





Abstract
Dilated cardiomyopathy (DCM) is defined by the presence of left ventricular dilation and systolic dysfunction in the absence of coronary artery disease, valvular disease, congenital heart disease, or altered haemodynamic conditions. Dilated cardiomyopathy can recognize multiple aetiologies, including infectious processes, effect of toxic substances, immunological mechanisms, and genetic causes. In recent years, many genes coding for proteins involved in the structure and function of the cardiomyocytes have been associated with the development of DCM, making the identification of familial forms increasingly frequent. At the same time, an ever-increasing use of cardiac magnetic resonance imaging has made it possible to identify early morpho-functional alterations in subjects with initial forms of the disease, or carriers of pathogenic genetic variants. The increasingly in-depth understanding of the genetic and molecular mechanisms operating in DCM has also favoured the development of new therapeutic strategies including drugs with molecular targets and gene therapies. In this panorama, screening of family members of patients affected by DCM represents an important tool for early diagnosis, treatment, and prognostic stratification. In relation to its clinical relevance and its complexity, it is important that family screening and follow-up of identified patients are carried out in units dedicated to the treatment and study of cardiomyopathies.
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is characterized by the presence of left ventricular dilatation and systolic dysfunction in the absence of coronary artery disease, valvular heart disease, congenital heart disease, or altered haemodynamic conditions capable of causing it. Left ventricular dilatation is defined by left ventricular end-diastolic dimensions or volumes corrected for body size, sex, and/or age: a left ventricular end-diastolic diameter >58 mm or an indexed left ventricular end-diastolic volume (iLVEDV) ≥75 mL/m2 in males and a left ventricular end-diastolic diameter >52 mm or an iLVEDV ≥62 mL/m2 in females. Global left ventricular systolic dysfunction is defined by a left ventricular ejection fraction (LVEF) <50%.1
It is important to remember that DCM can be caused by a set of extremely heterogeneous conditions with multiple aetiologies, including infectious processes (viruses, bacteria, and protozoa), the effect of toxic substances or immunological mechanisms, and genetic causes1 (Table 1). Similarly to the aetiology, the clinical presentation and prognosis can also be extremely different and can vary from forms with severe heart failure (in some cases advanced with the need for transplant or ventricular assistance), to ventricular arrhythmias and bradyarrhythmias, up to pauci-symptomatic—or asymptomatic forms. One explanation for this phenomenon can be traced back in part to the heterogeneity of the genetic architecture related to DCM. In recent years, numerous genes coding for a variety of cardio-myocyte cellular structures (sarcomere, Z-discs, desmosome, cytoskeleton, ion channels, and transcription and translation regulatory elements) have been associated with the development of DCM.2 It should be underlined that a genetic predisposition also plays an important role in favouring some toxic forms of DCM (alcoholic, chemotherapy induced) and peri-partum cardiomyopathy itself.3 In the absence of conclusive genetic information, from a purely clinical–anamnestic point of view, DCM is considered familial if (i) one or more first- or second-degree relatives have DCM or (ii) when there is a first-degree relative of any age with an established diagnosis of DCM suffering from sudden cardiac death.
Dilated cardiomyopathy: non-genetic causes
| Dilated cardiomyopathy: non-genetic aetiologies |
|---|
Post-myocarditis
|
| Toxic aetiologies: cocaine, amphetamines, ecstasy, cobalt, anabolic steroids, primary and secondary iron overloads |
| Peri-partum cardiomyopathy |
|
| Endocrinologic disorders: hypo- and hyperthyroidism, Cushing, Addison disease, pheochromocytoma, diabetes mellitus, acromegaly |
| Nutritional deficiencies: thiamine (beriberi), selenium, zinc, copper, carnitine |
| Electrolyte abnormalities: hypocalcaemia, hypophosphataemia |
| Dilated cardiomyopathy: non-genetic aetiologies |
|---|
Post-myocarditis
|
| Toxic aetiologies: cocaine, amphetamines, ecstasy, cobalt, anabolic steroids, primary and secondary iron overloads |
| Peri-partum cardiomyopathy |
|
| Endocrinologic disorders: hypo- and hyperthyroidism, Cushing, Addison disease, pheochromocytoma, diabetes mellitus, acromegaly |
| Nutritional deficiencies: thiamine (beriberi), selenium, zinc, copper, carnitine |
| Electrolyte abnormalities: hypocalcaemia, hypophosphataemia |
RSV, respiratory syncytial virus; HCV, hepatitis C virus; PVB-19, parvovirus B-19; CMV, cytomegalovirus; HIV-1, human immunodeficiency virus-1; HHV-6, human herpesvirus-6; EBV, Epstein–Barr virus; VZV, varicella zoster virus; HSV, herpes simplex virus; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; SLE, systemic lupus erythematosus; EGPA, eosinophilic granulomatosis with polyangiitis; RA, rheumatoid arthritis; CAR-T, chimeric antigen receptor T.
Dilated cardiomyopathy: non-genetic causes
| Dilated cardiomyopathy: non-genetic aetiologies |
|---|
Post-myocarditis
|
| Toxic aetiologies: cocaine, amphetamines, ecstasy, cobalt, anabolic steroids, primary and secondary iron overloads |
| Peri-partum cardiomyopathy |
|
| Endocrinologic disorders: hypo- and hyperthyroidism, Cushing, Addison disease, pheochromocytoma, diabetes mellitus, acromegaly |
| Nutritional deficiencies: thiamine (beriberi), selenium, zinc, copper, carnitine |
| Electrolyte abnormalities: hypocalcaemia, hypophosphataemia |
| Dilated cardiomyopathy: non-genetic aetiologies |
|---|
Post-myocarditis
|
| Toxic aetiologies: cocaine, amphetamines, ecstasy, cobalt, anabolic steroids, primary and secondary iron overloads |
| Peri-partum cardiomyopathy |
|
| Endocrinologic disorders: hypo- and hyperthyroidism, Cushing, Addison disease, pheochromocytoma, diabetes mellitus, acromegaly |
| Nutritional deficiencies: thiamine (beriberi), selenium, zinc, copper, carnitine |
| Electrolyte abnormalities: hypocalcaemia, hypophosphataemia |
RSV, respiratory syncytial virus; HCV, hepatitis C virus; PVB-19, parvovirus B-19; CMV, cytomegalovirus; HIV-1, human immunodeficiency virus-1; HHV-6, human herpesvirus-6; EBV, Epstein–Barr virus; VZV, varicella zoster virus; HSV, herpes simplex virus; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; SLE, systemic lupus erythematosus; EGPA, eosinophilic granulomatosis with polyangiitis; RA, rheumatoid arthritis; CAR-T, chimeric antigen receptor T.
The diagnostic approach to the patient with DCM is based on clinical and anamnestic evaluation including family history and family tree, electrocardiogram (ECG), and conventional echocardiogram, with advanced techniques for evaluating myocardial function such as speckle tracking and strain. However, the recent guidelines of the European Society of Cardiology (ESC) on the treatment of cardiomyopathies1 have placed the characterization of myocardial tissue through cardiac magnetic resonance imaging (CMR) at the centre of the diagnostic path of patients with cardiomyopathy. In addition to the clinical–instrumental evaluation mentioned above, CMR can offer not only a more precise definition of the morphology and function of the cardiac chambers but also a characterization of the myocardial tissue, in particular with the sequences obtained after contrast administration [late gadolinium enhancement (LGE)] and with T1 and T2 mapping sequences. Tissue characterization can provide important diagnostic and prognostic information in patients with DCM,1 allowing early identification of the presence of ventricular dysfunction and/or pathological substrate in patients with initial forms of the disease, or carriers of pathogenic genetic variants.4 As anticipated, genetic testing often represents the completion of the diagnostic process in patients with DCM, especially in familial forms and in forms with a more aggressive clinical presentation.
The rapid evolution of knowledge has also allowed the development of new therapeutic strategies including drugs with molecular targets and specific gene therapies for the different types of DCM.5 Increased diagnostic capabilities together with new therapeutic opportunities potentially capable of preventing complications or even the development of the phenotype have placed further emphasis on screening family members of patients affected by DCM. The aim of this article is therefore to provide a practical guide to clinical–instrumental and genetic screening of family members of patients affected by DCM.
Screening of the proband’s family members with dilated cardiomyopathy
Clinical evaluation, electrocardiogram, and echocardiography
In the presence of a diagnosis of DCM, the recent ESC guidelines recommend clinical–instrumental screening of all first-degree family members. A family history of DCM certainly represents a risk factor for the development of the disease in a family member subjected to screening. However, in the literature, family history alone is able to recognize a small portion of affected family members (34%), while many family members can only be identified through careful and systematic clinical follow-up.6 The ECG is able to evaluate many early alterations, which can provide important information on the aetiology and prognosis of patients.7 In particular, some peculiar characteristics, such as the presence of atrioventricular blocks, low QRS voltages, the early onset of atrial fibrillation and right bundle branch abnormality, may suggest the presence of a genetic substrate with a poor prognosis: desmosomal genes, lamin, filamin C, phospholamban, dystrophin, or RNA binding motif protein 20 (RBM20).7
Screening studies including echocardiography have estimated a prevalence of DCM of 2.0–4.6% in the initial screening of relatives of patients with DCM.8–10 In the same studies, a significantly greater number of subjects (12.0–24%) had left ventricular dilatation without systolic dysfunction at baseline assessment. During serial follow-up, 10–13% of family members subjected to screening developed a DCM. In these studies, the presence of left ventricular dilatation and reduced baseline systolic function values were significant predictors of the development of DCM.
In fact, mild non-diagnostic abnormalities potentially attributable to other concomitant factors such as sporting activity, arterial hypertension, and obesity are often detected in first-degree relatives. However, according to recent ESC guidelines,1 the presence of isolated dilatation of the left ventricle with preserved systolic function and the presence of a familial pathogenetic variant represent sufficient elements to make the diagnosis of DCM in a first-degree relative. In these subjects, in addition to the initial screening, clinical–instrumental follow-up is of great importance, since the penetrance of the disease can be variable and the pathological picture can present a significant dynamic variation.8–10
Recent studies have highlighted how speckle-tracking techniques are able to detect alterations in systolic and diastolic function earlier than classic parameters such as ejection fraction or tissue Doppler. Some authors have found reduced longitudinal strain values of the left ventricle in patients carrying pathogenic variants for DCM compared to healthy controls.11 This echocardiographic method could therefore identify the presence of subclinical ventricular dysfunction in family members of patients with DCM and therefore allow the phenotype to be characterized more precisely and earlier.
Cardiac magnetic resonance imaging
Cardiac magnetic resonance imaging is a non-invasive method capable of providing fundamental information to guide the diagnostic and therapeutic process of patients with cardiomyopathy. In fact, it represents the reference standard for the evaluation of endocavitary dimensions, thicknesses, and bi-ventricular systolic function, as well as the tool for obtaining information on tissue characterization such as the presence/absence of oedema, fibrosis, adipose infiltration, accumulation of iron or amyloid substance, and the extension of the extracellular space at the microscopic level. Tissue characterization highlights and quantifies the presence of micro- and macroscopic myocardial fibrosis, providing the clinician with useful information on arrhythmic risk stratification in patients with familial DCM.12
Currently, there is no mandatory indication to carry out a CMR in the context of screening family members affected by DCM, in the absence of clinical references or complex family genetic patterns. However, there are situations in which CMR plays a decisive role even in first- or second-degree relatives of patients suffering from DCM.
Cardiac magnetic resonance imaging plays an important role in the differential diagnosis between athlete’s heart and initial forms of DCM in family members who present isolated left ventricular dilatation and are also competitive athletes. More significant imaging findings include unbalanced left ventricular dilatation, left ventricular dysfunction, and the presence of LGE. However, in very early forms, the increase in T1 mapping or extracellular volume (ECV) constitutes an early marker of disease. In the hearts of competitive athletes, in fact, T1 or ECV values are typically at the lower limits of normal and sometimes slightly reduced.13 Another context in which CMR can play a crucial role is early diagnosis in family members carrying pathogenic variants: in these cases, CMR can detect initial changes in the size, function, and structure of the myocardial tissue such as the increase in endoventricular dimensions, the increase in T1, the increase in ECV, and/or the appearance of LGE (Figure 1).
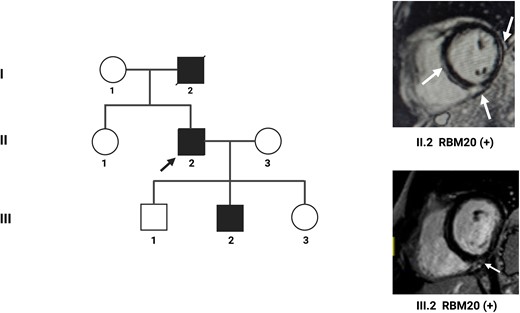
An example of familial dilated cardiomyopathy. A 56-year-old patient (II.2) is referred for frequent ventricular extrasystole. On the echocardiogram, dilated cardiomyopathy with moderate systolic dysfunction and left ventricular ejection fraction of 40% is diagnosed. Cardiac magnetic resonance imaging with contrast highlights numerous areas of late gadolinium enhancement at the level of the interventricular septum, the lateral wall, and the junction between the interventricular septum and the right ventricle (white arrows). Genetic analysis reveals a pathogenetic variant of the RBM20 gene. Family screening highlights mild dilatation of the left ventricle in the male child (III.2). Cardiac magnetic resonance imaging shows a small area of late gadolinium enhancement at the junction between the interventricular septum and the right ventricle (white arrow). Genetic analysis shows the same pathogenetic variant on the RBM20 gene detected in the father.
In some specific contexts, in addition to an advance in the diagnosis timing, CMR adds a non-negligible prognostic impact. For example, in family members of patients affected by lamin A/C or filamin C cardiomyopathy, the use of CMR as a screening element can recognize initial elements of the disease such as increased ECV, alterations in mid-basal septal strain with increased dispersion times, and the presence of mid-basal septal LGE or ‘ring-like’ pattern. These tissue characteristics may be present before the onset of the classic dilated-hypokinetic phenotype, evident on echocardiographic investigation, and are associated with an unfavourable prognosis.14
Genetic analysis
The genetic architecture of DCM patients is particularly complex and includes many genes coding for proteins involved in the structure of different cellular components.2 The result of the genetic analysis can profoundly influence the management of the patient with DCM in prognostic and therapeutic terms (for example for the implantation of a defibrillator in primary prevention) and for the impact on the management of family members (Table 2).
Malignant genotypes and risk factors for arrhythmic events
| Gene | Clinical and genetic variables | ECG | Imaging | Risk calculator |
|---|---|---|---|---|
| LMNA | Male sex | AV block | LVEF | 2019 LMNA-risk VTA1 |
| Non-missense variants | NSVT | LGE at CMR | ||
| FLNC truncating variants | Proband status (3) | Inferolateral T-wave inversion | LVEF < 45% | |
| LGE at CMR | ||||
| DSP | Proband status | LVEF < 45% | ||
| Variant location | LGE at CMR | |||
| TMEM43 | Male sex | >200 VEBs/24 h | LVEF < 45% | |
| Female sex with one or more risk factor | NSVT | LGE at CMR | ||
| RBM20 | Male sex | LVEF < 45% | ||
| LGE at CMR | ||||
| PLN | NSVT | LVEF < 45% | PLN risk calculator1 | |
| Multiple VEBs/24 h | LGE at CMR | |||
| Numbers of negative T-waves | ||||
| Low QRS voltages |
| Gene | Clinical and genetic variables | ECG | Imaging | Risk calculator |
|---|---|---|---|---|
| LMNA | Male sex | AV block | LVEF | 2019 LMNA-risk VTA1 |
| Non-missense variants | NSVT | LGE at CMR | ||
| FLNC truncating variants | Proband status (3) | Inferolateral T-wave inversion | LVEF < 45% | |
| LGE at CMR | ||||
| DSP | Proband status | LVEF < 45% | ||
| Variant location | LGE at CMR | |||
| TMEM43 | Male sex | >200 VEBs/24 h | LVEF < 45% | |
| Female sex with one or more risk factor | NSVT | LGE at CMR | ||
| RBM20 | Male sex | LVEF < 45% | ||
| LGE at CMR | ||||
| PLN | NSVT | LVEF < 45% | PLN risk calculator1 | |
| Multiple VEBs/24 h | LGE at CMR | |||
| Numbers of negative T-waves | ||||
| Low QRS voltages |
AV, atrioventricular; VT, ventricular tachycardia; VF, ventricular fibrillation; SCD, sudden cardiac death; LMNA, lamin A; NSVT, non-sustained ventricular tachycardia; LVEF, left ventricular ejection fraction; LGE, late gadolinium enhancement; CMR, cardiac magnetic resonance imaging; FLNC, filamin C; DSP, desmoplakin; TMEM43, transmembrane protein 43; VEB, ventricular ectopic beat; RBM20, RNA binding motif protein 20; PLN, phospholamban.
Malignant genotypes and risk factors for arrhythmic events
| Gene | Clinical and genetic variables | ECG | Imaging | Risk calculator |
|---|---|---|---|---|
| LMNA | Male sex | AV block | LVEF | 2019 LMNA-risk VTA1 |
| Non-missense variants | NSVT | LGE at CMR | ||
| FLNC truncating variants | Proband status (3) | Inferolateral T-wave inversion | LVEF < 45% | |
| LGE at CMR | ||||
| DSP | Proband status | LVEF < 45% | ||
| Variant location | LGE at CMR | |||
| TMEM43 | Male sex | >200 VEBs/24 h | LVEF < 45% | |
| Female sex with one or more risk factor | NSVT | LGE at CMR | ||
| RBM20 | Male sex | LVEF < 45% | ||
| LGE at CMR | ||||
| PLN | NSVT | LVEF < 45% | PLN risk calculator1 | |
| Multiple VEBs/24 h | LGE at CMR | |||
| Numbers of negative T-waves | ||||
| Low QRS voltages |
| Gene | Clinical and genetic variables | ECG | Imaging | Risk calculator |
|---|---|---|---|---|
| LMNA | Male sex | AV block | LVEF | 2019 LMNA-risk VTA1 |
| Non-missense variants | NSVT | LGE at CMR | ||
| FLNC truncating variants | Proband status (3) | Inferolateral T-wave inversion | LVEF < 45% | |
| LGE at CMR | ||||
| DSP | Proband status | LVEF < 45% | ||
| Variant location | LGE at CMR | |||
| TMEM43 | Male sex | >200 VEBs/24 h | LVEF < 45% | |
| Female sex with one or more risk factor | NSVT | LGE at CMR | ||
| RBM20 | Male sex | LVEF < 45% | ||
| LGE at CMR | ||||
| PLN | NSVT | LVEF < 45% | PLN risk calculator1 | |
| Multiple VEBs/24 h | LGE at CMR | |||
| Numbers of negative T-waves | ||||
| Low QRS voltages |
AV, atrioventricular; VT, ventricular tachycardia; VF, ventricular fibrillation; SCD, sudden cardiac death; LMNA, lamin A; NSVT, non-sustained ventricular tachycardia; LVEF, left ventricular ejection fraction; LGE, late gadolinium enhancement; CMR, cardiac magnetic resonance imaging; FLNC, filamin C; DSP, desmoplakin; TMEM43, transmembrane protein 43; VEB, ventricular ectopic beat; RBM20, RNA binding motif protein 20; PLN, phospholamban.
Genetic analysis is recommended in first-degree relatives of patients with DCM who present a pathogenic or likely pathogenic variant.2 The finding of the genetic variant may lead to the need for closer follow-up in the subject under examination, even in the absence of a manifest phenotype. Furthermore, the finding of some specific variants, such as those linked to desmosomal genes, can influence some elements of the clinical management of the subject even in the absence of a clear phenotype, for example with regard to suitability for competitive sports. On the contrary, the absence of the variant allows us to exclude with reasonable certainty the presence of the disease in the family member and to interrupt or at least significantly delay follow-up over time. In the case of identification of a variant of uncertain significance (VOUS) in the proband, genetic analysis should be considered only if there are other family members with a phenotype compatible with DCM, in order to characterize a possible familial segregation of the variant and determine its pathogenicity.2 The performance of a genetic test in paediatric patients can be considered whenever it can have a significant impact on the clinical management of the subject, for example when in the family there is a genetic variant associated with clinical onset in childhood or with a high incidence of malignant ventricular arrhythmias. However, it is always necessary to consider the potential psychological impact that the information deriving from this test can have on the minor and family members, especially in the event of a positive result.
In case of detection of a pathogenic or likely pathogenic variant for DCM in a proband, it is advisable to test first-degree relatives to search for the familial variant using a classic Sanger method. The use of more extensive panels and next-generation sequencing is not currently indicated, as it can potentially create problems relating to the detection of further variants that are difficult to interpret clinically.1 Obviously, if the pathogenic variant is found in a family member of the proband, it then becomes necessary to proceed with cascade screening of his family members too, with the particular limitations and precautions highlighted above in relation to the paediatric age.
The positive genetic test rate of family members of patients affected by DCM with a known pathogenic or likely pathogenic variant, in the literature, is around 40%. In a recent study,6 anamnestic and clinical information alone led to the identification of only a limited portion of cases of familial DCM, underlining the importance of a multidimensional evaluation (clinical, instrumental, and possibly genetic) in the close family members of patients with DCM. The recent ESC guidelines emphasize that age should not be a limiting factor in the use of genetic testing, considering the diagnostic implications for family members. However, in the case of limited economic resources, it is possible to use scores (for example the Madrid DCM Genotype Score) to identify patients with DCM with a high probability of a positive genotype and focus the genetic analysis on these subjects. In the Madrid score, elements suggestive of positive genetics are represented by the presence of a family history of DCM, the concomitance of myopathy, and the absence of left bundle branch abnormality and arterial hypertension.15
Where to perform screening and follow-up
The screening of first-degree family members should be carried out in Units for the study and treatment of Cardiomyopathies as defined by the recent ESC guidelines1 (Cardiomyopathy Units) with experience in clinical–anamnestic evaluation and in performing and interpreting multimodal imaging tests and genetic analysis. Regarding genetic analysis, it is important to underline the importance of pre- and post-test counselling, which must be carried out by specialists in medical genetics supported by cardiologists with experience in the management of patients with familial cardiomyopathies. It is in fact essential to provide correct information to the patient regarding the implications of the genetic test before its execution, as well as correct interpretation of the result and its prognostic and therapeutic implications. This counselling should possibly also make use of the support of a psychologist.
Similarly, the follow-up of family members, especially of carriers of pathogenic variants who have not yet developed a phenotype, should be performed in reference centres that can promptly detect, and possibly treat, the first phenotypic manifestations. Family member carriers of the genetic variant must undergo a complete clinical–instrumental evaluation including multimodal imaging and other tests based on the phenotype and genotype. These subjects, even if they do not present any phenotype, must also be included in a regular clinical–instrumental follow-up programme.
Individuals found not to carry the familial variant and who do not have a clinical phenotype can usually be excluded from follow-up, with the indication to perform a re-evaluation if they develop symptoms or new clinically relevant data emerge in the family. If genetic analysis has not been performed or has not identified a genetic cause in the family, follow-up of all first-degree relatives, including children, should be recommended. Follow-up times depend on the clinical picture, genotype, and family history of the patient. If in a family there are no other members affected by DCM besides the proband, in first-degree relatives aged ≥50 years with repeatedly normal findings on the ECG or cardiac imaging tests, an interruption or at least a less frequent follow-up may be considered.
Conclusions
Screening of family members of patients affected by DCM represents an important tool for the early diagnosis and possible treatment of other individuals affected by DCM.
Clinical–instrumental evaluation is accompanied by an increasing use of CMR and genetic analysis, which increasingly represents a diagnostic and prognostic element in this pathology. In relation to its clinical relevance and its complexity, family screening and follow-up of identified patients must be carried out in units dedicated to the treatment and study of cardiomyopathies.
Funding
None declared.
Data availability
No new data were generated or analysed in support of this research.
References
Author notes
Conflict of interest: none declared.

{kind=link}