





abstract
The class of new oral anticoagulants (NOACs) has been developed to provide reliable oral anticoagulation without the need for therapeutic drug monitoring. Based on phase I and II trials and pharmacokinetic and pharmacodynamic modeling, fixed drug doses have been selected for large phase III clinical trials for each currently available NOAC. In these trials, the use of the fixed dose without plasma level assessments was shown to be at least as effective and at least as safe as vitamin K antagonists with continuous therapeutic drug monitoring. Real world evidence reaffirms that the use of a fixed NOAC dose without plasma level assessment is safe and effective in a large variety of patients. Nevertheless, measurement of NOAC plasma levels can add information that may be useful in some clinical scenarios. This review discusses the possible use cases, the limitations, and the practical implementation of measuring NOAC plasma concentrations.
Introduction
First-generation oral anticoagulants: powerful drugs after haphazard discovery
In the developed world, ∼1–2% of the adult population is treated with oral anticoagulants.1 It is likely that the ongoing ageing of the population and an ever-increasing detection of many thrombotic conditions will only add to the overall use of oral anticoagulation.2
Historically, oral anticoagulation could only be achieved by using vitamin K antagonists (VKA). This class was developed following the serendipitous finding of bleeding disorders in animals who accidentally ingested natural sources of coumadin. The underlying pharmacological mechanism—inhibition of the hepatic vitamin K-dependent maturation of factors II, VII, IX, and X, resulting in non-functional precursors—was only discovered afterwards.3 This first generation of oral anticoagulants is the archetype of a drug with unpredictable pharmacokinetics (PK). The high intra- and interpatient variability can be explained by the many food and drug interactions, alterations in body composition, and pharmacogenetic differences.4 Furthermore, VKA are drugs with a narrow therapeutic index. As a result, a specific therapeutic range, based on the international normalized ratio (INR), has been identified to ensure net clinical benefit. The use of VKA requires continuous measurement of the pharmacodynamic (PD) effect with (empiric) adjustments of the dose as needed.5,6 This concept is widely recognized as ‘therapeutic drug monitoring’ (TDM).7
The unpredictable PK/PD of VKA is a major burden for many patients and physicians due to the repeated measurements and is associated with a continuous risk of either supra- or subtherapeutic levels. Therefore, for VKA, the quality of the TDM, measured by the time in therapeutic range (TTR), is of major importance for the quality of the therapy; studies have shown that VKA are associated with better outcomes when TTR is high, but that treatment with VKA can even result in greater mortality and higher risk of stroke compared to no treatment in patients with poor TTR.8 The limitations of VKA have hampered the broad uptake of oral anticoagulants.
Start of the new oral anticoagulant era: consistent and predictable
Vitamin K antagonists are effective drugs.9 Yet, their initial discovery and further development, predating many of the modern concepts of the coagulation cascade, have been without a clear plan.10 Conversely, new oral anticoagulants (NOACs) have been developed specifically with the goal of offering reliable oral anticoagulation without the need for TDM. In contrast to the discovery of VKA, NOAC development was deliberate and targeted.11 The first mention of dabigatran was in 2002.12 Initiation of research on edoxaban (or its predecessor) occurred in 1979, which led to the development of DX-9065a in 2003.13 Using a library of more than 200 000 molecules, rivaroxaban was selected via high throughput screening in 2005.14,15 Finally, Dupont laboratories worked on multiple molecules (1995), which was reduced to five compounds when BMS acquired Dupont in 2001; in 2005, apixaban was selected.16
In general, these molecules—while inherently very different—have several characteristics in common. They are orally active, reach peak plasma concentrations rapidly (within hours) and have shorter half-lives than VKA.17–20 In contrast to VKA, renal function does play a role in the clearance of these compounds. Perhaps most importantly, they have been shown to result in predictable exposure and pharmacodynamic effects. This means that there is a direct association between dose and plasma concentration and between (dose and) plasma concentration and pharmacodynamic effect (e.g. anti-Xa activity)21 (Figure 1).
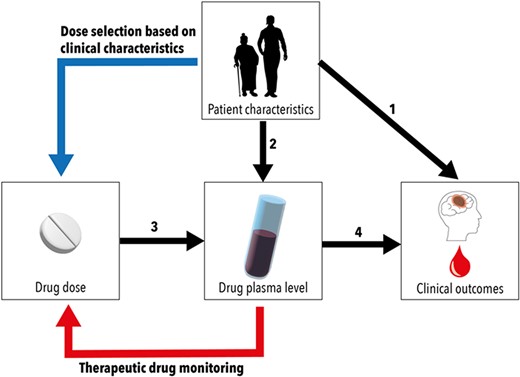
Relations between drug dose and clinical outcomes are influenced by patient characteristics and drug pharmacokinetics. Patient characteristics affect clinical outcomes directly (1) as well as indirectly, by influencing drug pharmacokinetics (2). The choice of drug dose determines drug plasma levels (3). This relation between drug dose and plasma levels can be predictable or unpredictable. The relation between plasma level and outcomes, e.g. efficacy and safety of a drug, determines the therapeutic range (4). In drugs with predictable pharmacokinetics, dose selection can be based on clinical characteristics (blue line). In drugs with unpredictable pharmacokinetics, therapeutic drug monitoring can be required to adapt the dose in order to maintain plasma levels in therapeutic range (red line).
Tailoring the dose: fixed-dose vs. pharmacokinetics-guided approach
While NOACs have been developed in response to the many difficulties related to the (safe) use of VKA, they do not resemble VKA that much. Rather, they have a PK/PD profile that is more similar to that of low-molecular-weight heparins (LMWH).22
The initial choice of fixed dose was based on Phases I and II trials and PK/PD modelling, and fixed dose (with or without a fixed-dose reduction based on pre-existing clinical criteria) has been evaluated in Phase III studies. In these Phase III studies, the use of a fixed dose of a NOAC without plasma level assessment was at least as effective and led to fewer major bleeds compared with VKA with continuous TDM to achieve optimal TTR. In such clinical trials, the TTR was frequently better than what is achieved in clinical practice. Real-world studies with fixed doses used in the absence of plasma level assessments have confirmed Phase III trial results.23 Hence, the fixed-dose approach is well supported and has been adopted in the respective NOAC labels, as is the case for most therapeutics.24,25
Although NOACs have been designed to be used without plasma level assessment, and their use in a large variety of patients has been shown to be safe and effective without plasma level assessments, measurement of NOAC plasma levels can add information that may be useful in some clinical scenarios. This review discusses the possible use cases, the limitations, and the practical implementation of measuring NOAC plasma concentrations.
New oral anticoagulant plasma level assessments: the value
New oral anticoagulant measurements as part of drug development
It is perfectly feasible to determine NOAC concentrations.26 This was extensively done in Phases I and II investigations to better understand the associations between dose, exposure, impact on clotting parameters, and clinical outcomes, such as major bleeds. All these measurements supported the concept that NOACs could be used in fixed doses in subsequent clinical trials. Different approaches were pursued to estimate the best possible fixed dose for each of the four NOACs that are currently marketed in Europe [e.g. the Phase IIb study of edoxaban vs. warfarin for stroke prevention in atrial fibrillation (SPAF) by Weitz et al.27].
Finally, taking into account the entire Phase III evidence base, it is now accepted that, in general, NOACs exhibit a consistent and predictable association between dose and clinical outcome.28 In that regard, the Phase II studies seem to have found the ‘sweet spot’ of the NOAC fixed doses for most patients.21
New oral anticoagulant pharmacokinetics parameters are associated with clinical outcomes
In Phase II studies, associations were observed between NOAC PK parameters and selected outcomes. For example, an increase in the total drug exposure, characterized by the area-under-the-curve at steady state (AUCss), was associated with more bleeds in the apixaban developmental programme (more so than Cmin and Cmax).29 Importantly, no single PK parameter was found to correlate with outcomes for all NOACs, e.g. rather than the AUCss of dabigatran its Cmax was associated with the bleeding rate.30
Likewise, PK associations were also found in post hoc analyses of the landmark NOAC trials in atrial fibrillation (AF).31–34 Both clinical characteristics and NOAC exposure were identified as determinants for clinical outcomes. Importantly, clinical characteristics both influence exposure and clinical outcome. For example, a higher age will not only increase the risk of stroke in AF but will also aggravate the bleeding risk and impact plasma concentrations. It is a difficult exercise to estimate the best possible plasma concentration for a given age (or other confounder) in order to both reduce the stroke and bleeding risks in AF. It is crucial to recognize that patients were randomized to fixed doses, and not to plasma ranges. Based on these PK analyses, no additional covariates, compared to the ones already included in the dosing protocol, were uncovered.31–34
Plasma concentrations were not determined in all patients; the measurements were analysed post hoc and did not result in any therapy changes. Furthermore, in the heavily publicized Reilly paper,35 the PK analysis was based on a single trough measurement at 1 month after randomization in RE-LY.
It is important to consider that for none of the NOACs a therapeutic range has been identified. Rather, we have access to data on observed (or on-therapy) ranges based on plasma values determined at (mostly) one time point in selected study participants, who were clinically stable and received fixed doses at the time. Bearing in mind these limitations, the major value at the moment of the available data is that: observed plasma ranges were broad (and reassuring); there seems to be a threshold below which the stroke risk increases in AF; accordingly, higher plasma values were associated with more bleeds. Also, reassuring was that plasma values did not correspond with the risk of intracranial bleeds.
New oral anticoagulant use cases
Below we have worked out several use cases for the clinical practitioner, based on the patient profile, and potential value that a plasma value might have in shared decision making.
‘Standard’, chronic use
Patients similar to Phase III study patients
This constitutes trial eligible patients, i.e. those who would fall within in- and exclusion criteria, or the use according to the product label. In these cases, Phase III studies have robustly confirmed the efficacy and safety of NOACs without any need for plasma level assessments. If plasma levels would be outside the expected values in these patients, it is unclear whether changing to a dose that has not been studied would actually improve outcome.
Patients with one or several clinical features that may strongly influence new oral anticoagulant pharmacokinetics
If the patients treated with a NOAC would not have qualified for inclusion in one of the landmark trials, then assessing exposure might have some merit in specific circumstances. Although real-world evidence is reassuring on a population level, the individual safety and efficacy in patients with risk factors for altered PK/PD are difficult to assess. Even then, we would suggest refraining from over-relying on plasma measurement.
Only in the setting of a high pretest likelihood of reaching an extremely high or low plasma value [outside the 90–95% confidence interval (CI) of the on-therapy ranges], we suggest performing a plasma assessment. Moreover, not all changes in exposure are relevant to the patient. A change in the pharmacodynamic response is more likely when a covariate (e.g. weight or renal function) has a ‘meaningful’ impact on NOAC exposure (<50% or +100% AUC).31 For most drugs, crossing such an AUC threshold would then commonly be considered to be clinically relevant.36 In Phase II studies, these covariates—as a rule—have been identified, and they have been incorporated into the dose reduction criteria (in part) to alter the NOAC dose in response to patient characteristics.
Below, a selection of the most clinically relevant covariates has been listed.
Renal function
All NOACs are renally cleared to some extent (dabigatran 80%, rivaroxaban 35%, apixaban 27%, and edoxaban 50%). Renal impairment will hence lower the clearance (CLrenal) of all NOACs to some degree. As a result, AUC and t1/2 will be increased moderately. In the landmark AF trials, renal function has been included in the dose criteria for each NOAC. In the case of mild to moderate renal impairment, the NOAC dose should be evaluated for reduction.
The European drug label/SmPC precludes use in patients with severe renal insufficiency (Creatinine Clearance (CrCl) < 30 mL/min for dabigatran; CrCl < 15 mL/min for apixaban, edoxaban, and rivaroxaban with the CrCl estimated by the Cockcroft–Gault equation). However, well-studied alternatives are lacking, and furthermore, potential alternatives might also suffer from specific drawbacks and risks in these patients.
In general, a reduced renal function increases both the bleeding risk as well as the thrombotic risk even beyond the effect on the pharmacology of antithrombotic drugs. In end-stage kidney disease, there is a disproportionately high bleeding risk due to various reasons, which also include uraemia-induced platelet dysfunction. In end-stage renal disease, the bleeding risk is also strongly increased with VKAs, which do not rely on renal function for their metabolism. As a consequence, warfarin use in dialysis-dependent adults with AF has not been associated with any net clinical benefit.
Drug–drug interactions
Drug–drug interactions do occur in NOAC users and can be significant, but they only rarely result in meaningful therapy changes, particularly when compared with VKA.37 Major mechanisms include inhibition or induction of CYP3A4 and/or P-glycoprotein (PgP). All NOACs are PgP-substrates, and anti-Xa inhibitors undergo CYP3A4-dependent metabolization as well (apixaban, rivaroxaban > edoxaban). Not all NOACs are exactly alike. Dabigatran is most dependent on renal clearance and PgP, taking into account that only the etexilate is a substrate for PgP. Edoxaban is only metabolized very limited by CYP3A4. Apixaban and rivaroxaban largely undergo metabolic clearance to the same degree.24,37
For all NOAC, multiple clearance pathways exist. This means that a NOAC has multiple escape routes available to be eliminated from the body. Hence, in clinical practice, only the co-administration with potent interacting drugs (at least: AUC −50% or +100%) should be avoided or managed with caution.38 The NOAC SmPCs provide a good basic source of multiple agents that should not be combined or should be used with caution (e.g. the pan-inducer rifampicin or the strong combined CYP3A4-PgP inhibitor ketoconazole), but the exact effect in a given patient remains difficult to predict. Based on the patient profile, selection of a NOAC with the lowest potential for drug–drug interactions can be recommended. Nevertheless, especially in the case of the use of multiple strong drug inhibitors and/or inducers, NOAC plasma values might allow the prescriber to determine whether the individual patient is an outlier and whether to subsequently alter the drug regimen.
Advanced age and/or frailty
Advanced age has been associated with more thrombotic and bleeding events. Many clinically relevant outcomes are compounded in high age across multiple indications (e.g. heart failure hospitalizations in patients with heart failure with reduced ejection fraction). In general, older people take more drugs, have more concomitant diseases, and have a reduced physiological reserve. Additionally, changes in body composition, PK and PD response to many drugs render older people more vulnerable to drug-related harm.39 With regard to anticoagulation use, this means that older adults might experience more harm.40 Importantly, given the higher baseline stroke risk, the absolute risk reduction of NOACs compared to VKA is at least retained (or larger).
Higher age has been associated with increased exposure to NOACs.31–34 Only after RE-LY was completed, age was identified as an important covariate and included as part of the dose reduction criteria in the European label. For apixaban, age (>80 years) was already part of the trial protocol (ARISTOTLE).41 Importantly, a high chronological age in and of itself does not preclude the use of NOACs or does not inherently mean that plasma concentrations should be measured. Even in the landmark trials, older adults were enrolled who were multimorbid, polymedicated, and renally impaired.42 Rather, in patients with a very high functional age (e.g. functional dependency, severe dementia) and measures of frailty (e.g. >30 s needed for the Timed Up And Go test) plasma measurements might be rarely indicated, mostly to exclude outliers. Most likely, such patients will already receive a reduced NOAC dose based on patient characteristics.
Altered absorption
All NOACs are taken orally and then undergo a pharmaceutical phase during which the drug substance is dissolved in the stomach and the first part of the duodenum. Absorption then takes place largely in the duodenum. The extent of dissolution, permeability, and resulting bioavailability is not uniform among NOACs (dabigatran 3–7%, rivaroxaban >90% in fed state, apixaban 50%, and edoxaban 62%).43 Concomitant food intake is indicated to improve gastro-intestinal (GI) uptake for the therapeutic doses of rivaroxaban (15 and 20 mg), probably by prolonging GI residence time.44
Appropriate NOAC therapy should be verified in all clinical situations which might limit the GI uptake of NOACs, e.g. in patients with short bowel or GI fistulation, post-bariatric surgery, or even in selected patients with substantial chronic GI inflammation. Here, there is certainly room to establish appropriate exposure by performing at least one plasma measurement.
Weight
Weight can impact NOAC exposure and some might assume that extremes of weight might hence influence exposure to a larger—even clinically relevant—degree. There might be a potential increase in the risk of bleeding in patients with lower weight (e.g. <40–50 kg) and conversely a higher risk of breakthrough thrombotic events in higher weight regions (e.g. >120 kg). However, several meta-analyses of landmark trials in AF have suggested the conservation of benefit of NOACs compared to VKA across a broad weight range.45–47 Furthermore, the latest update of the International Society on Thrombosis and Haemostasis on use of NOACs in VTE recommended to rather rely on the published trial data, i.e. to use the tested doses. Importantly, they suggested to refrain from regularly determining peak or trough plasma levels, given the lack of trial data to inform downstream clinical decisions.48 In the majority of patients with a lower or higher weight, it is hence not necessary to perform plasma level monitoring. However, in some patients with extremely deviating weights (e.g. anorexia, cachexia, or severe obesity) it may be considered to perform plasma level assessment of the NOAC to verify whether plasma levels fall within range, particularly if other factors that may influence NOAC exposure are present, such as a reduced kidney function or potentially interfering concomitant drugs.
Combination
The combined/integrated effects of several of the aforementioned covariates are often difficult to predict. Commonly, such patients were frequently excluded from the landmark trials.49 It might be rational to confirm at least whether plasma exposure is comparable to observed on-therapy ranges from said trials. In patients with one or more of these features, a single ‘spot’ plasma level assessment to check whether the plasma levels are within the expected values can help to reassure clinicians.
The optimal approach when levels are outside of the expected values is unknown, however, as there are no studies that have compared the use of other—unapproved—doses of NOACs in these situations. A conservative approach would be to conclude that the use of that particular NOAC in that individual patient is not recommended, and that an alternative strategy should be considered. This can include switching to a NOAC with different pharmacokinetic profile, or a switch to an alternative anticoagulant therapy. In select cases, and after careful consideration by an experienced physician, off-label dosing of NOACs could be considered. This mostly pertains to relative underdosing in specific cases, bearing in mind the recent results from ELDERCARE-AF which showed that a lower dose (resulting in a similar AUC) still conferred substantial protection against stroke as compared with no anticoagulation.50 The size of the effect (placebo vs. edoxaban 15 mg od: 6.7%/y vs. 2.3%/y) was comparable to that of VKA vs. placebo in the meta-analysis by Hart et al.51 If anything, ELDERCARE-AF trial also confirmed the very high stroke burden in older adults. If the alternative would be to discontinue all anticoagulants altogether (owing to very high age or a history of GI bleeds), using a lower dose might be the wiser option.
Emergencies and other special situations: residual new oral anticoagulant plasma levels
In several acute situations, assessment of plasma levels could help to guide treatment, e.g. drug intoxications, comatose patients requiring urgent or semi-urgent intervention, semi-urgent interventions in patients with expected prolonged half-lives (most commonly owing to renal impairment). Here, plasma levels can be used to exclude residual anticoagulant activity (e.g. <30 or <50 ng/mL).31
The largest prospective study of periprocedural use of NOACs, the PAUSE study, showed considerable variation of periprocedural plasma levels at the time of intervention following a standardized interruption approach.52 However, plasma levels at time of intervention did not correlate with the procedural bleeding risk.53 Therefore, there is no need to routinely measure NOAC levels if an appropriate timing of interruption has been used.24
New oral anticoagulant plasma level assessments: limitations and practical considerations
Below, we have worked out several elements that need to be considered when planning for, performing, and finally interpreting NOAC plasma monitoring.
Basis for a pharmacokinetics-guided approach
A PK-guided approach is only useful when the following criteria have been met. It has to concern a drug with considerable inter/intrapatient variability (precluding the use of fixed dose) and a narrow therapeutic index. There has to be a predefined range (‘sweet spot’ for efficacy and safety) to compare the patient’s values to. This range should best be validated in a randomized controlled trial (RCT). Then we have to decide which type of patients are more likely to derive a benefit from plasma monitoring. Also, we have to be able to detect the concentration. It should be clear what type of sample should best be used (trough or peak or both; full AUC) and how frequently it should be retested. It is crucial to ensure that inadequate sampling does not confound the test result. If possible, relevant covariates that might impact net clinical benefit of the NOAC should be evaluated as well. If the patient is then finally identified as an outlier, we should have to decide how to change the dose in such a manner that the next result will fall within range AND that this approach will result in improved outcomes.
Bearing in mind the above and taking into account the logistics of performing plasma measurement, trough values are likely to be preferred for most cases when evaluating plasma NOAC exposure, as excluding significant accumulation of NOACs is often the main concern. In contrast, when adequate absorption is assessed (e.g. in patients with short bowel), peak levels would be more informative. The observed results can then be compared to the on-therapy ranges as observed in the relevant clinical trials. Suitable patient profiles have been discussed in the chapter above, to establish the exposure in on-label use with presence of strong covariates and/or to exclude residual anticoagulant activity in specific situations.
Additional information on technical aspects of sampling, laboratory analysis, and further information on interpretation have been summarized in Table 1.
Potential limitations and solutions in measurement of plasma levels
| Limitations | Potential solutions | |
|---|---|---|
| Patient selection | Most patients do not have an indication for plasma monitoring. | Limit the assessment to patients with a high pretest risk of falling outside the observed on-therapy ranges and/or to exclude residual anticoagulant activity in (semi)urgent situations. |
| Sampling |
| Trough levels are theoretically more ‘stable’ in chronic NOAC use. If information needed about absorption (e.g. short bowel) is important, peak levels may be considered. |
| Analysis | Availability of laboratory tests.26 |
|
| Clinical consequences | The clinical effect of adapting a dose based on plasma levels has not been studied. There is no certainty that administering an altered dose will result in improved efficacy and/or safety. | The greatest caution should be made in adapting clinical doses based on PK measurements.24 |
| Retesting frequency | Several studies have shown considerable between-person and within-person variability between serial measurements, with values changing from within-range to out-of-range values between measurements in otherwise stable patients. |
| Limitations | Potential solutions | |
|---|---|---|
| Patient selection | Most patients do not have an indication for plasma monitoring. | Limit the assessment to patients with a high pretest risk of falling outside the observed on-therapy ranges and/or to exclude residual anticoagulant activity in (semi)urgent situations. |
| Sampling |
| Trough levels are theoretically more ‘stable’ in chronic NOAC use. If information needed about absorption (e.g. short bowel) is important, peak levels may be considered. |
| Analysis | Availability of laboratory tests.26 |
|
| Clinical consequences | The clinical effect of adapting a dose based on plasma levels has not been studied. There is no certainty that administering an altered dose will result in improved efficacy and/or safety. | The greatest caution should be made in adapting clinical doses based on PK measurements.24 |
| Retesting frequency | Several studies have shown considerable between-person and within-person variability between serial measurements, with values changing from within-range to out-of-range values between measurements in otherwise stable patients. |
NOAC, new oral anticoagulant; LC-MS/MS, liquid chromatography with tandem mass spectrometry; PK, pharmacokinetics; t1/2, half-life.
Potential limitations and solutions in measurement of plasma levels
| Limitations | Potential solutions | |
|---|---|---|
| Patient selection | Most patients do not have an indication for plasma monitoring. | Limit the assessment to patients with a high pretest risk of falling outside the observed on-therapy ranges and/or to exclude residual anticoagulant activity in (semi)urgent situations. |
| Sampling |
| Trough levels are theoretically more ‘stable’ in chronic NOAC use. If information needed about absorption (e.g. short bowel) is important, peak levels may be considered. |
| Analysis | Availability of laboratory tests.26 |
|
| Clinical consequences | The clinical effect of adapting a dose based on plasma levels has not been studied. There is no certainty that administering an altered dose will result in improved efficacy and/or safety. | The greatest caution should be made in adapting clinical doses based on PK measurements.24 |
| Retesting frequency | Several studies have shown considerable between-person and within-person variability between serial measurements, with values changing from within-range to out-of-range values between measurements in otherwise stable patients. |
| Limitations | Potential solutions | |
|---|---|---|
| Patient selection | Most patients do not have an indication for plasma monitoring. | Limit the assessment to patients with a high pretest risk of falling outside the observed on-therapy ranges and/or to exclude residual anticoagulant activity in (semi)urgent situations. |
| Sampling |
| Trough levels are theoretically more ‘stable’ in chronic NOAC use. If information needed about absorption (e.g. short bowel) is important, peak levels may be considered. |
| Analysis | Availability of laboratory tests.26 |
|
| Clinical consequences | The clinical effect of adapting a dose based on plasma levels has not been studied. There is no certainty that administering an altered dose will result in improved efficacy and/or safety. | The greatest caution should be made in adapting clinical doses based on PK measurements.24 |
| Retesting frequency | Several studies have shown considerable between-person and within-person variability between serial measurements, with values changing from within-range to out-of-range values between measurements in otherwise stable patients. |
NOAC, new oral anticoagulant; LC-MS/MS, liquid chromatography with tandem mass spectrometry; PK, pharmacokinetics; t1/2, half-life.
What levels are ‘expected values’ for different new oral anticoagulant doses and indications?
In sum, reported plasma values are descriptive and depend on the study, the enrolled study population, and the investigated drug dose. They concern post hoc findings, which did not impact dosing in any of the RCTs. Furthermore, associations between plasma values and clinical outcomes were observational in nature by definition. Because of the differences in the study populations between patients with venous thromboembolism (VTE) and with AF, the same drug dose can have different expected ranges for different indications (Table 2).
Expected ranges of NOAC plasma levels by dose and clinical indication
| Molecule | Test | Peak54,55 | Dose | Indication | Trough/peak | Plasma level (ng/mL) | Ref. |
|---|---|---|---|---|---|---|---|
| Dabigatran20,56 | dTT | 1.5–3 h | 150 mg bid | AF | Trough | 91 (25–75th percentile: 61–143) | SPC |
| Peak | 175 (25–75th percentile: 117–275) | SPC | |||||
| VTE | Trough | 60 (25–75th percentile: 39–95) | SPC | ||||
| Peak | 175 (25–75th percentile: 117–275) | SPC | |||||
| 110 mg bid | AF | Trough | 63 (5–95th percentile: 62–64) | 34 | |||
| Peak | No data found | ||||||
| VTE | Trough | No data found | |||||
| Peak | No data found | ||||||
| Rivaroxaban19,57,58 | Chromogene anti-FXA assay | 2–3 h | 20 mg od | AF | Trough | 44 (5–95th percentile: 22–137) | 19 |
| Peak | 249 (5–95th percentile: 184–343) | 19 | |||||
| VTE | Trough | 26 (5–95th percentile: 6–87) | 19 | ||||
| Peak | 270 (5–95th percentile: 189–419) | 19 | |||||
| 15 mg od | AF | Trough | 57 (5–95th percentile: 18–136) | 19 | |||
| Peak | 229 (5–95th percentile: 178–313) | 19 | |||||
| 10 mg od | VTE prevention after joint surgery | Trough | 9 (5–95th percentile: 1–38) | 19 | |||
| Peak | 125 (5–95th percentile: 91–196) | 19 | |||||
| 10 mg od | VTE secondary prevention | Trough | 14 (min/max: 4–51) | SPC | |||
| 10 mg od | VTE secondary prevention | Peak | 101 (min/max: 7–273) | SPC | |||
| 2.5 mg bid | Vascular prevention in chronic CAD/PAD | Trough | 17 (5–95th percentile: 6–37) | 19 | |||
| Peak | 46 (5–95th percentile: 28–70) | 19 | |||||
| Apixaban16,17,59–61 | Chromogene anti-FXA assay | 3–4 h | 5 mg bid | AF | Trough | 103 (5–95th percentile: 41–230) | SPC17 |
| Peak | 171 (5–95th percentile: 91–321) | SPC17 | |||||
| 10 mg bid | VTE (initial treatment) | Trough | 120 (5–95th percentile: 41–335) | SPC17 | |||
| Peak | 251 (5–95th percentile: 111–572) | SPC17 | |||||
| 5 mg bid | VTE | Trough | 63 (5–95th percentile: 22–177) | SPC17 | |||
| Peak | 132 (5–95th percentile: 59–302) | SPC17 | |||||
| 2.5 mg bid | AF | Trough | 79 (5–95th percentile: 34–162) | SPC17 | |||
| Peak | 123 (5–95th percentile: 69–221) | SPC17 | |||||
| VTE | Trough | 32 (5–95th percentile: 11–90) | SPC17 | ||||
| Peak | 67 (5–95th percentile: 153) | SPC17 | |||||
| Edoxaban62 | Chromogene anti-FXA assay | 1–2 h | 60 mg od | AF | Trough | 36 (25–75th percentile: 19–62) | 63 |
| Peak | 170 (1.5*IQR125–245) | 27 | |||||
| VTE | Trough | 19 (25–75th percentile: 10–39) | 64 | ||||
| Peak | 234 (25–75th percentile: 149–317) | 64 | |||||
| 30 mg od | AF | Trough | 27 (25–75th percentile: 15–44.6) | 63 | |||
| Peak | 85 (57–115) | 27 | |||||
| VTE | Trough | 16 (25–75th percentile: 8–32) | 64 | ||||
| Peak | 164 (25–75th percentile: 99–225) | 64 | |||||
| 15 mg od | AF | Trough | 12 (25–75th percentile: 7–21) | 63 | |||
| Peak | No data found | ||||||
| Molecule | Test | Peak54,55 | Dose | Indication | Trough/peak | Plasma level (ng/mL) | Ref. |
|---|---|---|---|---|---|---|---|
| Dabigatran20,56 | dTT | 1.5–3 h | 150 mg bid | AF | Trough | 91 (25–75th percentile: 61–143) | SPC |
| Peak | 175 (25–75th percentile: 117–275) | SPC | |||||
| VTE | Trough | 60 (25–75th percentile: 39–95) | SPC | ||||
| Peak | 175 (25–75th percentile: 117–275) | SPC | |||||
| 110 mg bid | AF | Trough | 63 (5–95th percentile: 62–64) | 34 | |||
| Peak | No data found | ||||||
| VTE | Trough | No data found | |||||
| Peak | No data found | ||||||
| Rivaroxaban19,57,58 | Chromogene anti-FXA assay | 2–3 h | 20 mg od | AF | Trough | 44 (5–95th percentile: 22–137) | 19 |
| Peak | 249 (5–95th percentile: 184–343) | 19 | |||||
| VTE | Trough | 26 (5–95th percentile: 6–87) | 19 | ||||
| Peak | 270 (5–95th percentile: 189–419) | 19 | |||||
| 15 mg od | AF | Trough | 57 (5–95th percentile: 18–136) | 19 | |||
| Peak | 229 (5–95th percentile: 178–313) | 19 | |||||
| 10 mg od | VTE prevention after joint surgery | Trough | 9 (5–95th percentile: 1–38) | 19 | |||
| Peak | 125 (5–95th percentile: 91–196) | 19 | |||||
| 10 mg od | VTE secondary prevention | Trough | 14 (min/max: 4–51) | SPC | |||
| 10 mg od | VTE secondary prevention | Peak | 101 (min/max: 7–273) | SPC | |||
| 2.5 mg bid | Vascular prevention in chronic CAD/PAD | Trough | 17 (5–95th percentile: 6–37) | 19 | |||
| Peak | 46 (5–95th percentile: 28–70) | 19 | |||||
| Apixaban16,17,59–61 | Chromogene anti-FXA assay | 3–4 h | 5 mg bid | AF | Trough | 103 (5–95th percentile: 41–230) | SPC17 |
| Peak | 171 (5–95th percentile: 91–321) | SPC17 | |||||
| 10 mg bid | VTE (initial treatment) | Trough | 120 (5–95th percentile: 41–335) | SPC17 | |||
| Peak | 251 (5–95th percentile: 111–572) | SPC17 | |||||
| 5 mg bid | VTE | Trough | 63 (5–95th percentile: 22–177) | SPC17 | |||
| Peak | 132 (5–95th percentile: 59–302) | SPC17 | |||||
| 2.5 mg bid | AF | Trough | 79 (5–95th percentile: 34–162) | SPC17 | |||
| Peak | 123 (5–95th percentile: 69–221) | SPC17 | |||||
| VTE | Trough | 32 (5–95th percentile: 11–90) | SPC17 | ||||
| Peak | 67 (5–95th percentile: 153) | SPC17 | |||||
| Edoxaban62 | Chromogene anti-FXA assay | 1–2 h | 60 mg od | AF | Trough | 36 (25–75th percentile: 19–62) | 63 |
| Peak | 170 (1.5*IQR125–245) | 27 | |||||
| VTE | Trough | 19 (25–75th percentile: 10–39) | 64 | ||||
| Peak | 234 (25–75th percentile: 149–317) | 64 | |||||
| 30 mg od | AF | Trough | 27 (25–75th percentile: 15–44.6) | 63 | |||
| Peak | 85 (57–115) | 27 | |||||
| VTE | Trough | 16 (25–75th percentile: 8–32) | 64 | ||||
| Peak | 164 (25–75th percentile: 99–225) | 64 | |||||
| 15 mg od | AF | Trough | 12 (25–75th percentile: 7–21) | 63 | |||
| Peak | No data found | ||||||
Expected ranges of NOAC plasma levels by dose and clinical indication
| Molecule | Test | Peak54,55 | Dose | Indication | Trough/peak | Plasma level (ng/mL) | Ref. |
|---|---|---|---|---|---|---|---|
| Dabigatran20,56 | dTT | 1.5–3 h | 150 mg bid | AF | Trough | 91 (25–75th percentile: 61–143) | SPC |
| Peak | 175 (25–75th percentile: 117–275) | SPC | |||||
| VTE | Trough | 60 (25–75th percentile: 39–95) | SPC | ||||
| Peak | 175 (25–75th percentile: 117–275) | SPC | |||||
| 110 mg bid | AF | Trough | 63 (5–95th percentile: 62–64) | 34 | |||
| Peak | No data found | ||||||
| VTE | Trough | No data found | |||||
| Peak | No data found | ||||||
| Rivaroxaban19,57,58 | Chromogene anti-FXA assay | 2–3 h | 20 mg od | AF | Trough | 44 (5–95th percentile: 22–137) | 19 |
| Peak | 249 (5–95th percentile: 184–343) | 19 | |||||
| VTE | Trough | 26 (5–95th percentile: 6–87) | 19 | ||||
| Peak | 270 (5–95th percentile: 189–419) | 19 | |||||
| 15 mg od | AF | Trough | 57 (5–95th percentile: 18–136) | 19 | |||
| Peak | 229 (5–95th percentile: 178–313) | 19 | |||||
| 10 mg od | VTE prevention after joint surgery | Trough | 9 (5–95th percentile: 1–38) | 19 | |||
| Peak | 125 (5–95th percentile: 91–196) | 19 | |||||
| 10 mg od | VTE secondary prevention | Trough | 14 (min/max: 4–51) | SPC | |||
| 10 mg od | VTE secondary prevention | Peak | 101 (min/max: 7–273) | SPC | |||
| 2.5 mg bid | Vascular prevention in chronic CAD/PAD | Trough | 17 (5–95th percentile: 6–37) | 19 | |||
| Peak | 46 (5–95th percentile: 28–70) | 19 | |||||
| Apixaban16,17,59–61 | Chromogene anti-FXA assay | 3–4 h | 5 mg bid | AF | Trough | 103 (5–95th percentile: 41–230) | SPC17 |
| Peak | 171 (5–95th percentile: 91–321) | SPC17 | |||||
| 10 mg bid | VTE (initial treatment) | Trough | 120 (5–95th percentile: 41–335) | SPC17 | |||
| Peak | 251 (5–95th percentile: 111–572) | SPC17 | |||||
| 5 mg bid | VTE | Trough | 63 (5–95th percentile: 22–177) | SPC17 | |||
| Peak | 132 (5–95th percentile: 59–302) | SPC17 | |||||
| 2.5 mg bid | AF | Trough | 79 (5–95th percentile: 34–162) | SPC17 | |||
| Peak | 123 (5–95th percentile: 69–221) | SPC17 | |||||
| VTE | Trough | 32 (5–95th percentile: 11–90) | SPC17 | ||||
| Peak | 67 (5–95th percentile: 153) | SPC17 | |||||
| Edoxaban62 | Chromogene anti-FXA assay | 1–2 h | 60 mg od | AF | Trough | 36 (25–75th percentile: 19–62) | 63 |
| Peak | 170 (1.5*IQR125–245) | 27 | |||||
| VTE | Trough | 19 (25–75th percentile: 10–39) | 64 | ||||
| Peak | 234 (25–75th percentile: 149–317) | 64 | |||||
| 30 mg od | AF | Trough | 27 (25–75th percentile: 15–44.6) | 63 | |||
| Peak | 85 (57–115) | 27 | |||||
| VTE | Trough | 16 (25–75th percentile: 8–32) | 64 | ||||
| Peak | 164 (25–75th percentile: 99–225) | 64 | |||||
| 15 mg od | AF | Trough | 12 (25–75th percentile: 7–21) | 63 | |||
| Peak | No data found | ||||||
| Molecule | Test | Peak54,55 | Dose | Indication | Trough/peak | Plasma level (ng/mL) | Ref. |
|---|---|---|---|---|---|---|---|
| Dabigatran20,56 | dTT | 1.5–3 h | 150 mg bid | AF | Trough | 91 (25–75th percentile: 61–143) | SPC |
| Peak | 175 (25–75th percentile: 117–275) | SPC | |||||
| VTE | Trough | 60 (25–75th percentile: 39–95) | SPC | ||||
| Peak | 175 (25–75th percentile: 117–275) | SPC | |||||
| 110 mg bid | AF | Trough | 63 (5–95th percentile: 62–64) | 34 | |||
| Peak | No data found | ||||||
| VTE | Trough | No data found | |||||
| Peak | No data found | ||||||
| Rivaroxaban19,57,58 | Chromogene anti-FXA assay | 2–3 h | 20 mg od | AF | Trough | 44 (5–95th percentile: 22–137) | 19 |
| Peak | 249 (5–95th percentile: 184–343) | 19 | |||||
| VTE | Trough | 26 (5–95th percentile: 6–87) | 19 | ||||
| Peak | 270 (5–95th percentile: 189–419) | 19 | |||||
| 15 mg od | AF | Trough | 57 (5–95th percentile: 18–136) | 19 | |||
| Peak | 229 (5–95th percentile: 178–313) | 19 | |||||
| 10 mg od | VTE prevention after joint surgery | Trough | 9 (5–95th percentile: 1–38) | 19 | |||
| Peak | 125 (5–95th percentile: 91–196) | 19 | |||||
| 10 mg od | VTE secondary prevention | Trough | 14 (min/max: 4–51) | SPC | |||
| 10 mg od | VTE secondary prevention | Peak | 101 (min/max: 7–273) | SPC | |||
| 2.5 mg bid | Vascular prevention in chronic CAD/PAD | Trough | 17 (5–95th percentile: 6–37) | 19 | |||
| Peak | 46 (5–95th percentile: 28–70) | 19 | |||||
| Apixaban16,17,59–61 | Chromogene anti-FXA assay | 3–4 h | 5 mg bid | AF | Trough | 103 (5–95th percentile: 41–230) | SPC17 |
| Peak | 171 (5–95th percentile: 91–321) | SPC17 | |||||
| 10 mg bid | VTE (initial treatment) | Trough | 120 (5–95th percentile: 41–335) | SPC17 | |||
| Peak | 251 (5–95th percentile: 111–572) | SPC17 | |||||
| 5 mg bid | VTE | Trough | 63 (5–95th percentile: 22–177) | SPC17 | |||
| Peak | 132 (5–95th percentile: 59–302) | SPC17 | |||||
| 2.5 mg bid | AF | Trough | 79 (5–95th percentile: 34–162) | SPC17 | |||
| Peak | 123 (5–95th percentile: 69–221) | SPC17 | |||||
| VTE | Trough | 32 (5–95th percentile: 11–90) | SPC17 | ||||
| Peak | 67 (5–95th percentile: 153) | SPC17 | |||||
| Edoxaban62 | Chromogene anti-FXA assay | 1–2 h | 60 mg od | AF | Trough | 36 (25–75th percentile: 19–62) | 63 |
| Peak | 170 (1.5*IQR125–245) | 27 | |||||
| VTE | Trough | 19 (25–75th percentile: 10–39) | 64 | ||||
| Peak | 234 (25–75th percentile: 149–317) | 64 | |||||
| 30 mg od | AF | Trough | 27 (25–75th percentile: 15–44.6) | 63 | |||
| Peak | 85 (57–115) | 27 | |||||
| VTE | Trough | 16 (25–75th percentile: 8–32) | 64 | ||||
| Peak | 164 (25–75th percentile: 99–225) | 64 | |||||
| 15 mg od | AF | Trough | 12 (25–75th percentile: 7–21) | 63 | |||
| Peak | No data found | ||||||
When are plasma levels useful?
After decades of emphasizing the importance of drug monitoring of VKAs, measuring the effect of anticoagulants has been engrained in clinical practice. Nevertheless, the target-specific NOACs have been developed to provide reliable PK/PD profiles, and the predictable effect of fixed doses without drug monitoring has been validated in clinical trials with thousands of patients.28 For the large majority of patients, use of the approved doses (taking into account clinical factors for dose adjustment as per the product’s label) has been shown to provide a good balance of safety and efficacy, and to offer additional advantages over VKA in terms of clinical outcomes without the need for drug monitoring. Importantly, use of non-tested doses in those patients could cause more harm than benefit.65 Therefore, in most patients, plasma monitoring is not indicated.24
However, in patients with a factor or (in particular) a combination of factors that are expected to strongly influence NOAC absorption, metabolization, or elimination; and where NOAC is still deemed the preferred choice, a spot assessment can help to check whether the plasma levels of a carefully timed sample fall within the expected values. Knowledge of timing and of the correct expected ranges is crucial. For most cases, trough levels are best suited since they are more stable and better represent drug accumulation (to exclude overtreatment), but peak levels can be useful to ensure proper drug absorption (to exclude undertreatment).
The AUCss provides the most accurate information, but this is not feasible in daily clinical practice. It is important to realize that the best approach when levels are beyond the expected values is unknown. This indicates, however, that the information about efficacy and safety of NOACs as studied in the Phase III trials cannot directly be applied to this patient.
In acute situations, measuring plasma levels can help to assess optimal timing of interventions. However, most elective and even urgent invasive procedures do not require the use of plasma levels.
In conclusion, plasma measurements should not be performed in most patients. In rare circumstances, knowing about exposure might be informative. Changing the dose based on measured exposure is not recommended.
Funding
This paper was published as part of a supplement financially supported by Daiichi Sankyo Europe GmbH.
Conflict of interest: TV has received honoraria for participation in advisory boards or as part of a speaker bureau from Bayer, Boehringer Ingelheim, BMS/Pfizer, Daiichi Sankyo, Leo Pharma, Sanofi Aventis.

{kind=link}