardiomyopathy (HCM) is a myocardial disease characterized by left ventricular hypertrophy not solely explained by hemodynamic overload.1 This condition can lead to functional disability from heart failure and stroke, and represents one of the most frequent causes of sudden cardiac death (SCD) in young age.2,3 In the last decades, numerous prognostic factors have been investigated.2,4 Hyperhomocysteinaemia, defined as elevation of plasma homocysteine concentration, has been associated with stroke and arrhythmias and proposed as an independent cardiovascular disease risk factor.5 The C677T polymorphism for the MTHFR gene is associated with reduced enzyme activity and increased homocysteine levels. The clinical role of hyperhomocysteinaemia in HCM patients remains unexplored.
The aim of this pilot study was to evaluate the prevalence and clinical implications of hyperhomocysteinaemia in a cohort of patients with HCM and MTHFR C677T polymorphism.
Methods
A total of 65 consecutive patients with a diagnosis of HCM1 and MTHFR C677T polymorphism were retrospectively evaluated at the Cardiomyopathies and Heart Failure Clinic of the University of Campania ‘Luigi Vanvitelli’ between January 2008 and January 2019.
Patients were enrolled after informed consent was obtained. All patients were clinically evaluated and received electrocardiography (ECG), echocardiography, blood tests, 24-h ECG monitoring, exercise stress test and, if required, cardiac magnetic resonance and/or genetic analysis. Clinical-instrumental evaluation was repeated every 6–12 months, and, in this context, patients were asked about the occurrence of adverse events during this period.
The primary endpoint was a composite of all-cause death, decompensated heart failure, syncope, arrhythmia, implantable cardioverter-defibrillator implantation, or all-cause hospitalization. Secondary endpoints were the individual components of the primary endpoint. To assess the association between homocysteine levels and primary endpoint, a multivariable logistic regression analysis was performed. A p-value < 0.05 was considered significant.
Results
The mean age at HCM diagnosis of the total cohort was 30.5 ± 19.2 years. Out of 65 participants, 52% had a mutation in sarcomeric genes and 49% showed plasma homocysteine levels >12 µmol/L (hyperhomocysteinaemia). In patients with hyperhomocysteinaemia, plasma homocysteine levels were higher compared with those without (20.6 ± 8 vs. 9.1 ± 1.9, p-value < 0.001), while mean age at HCM diagnosis, family history of HCM and SCD, genetic variants and degree of hypertrophy or left ventricular outflow tract obstruction did not differ statistically significantly between the two groups.
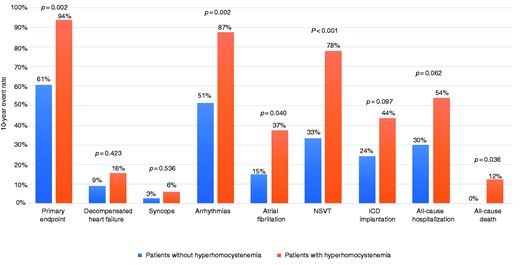
Rate of adverse events at 10-year follow-up in patients with or without hyperhomocysteinaemia.
NSVT: non-sustained ventricular tachycardia.
After a multivariate analysis adjustment for age and sex, hyperhomocysteinaemia was found to be an independent predictor for the primary endpoint (odds ratio (OR) 9.4, 95% confidence interval (CI) 1.7–51.5, p-value = 0.010), arrhythmias (OR 8.9, 95% CI 2.1–40.6, p-value = 0.003), NSVT (OR 9.8, 2.1–27.8, p-value = 0.002) and all-cause hospitalization (OR 3.8, 1.1–12.9, p-value = 0.030).
Discussion
Many studies have addressed prognostic factors in HCM.2 Nevertheless, risk stratification is still incomplete, and the investigation of disease modifier (phenotype and prognosis) represents an important challenge in cardiomyopathies, both in adults and in children.5
In our cohort of patients with both HCM and MTHFR C677T polymorphism, about 50% showed hyperhomocysteinaemia. This status appears to predispose to higher risk of cardiovascular events, emerging as a possible contributor to adverse outcomes in this heterogeneous population. Our results would have been strengthened by survival analysis, which was not performed due to missing data on time-to-events, a limitation of the present study.
Whether these results will be confirmed in larger, longitudinal studies, a systematic evaluation of plasma homocysteine levels in HCM patients could be of clinical interest, allowing to identify those patients potentially at increased risk for cardiovascular events and who could benefit from specific management.6
Conclusions
In patients with HCM and MTHFR C677T polymorphism, hyperhomocysteinaemia is associated with a significantly higher rate of adverse events during follow-up and emerged as a potential modifier of clinical outcome.
AE and EM contributed equally.
Acknowledgements
We acknowledge Mrs Giuseppina Cacciuottolo, Giuseppina Tabasco, Carmela Pica, Margherita Chianese for their precious contribution.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.

{kind=link}
Comments