





Abstract
Arrhythmogenic cardiomyopathy (ACM) is a cardiac disorder characterized by structural alterations of the myocardium, which predisposes individuals to ventricular arrhythmias and increases the risk of sudden cardiac death. Initially described as arrhythmogenic right ventricular cardiomyopathy, the involvement of the left ventricle (LV) has been subsequently recognized, leading to the classification of various phenotypes under LV non-dilated cardiomyopathy. The clinical spectrum of ACM ranges from life-threatening ventricular arrhythmias to overt heart failure, sometimes presenting with acute myocarditis-like episodes and extracardiac symptoms, further contributing to the disease’s heterogeneity. Diagnosis relies on imaging modalities, such as echocardiogram and cardiac magnetic resonance imaging, to detect areas of fibro-fatty replacement and/or non-ischemic ventricular scarring, integrated with genetic analysis. The 2023 European Society of Cardiology guidelines on Cardiomyopathies underscore the importance of a comprehensive diagnostic approach, combining imaging and genetics for arrhythmic risk stratification and comprehensive patient management. Growing evidence on genotype–phenotype correlation, along with the validation of specific predictive scores, is improving ACM clinical management and promoting personalized treatment tailored to individual and familial characteristics.
From arrhythmogenic right ventricular cardiomyopathy to non-dilated left ventricular cardiomyopathy
Arrhythmogenic cardiomyopathy (ACM) is a myocardial disorder characterized by structural and functional abnormalities of cardiomyocytes, associated with an increased predisposition for malignant ventricular arrhythmias and sudden cardiac death (SCD). Historically, ACM was recognized predominantly as arrhythmogenic right ventricular cardiomyopathy (ARVC). This form primarily involves the right ventricle (RV) with dilation, dysfunction, and/or wall motion alterations, associated with malignant ventricular arrhythmias originating from the RV. The structural and electrical alterations, resulting from fibro-fatty replacement of cardiomyocytes, were initially thought to be confined to the ‘triangle of dysplasia’ (between the pulmonary infundibulum, infero-posterior wall, and apex of the RV). The first diagnostic criteria were defined in 1994, with a subsequent revision in 2010 aimed at improving diagnostic accuracy by including quantitative imaging data for morpho-functional assessment and criteria related to family history/genetics.1 However, the spread of contrast-enhanced cardiac magnetic resonance (CMR) and genetic analysis in clinical practice has contributed to a broader understanding of ACM, with evidence of fibrotic or fibro-fatty replacement of the myocardium in both ventricles and a clinical and genetic overlap between left ventricular (LV) and RV cardiomyopathies (Figure 1). This paradigm shift in ARVC has been incorporated into the 2023 European Society of Cardiology (ESC) guidelines on Cardiomyopathies, which explicitly distinguishes it from the LV forms, now grouped into a broader concept of LV non-dilated cardiomyopathy (NDLVC) and dilated cardiomyopathy (DCM).2,3 Non-dilated cardiomyopathy is an umbrella term, which includes arrhythmogenic forms with predominant involvement of the LV, up to the hypokinetic LV cardiomyopathies, recognizing that ventricular arrhythmias and SCD risk can manifest in a broader range of phenotypes, not exclusively limited to the RV. For the first time compared to previous classifications of cardiomyopathies, NDLVC considers non-ischemic ventricular scarring and fibro-fatty replacement in the LV as morphological diagnostic criteria, regardless of regional wall motion abnormalities or LV ejection fraction (LVEF). Despite the introduction of the NDLVC entity, many clinicians and researchers continue to use the term ACM. Regardless of nomenclature (ACM or NDLVC), the starting phenotype must systematically initiate a ‘step-by-step’ diagnostic–therapeutic approach in cardiomyopathies, in which the different phenotypic expressions represent the first step in clinical management, enhancing the importance of the aetiological diagnosis, and prompting us to invasive techniques when this can have a prognostic impact2 (Figure 2).
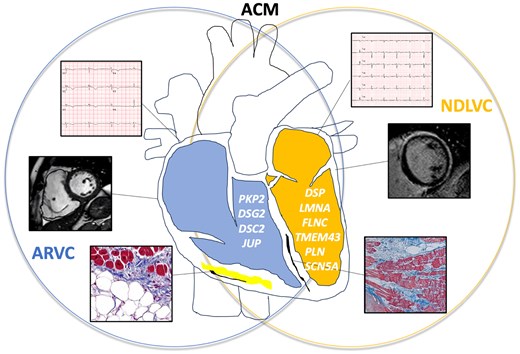
arrhythmogenic cardiomyopathy includes right-sided (arrhythmogenic right ventricular cardiomyopathy, blue), left-sided (non-dilated left ventricular cardiomyopathy, yellow), and biventricular forms, with clinical, instrumental, and genetic overlap. Arrhythmogenic right ventricular cardiomyopathy is characterized by regional kinetics abnormalities (akinesia, dyskinesia, or aneurysm) associated with right ventricle dilation or reduced global systolic function, due to fibro-adipose infiltration. ECG findings typically include inverted T waves extending to V3 or beyond, sometimes accompanied by epsilon waves. Genes typically associated with arrhythmogenic right ventricular cardiomyopathy are PKP2, DSG2, DSC2, and JUP. NDLVC is characterized by left ventricle global systolic dysfunction without dilation and/or presence of more or less extensive late gadolinium enhancement (up to a ‘ring-like’ late gadolinium enhancement pattern), due to fibrous replacement of the myocardium. ECG findings typically include inverted T waves in V4–V6 leads and low QRS voltages. Genes typically associated with non-dilated left ventricular cardiomyopathy are DSP, LMNA, FLNC, TMEM43, PLN, and SCN5A. ACM, arrhythmogenic cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; NDLVC, non-dilated left ventricular cardiomyopathy; DSC2, desmocollin2; DSG2, desmoglein2; DSP, desmoplakin; FLNC, filamin C; JUP, plakoglobin; LMNA, lamin A/C; PLN, phospholamban; PKP2, plakophilin; SCN5A, sodium channel alpha subunit; TMEM43, transmembrane protein 43.
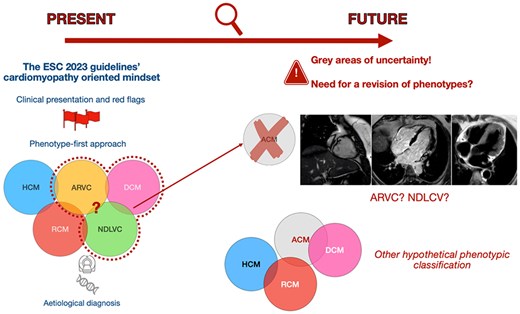
Evolution of the diagnostic approach to cardiomyopathies. On the left, the approach proposed by the European Society of Cardiology 2023 guidelines on cardiomyopathies is illustrated, which starts from clinical red flags to identify the phenotype and uses genetic testing and magnetic resonance imaging to arrive at an aetiological diagnosis. On the right, the controversies on the phenotypes defined in the guidelines are shown through cardiac magnetic resonance images of an individual with desmoplakin cardiomyopathy. These images highlight the overlapping zones and grey areas between arrhythmogenic right ventricular cardiomyopathy and non-dilated left ventricular cardiomyopathy, previously included under the umbrella term of arrhythmogenic cardiomyopathy. Reproduced with permission of the authors. ACM, arrhythmogenic cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; NDLVC, non-dilated left ventricular cardiomyopathy; RCM, restrictive cardiomyopathy.
From phenotype to clinical manifestations
Arrhythmogenic cardiomyopathy can present with a broad spectrum of clinical manifestations, ranging from arrhythmias to heart failure (HF), with the severity depending on the extent of ventricular involvement, disease progression, genetic background, and other factors not yet fully understood. On the one hand, in the early or subclinical phase, ACM may be asymptomatic, sometimes diagnosed incidentally through clinical or genetic screening of individuals with a positive family history. Already in this phase, electrocardiographic alterations (such as low voltages, negative T waves, and frequent ventricular extrasystoles) may be present as early warning signs for further diagnostic evaluation.4 Cardiac magnetic resonance, with its ability to detect early fibrous or fibro-adipose replacement, is crucial for early identification.5
On the other hand, ventricular arrhythmias [including ventricular extrasystoles >500/24 h, non-sustained ventricular tachycardias (NSVT), and sustained ventricular tachycardias (sVT), as well as ventricular fibrillation and SCD] may be the first manifestation of the disease. Arrhythmias may present as palpitations and episodes of syncope or pre-syncope and may be triggered by physical effort. Family history, particularly regarding major arrhythmic events and SCD, must always be investigated and explored in depth for risk stratification in ACM patients.3
With disease progression, myocardial structural involvement becomes more evident, and the fibrous or fibro-fatty replacement, initially localized, may extend to biventricular forms, leading to biventricular dilation and dysfunction. These changes may be associated with the classic HF signs and symptoms, with possible progression to advanced HF.
In a minority of cases, especially in younger individuals, chest pain may represent the initial symptom, often accompanied by elevated troponin levels and ECG alterations, mimicking acute myocarditis, in the so-called ‘hot phases’ of ACM. The underlying pathophysiological mechanisms are unclear but may involve inflammatory or apoptotic processes.6 Finally, recessive genetic forms of ACM, such as in Naxos and Carvajal syndrome, can present with distinctive extracardiac features, such as palmoplantar keratosis and woolly hair.
From phenotype to multimodal imaging
Multimodal imaging plays a pivotal role in the diagnosis and follow-up of ACM patients.2 Echocardiography remains the first-level diagnostic tool, allowing the first evaluation of the phenotype in a rapid and non-invasive way. Traditional diagnostic criteria for ARVC include the identification of regional wall motion abnormalities (akinesia, dyskinesia, or aneurysm) in the presence of RV dilation or reduction of the global systolic function.1 In cases of suboptimal imaging quality, contrast-enhanced echocardiography can enhance visualization of the endocardial border. Additionally, the echocardiographic study must equally carefully consider the LV,7 evaluating both structural and functional abnormalities.8 However, echocardiography has limited sensitivity in identifying LV-dominant variants, as tissue changes in the LV may not always lead to significant regional or global functional abnormalities, due to their epicardial or intramural distribution. In this context, advanced echocardiographic techniques, such as myocardial strain imaging, can improve the detection of LV involvement. Similarly, a reduction in RV strain has been described in patients affected by ACM and associated with disease progression.9 Finally, myocardial strain imaging is particularly valuable in identifying subclinical disease, particularly in relatives of affected individuals or asymptomatic carriers of genetic variants associated with ACM.
Cardiac magnetic resonance is the gold standard for ACM diagnosis. Given the echocardiography’s relatively low negative predictive value, CMR is essential even in the presence of a normal echocardiographic examination. The modality provides detailed morphological and functional assessment, including cardiac chamber dimensions, wall thickness, regional kinetics, and global systolic function of the ventricles. Cardiac magnetic resonance also allows for precise quantification of flows, which helps in the differential diagnosis of RV dilation associated with congenital anomalies with left-to-right shunt. Cardiac magnetic resonance also enables precise tissue characterization, identifying areas of fibrosis or fibro-adipose replacement, which are hallmark features of ACM. According to the European Task Force criteria for ACM diagnosis, the presence of morpho-functional alterations (using specific nomograms for age, sex, body surface, and athletes) and structural alterations of the ventricles should be evaluated.8 In particular, late gadolinium enhancement (LGE) in one or more segments of the RV, confirmed in two orthogonal planes, is a minor diagnostic criterion for ARVC. For ALVC (according to ESC guidelines often included in NDLVC), LGE with a ‘ring-like’ pattern (subepicardial or intramyocardial LGE in at least three consecutive segments, confirmed in two orthogonal planes) is a key diagnostic feature, characteristically associated with pathogenic variants of the DSP, FLNC, and PLN genes.8 In comparison, LGE with a non-ischemic pattern (subepicardial or intramyocardial) in one or two segments of the free wall, the interventricular septum, or both is considered a minor criterion.
In addition to its importance in diagnosis, CMR offers important prognostic information, with LV involvement being an independent predictor of major arrhythmic events.10,11
Other imaging techniques have a more limited role in ACM diagnosis. Computed tomography (CT), in addition to evaluating coronary anatomy, can provide morpho-functional information and tissue characterization by identifying hypodense areas of fat infiltration and scar areas through the late iodine enhancement (LIE) technique.12 However, CT is constrained by the exposure to ionizing radiation and iodinated contrast medium, limiting its use in patients with a poor echocardiographic window or contraindications to CMR.
Positron emission tomography (PET) is used in differential diagnosis with inflammatory forms like cardiac sarcoidosis.
Finally, due to its invasive nature, endomyocardial biopsy (EMB) in ACM is limited to selected cases, particularly for a targeted differential diagnosis (e.g. sarcoidosis). Endomyocardial biopsy can also be a valuable tool in the suspicion of ‘hot phases’ of ACM, for correctly classifying young subjects with recurrent myocarditis-like episodes or chronic troponin release, and evaluating the initiation of targeted immunosuppressive therapy.
From phenotype to genotype
Genetic analysis is an essential tool for an adequate diagnostic–therapeutic pathway in ACM. Pathogenic or likely pathogenic genetic variants are identified in ∼50% of ACM cases, and typically follow a Mendelian inheritance. Recent studies have also highlighted the role of non-Mendelian inheritance, arising from combinations of several less rare variants, in the pathogenesis of familial ACM where a rare variant is not identified but polygenic risks are implicated.2 The complexity of the genetic architecture, along with the possible interaction between genes and introns, and the possible role of variants of uncertain significance, together with incomplete penetrance and variable phenotypic expressivity, make ACM an extremely heterogeneous disorder and complicate genotype–phenotype correlation studies. Finally, some environmental factors can further influence the disease phenotypic manifestations. For example, intense sporting activity represents a predisposing factor not only for ACM development but also for life-threatening ventricular arrhythmias.13 Despite these complexities, identifying a rare pathogenetic variant remains crucial in supporting the clinical diagnosis, defining the prognosis, and guiding therapeutic choices. Furthermore, cascade screening of relatives of affected genotype-positive individuals allows the identification of phenotype-negative carriers, at higher risk of developing ACM, to be followed with close follow-up.14
Historically, ACM has been associated with variants in genes encoding desmosomal proteins, which are fundamental for intercellular adhesion and communication. The first genetic variants were identified in two syndromes with cardiac and cutaneous manifestations: Naxos syndrome, associated with the plakoglobin (JUP) gene, and Carvajal syndrome, associated with the desmoplakin (DSP) gene. Both syndromes exhibit an autosomal recessive inheritance pattern, in contrast to most ACM cases, which are inherited in an autosomal dominant inheritance pattern with incomplete penetrance and variable and age-related expressivity. Starting from these initial discoveries in the JUP and DSP genes, additional ACM-associated genes have been identified, including plakophilin2 (PKP2), desmocollin2 (DSC2), and desmoglein2 (DSG2). Among the desmosomal genes, PKP2 is the most frequently involved, particularly in ARVC. Variants in DSC2, DSG2 and particularly DSP genes are more commonly linked to biventricular or isolated LV forms. In ∼3.5% of cases, the same desmosomal genes can be associated with forms of ‘overlap’ of ACM and DCM.15 Arrhythmogenic cardiomyopathy caused by DSP variants has a peculiar phenotypic manifestation, with a progressive fibrotic replacement of cardiomyocytes before significant LV systolic dysfunction develops.
Although initially considered a disorder primarily caused by desmosomal genes, increasing evidence has identified non-desmosomal genes in ACM pathogenesis. These include genes encoding nuclear proteins (lamin A/C, LMNA, and transmembrane protein 43, TMEM43), cytoskeletal structures (filamin C, FLNC), RNA-binding proteins such as splicing modulators (RBM20), and those involved in the regulation of calcium ionic currents (phospholamban, PLN). Although more rarely, genes typically associated with DCM (titin, TTN) or channelopathies (SCN5A, associated with Brugada syndrome and long QT syndrome) can also manifest with ACM features. Characteristics associated with the main desmosomal and non-desmosomal genes responsible for ACM are summarized in Table 1.
Characteristics of the main desmosomal and non-desmosomal genes associated with arrhythmogenic cardiomyopathy
| ECG(4) | Arrhythmias | Cardiac magnetic resonance imaging2,5 | Clinical phenotype16 | Risk of sudden cardiac death Arrhythmic risk predictors Prognostic scores | ||
|---|---|---|---|---|---|---|
| Desmosomal Genes (50% of cases) | PKP2 DSG2 DSC2 DSP JUP |
| Frequent ventricular extrasystoles Major ventricular arrhythmias NSVT | LGE with non-ischemic or circumferential pattern in LV (‘Ring-like’ pattern) LGE in isolated RV |
| Annual risk of sudden cardiac death, depending on risk factors (>10% if ACR due to VF or sVT, 1–10% if severe RV or LV dysfunction, NSVT, induction of major ventricular arrhythmias at electrophysiological study, syncope, ventricular extrasystoles >1000/24 h, male sex, negative T wave extension, presence of multiple desmosomal mutations)8
|
| Non-desmosomal genes | LMNA |
| Supraventricular arrhythmias (atrial fibrillation, atrial flutter, atrial tachycardia) Frequent ventricular extrasystoles Major ventricular arrhythmias | LGE with non-ischemic pattern |
|
|
| FLNC |
| Frequent ventricular extrasystoles Major ventricular arrhythmias | LGE with non-ischemic or circumferential pattern (‘Ring-like’ pattern) |
| Sudden cardiac death risk per year: 5–10%2 Presence of LGE LVEF < 45% Male sex Older age NSVT Previous syncope
| |
| TMEM43 | Major ventricular arrhythmias | LGE with non-ischemic pattern |
| Sudden cardiac death risk per year: 5–10%2 Male sex Female sex + one additional factor among LVEF <45%, NSVT, LGE, > 200 ventricular extrasystoles on 24-h Holter ECG | ||
| RBM20 | Major ventricular arrhythmias | LGE with non-ischemic pattern |
| Annual risk of sudden cardiac death: 3–5%2 Male sex Presence of LGE LVEF < 45% | ||
| PLN |
| Major ventricular arrhythmias | LGE with non-ischemic or circumferential pattern (‘Ring-like’ pattern) | ABVC | Annual risk of sudden cardiac death: 3–5%2 Presence of LGE LVEF < 45% NSVT
| |
| ECG(4) | Arrhythmias | Cardiac magnetic resonance imaging2,5 | Clinical phenotype16 | Risk of sudden cardiac death Arrhythmic risk predictors Prognostic scores | ||
|---|---|---|---|---|---|---|
| Desmosomal Genes (50% of cases) | PKP2 DSG2 DSC2 DSP JUP |
| Frequent ventricular extrasystoles Major ventricular arrhythmias NSVT | LGE with non-ischemic or circumferential pattern in LV (‘Ring-like’ pattern) LGE in isolated RV |
| Annual risk of sudden cardiac death, depending on risk factors (>10% if ACR due to VF or sVT, 1–10% if severe RV or LV dysfunction, NSVT, induction of major ventricular arrhythmias at electrophysiological study, syncope, ventricular extrasystoles >1000/24 h, male sex, negative T wave extension, presence of multiple desmosomal mutations)8
|
| Non-desmosomal genes | LMNA |
| Supraventricular arrhythmias (atrial fibrillation, atrial flutter, atrial tachycardia) Frequent ventricular extrasystoles Major ventricular arrhythmias | LGE with non-ischemic pattern |
|
|
| FLNC |
| Frequent ventricular extrasystoles Major ventricular arrhythmias | LGE with non-ischemic or circumferential pattern (‘Ring-like’ pattern) |
| Sudden cardiac death risk per year: 5–10%2 Presence of LGE LVEF < 45% Male sex Older age NSVT Previous syncope
| |
| TMEM43 | Major ventricular arrhythmias | LGE with non-ischemic pattern |
| Sudden cardiac death risk per year: 5–10%2 Male sex Female sex + one additional factor among LVEF <45%, NSVT, LGE, > 200 ventricular extrasystoles on 24-h Holter ECG | ||
| RBM20 | Major ventricular arrhythmias | LGE with non-ischemic pattern |
| Annual risk of sudden cardiac death: 3–5%2 Male sex Presence of LGE LVEF < 45% | ||
| PLN |
| Major ventricular arrhythmias | LGE with non-ischemic or circumferential pattern (‘Ring-like’ pattern) | ABVC | Annual risk of sudden cardiac death: 3–5%2 Presence of LGE LVEF < 45% NSVT
| |
ACR, cardiocirculatory arrest; ABVC, arrhythmogenic biventricular cardiomyopathy; ALVC, arrhythmogenic left ventricular cardiomyopathy; AV, atrioventricular; ACM, arrhythmogenic cardiomyopathy; CMP, cardiomyopathy; DCM, dilated cardiomyopathy; LVEF, left ventricular ejection fraction; VF, ventricular fibrillation; LGE, late gadolinium enhancement; LV, left ventricle; NSVT, non-sustained ventricular tachycardia; sVT, sustained ventricular tachycardia; RV, right ventricle.
Characteristics of the main desmosomal and non-desmosomal genes associated with arrhythmogenic cardiomyopathy
| ECG(4) | Arrhythmias | Cardiac magnetic resonance imaging2,5 | Clinical phenotype16 | Risk of sudden cardiac death Arrhythmic risk predictors Prognostic scores | ||
|---|---|---|---|---|---|---|
| Desmosomal Genes (50% of cases) | PKP2 DSG2 DSC2 DSP JUP |
| Frequent ventricular extrasystoles Major ventricular arrhythmias NSVT | LGE with non-ischemic or circumferential pattern in LV (‘Ring-like’ pattern) LGE in isolated RV |
| Annual risk of sudden cardiac death, depending on risk factors (>10% if ACR due to VF or sVT, 1–10% if severe RV or LV dysfunction, NSVT, induction of major ventricular arrhythmias at electrophysiological study, syncope, ventricular extrasystoles >1000/24 h, male sex, negative T wave extension, presence of multiple desmosomal mutations)8
|
| Non-desmosomal genes | LMNA |
| Supraventricular arrhythmias (atrial fibrillation, atrial flutter, atrial tachycardia) Frequent ventricular extrasystoles Major ventricular arrhythmias | LGE with non-ischemic pattern |
|
|
| FLNC |
| Frequent ventricular extrasystoles Major ventricular arrhythmias | LGE with non-ischemic or circumferential pattern (‘Ring-like’ pattern) |
| Sudden cardiac death risk per year: 5–10%2 Presence of LGE LVEF < 45% Male sex Older age NSVT Previous syncope
| |
| TMEM43 | Major ventricular arrhythmias | LGE with non-ischemic pattern |
| Sudden cardiac death risk per year: 5–10%2 Male sex Female sex + one additional factor among LVEF <45%, NSVT, LGE, > 200 ventricular extrasystoles on 24-h Holter ECG | ||
| RBM20 | Major ventricular arrhythmias | LGE with non-ischemic pattern |
| Annual risk of sudden cardiac death: 3–5%2 Male sex Presence of LGE LVEF < 45% | ||
| PLN |
| Major ventricular arrhythmias | LGE with non-ischemic or circumferential pattern (‘Ring-like’ pattern) | ABVC | Annual risk of sudden cardiac death: 3–5%2 Presence of LGE LVEF < 45% NSVT
| |
| ECG(4) | Arrhythmias | Cardiac magnetic resonance imaging2,5 | Clinical phenotype16 | Risk of sudden cardiac death Arrhythmic risk predictors Prognostic scores | ||
|---|---|---|---|---|---|---|
| Desmosomal Genes (50% of cases) | PKP2 DSG2 DSC2 DSP JUP |
| Frequent ventricular extrasystoles Major ventricular arrhythmias NSVT | LGE with non-ischemic or circumferential pattern in LV (‘Ring-like’ pattern) LGE in isolated RV |
| Annual risk of sudden cardiac death, depending on risk factors (>10% if ACR due to VF or sVT, 1–10% if severe RV or LV dysfunction, NSVT, induction of major ventricular arrhythmias at electrophysiological study, syncope, ventricular extrasystoles >1000/24 h, male sex, negative T wave extension, presence of multiple desmosomal mutations)8
|
| Non-desmosomal genes | LMNA |
| Supraventricular arrhythmias (atrial fibrillation, atrial flutter, atrial tachycardia) Frequent ventricular extrasystoles Major ventricular arrhythmias | LGE with non-ischemic pattern |
|
|
| FLNC |
| Frequent ventricular extrasystoles Major ventricular arrhythmias | LGE with non-ischemic or circumferential pattern (‘Ring-like’ pattern) |
| Sudden cardiac death risk per year: 5–10%2 Presence of LGE LVEF < 45% Male sex Older age NSVT Previous syncope
| |
| TMEM43 | Major ventricular arrhythmias | LGE with non-ischemic pattern |
| Sudden cardiac death risk per year: 5–10%2 Male sex Female sex + one additional factor among LVEF <45%, NSVT, LGE, > 200 ventricular extrasystoles on 24-h Holter ECG | ||
| RBM20 | Major ventricular arrhythmias | LGE with non-ischemic pattern |
| Annual risk of sudden cardiac death: 3–5%2 Male sex Presence of LGE LVEF < 45% | ||
| PLN |
| Major ventricular arrhythmias | LGE with non-ischemic or circumferential pattern (‘Ring-like’ pattern) | ABVC | Annual risk of sudden cardiac death: 3–5%2 Presence of LGE LVEF < 45% NSVT
| |
ACR, cardiocirculatory arrest; ABVC, arrhythmogenic biventricular cardiomyopathy; ALVC, arrhythmogenic left ventricular cardiomyopathy; AV, atrioventricular; ACM, arrhythmogenic cardiomyopathy; CMP, cardiomyopathy; DCM, dilated cardiomyopathy; LVEF, left ventricular ejection fraction; VF, ventricular fibrillation; LGE, late gadolinium enhancement; LV, left ventricle; NSVT, non-sustained ventricular tachycardia; sVT, sustained ventricular tachycardia; RV, right ventricle.
The genetic background of the affected individuals also plays a significant role in therapeutic choices, providing prognostic insights, and contributing to the multi-parametric stratification of arrhythmic risk, together with family, clinical, and multimodal imaging data. Historically, LVEF below 35% has been the key criterion for implantable cardioverter defibrillator (ICD) implantation in primary prevention in the context of DCM and NDLVC. In recent years, however, it is increasingly clear that LVEF alone is insufficient for accurate arrhythmic risk stratification in DCM and NDLVC patients, in particular in carriers of variants affecting arrhythmic genes such as LMNA, FLNC, TMEM43, RBM20, PLN, and DSP.2 In this sense, the recent ESC guidelines on cardiomyopathies recommend ICD implantation in primary prevention in patients of arrhythmic genotypes, regardless of the LVEF value, in the presence of additional risk factors, such as a history of syncope and LGE on CMR, with a class of recommendation IIa.2 Furthermore, for specific genotypes, such as LMNA, DSP, and PLN, predictive risk scores for significant ventricular arrhythmias during follow-up have been developed and validated. In the case of LMNA, the score evaluates the risk of major ventricular arrhythmias within 5 years, taking into account male sex, history of atrioventricular blocks and NSVT, non-missense mutations, and LVEF.19 Similarly, the risk score for patients with DSP-related cardiomyopathy18 predicts major arrhythmic events in the next 5 years, classifying patients into low, intermediate, and high risk, including as risk factors female sex (in contrast with the LMNA risk score), history of NSVT, burden of extrasystoles in 24 h, moderate-to-severe RV dysfunction, and LVEF < 50%. The 5-year arrhythmic risk score for PLN gene (p.Arg14del variant) includes the following factors: history of NSVT, negative T waves in anterior and inferior leads, number of ventricular extrasystoles on 24-h Holter-ECG and LVEF.20 For FLNC truncating variants, a recently developed risk score predicts major ventricular arrhythmic SCD or life-threatening ventricular arrhythmias, incorporating five key variables: male sex, NSVT, previous syncope, age, and LVEF. For ARVC, a specific predictive risk score for significant ventricular arrhythmias has been developed,17 particularly for patients with PKP2 mutations, although it may overestimate risk in low-risk classes and perform suboptimally in carriers of other genes or genotype-negative individuals. In general, both in carriers of genetic variants in arrhythmic genes and in genotype-negative cases, recent unexplained syncope and a family history of SCD or life-threatening ventricular arrhythmias constitute fundamental anamnestic elements to be integrated into the overall stratification of arrhythmic risk in ACM patients.3
Finally, genetic testing integrated with clinical data provides important information that guides therapeutic and prognostic decisions, not only for the probands but also for their family. When a pathogenic variant is identified in the proband, cascade testing should be offered to first-degree relatives. Detecting a pathogenic variant in relatives, even if phenotype-negative, allows us to identify subjects at greater risk of developing ACM over time. It is important to inform relatives of potential implications in terms of early surveillance, preventive interventions, and lifestyle modifications. In relatives, therefore, an initial evaluation with electrocardiogram and cardiac imaging (echocardiogram and possibly CMR) is recommended, sometimes associated with Holter-ECG and stress test for the arrhythmic burden, to be repeated every 1–2 years.2
Conclusions
Arrhythmogenic cardiomyopathy represents a myocardial disorder with very heterogeneous clinical manifestations, ranging from asymptomatic forms to SCD. Starting from the classic RV phenotype, it has progressively included variants involving the LV or both ventricles. The evolution of the phenotypic spectrum of ACM reflects the progress of diagnostic knowledge, particularly multimodal imaging, and genetic analysis, which are effective tools in the early identification of the disease and in refining risk stratification through an integrated approach.
Funding
No funding provided.
Data availability
No new data were generated or analysed in support of this research.
Disclaimer
This paper was originally published as ‘Espressioni fenotipiche e manifestazioni cliniche della cardiomiopatia aritmogena’, in the Volume degli Atti del Congresso “Conoscere e Cuare il Cuore 2025”, published by Centro per la Lotta contro l'Infarto for distribution at the CCC Conference. This paper was translated by Dr. Mario Albertucci, representative of the CLI Foundation, and republished with permission.
References
Author notes
Conflict of interest: none declared.

{kind=link}
{kind=link}