





Abstract
Both experimental and clinical findings linking vitamin D to cardiovascular (CV) risk have prompted consideration of its supplementation to improve overall health. Yet several meta-analyses do not provide support for the clinical effectiveness of this strategy. Meanwhile, the understanding of the roles of vitamin D in the pathophysiology of CV diseases has evolved. Specifically, recent work has revealed some non-classical pleiotropic effects of vitamin D, increasing the complexity of vitamin D signalling. Within particular microenvironments (e.g. dysfunctional adipose tissue and atherosclerotic plaque), vitamin D can act locally at cellular level through intracrine/autocrine/paracrine feedforward and feedback circuits. Within atherosclerotic tissues, ‘local’ vitamin D levels may influence relevant systemic consequences independently of its circulating pool. Moreover, vitamin D links closely to other signalling pathways of CV relevance including those driving cellular senescence, ageing, and age-related diseases—among them CV conditions. This review updates knowledge on vitamin D biology aiming to clarify the widening gap between experimental and clinical evidence. It highlights the potential reverse causation confounding correlation between vitamin D status and CV health, and the need to consider novel pathophysiological concepts in the design of future clinical trials that explore the effects of vitamin D on atherosclerosis and risk of CV events.
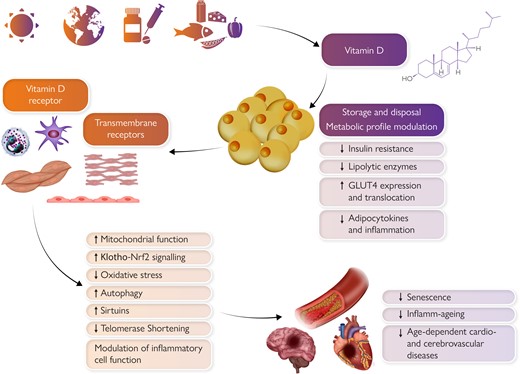
Pleiotropic effects of vitamin D on the cardiovascular system. Sunlight exposure, exogenous dietary, and pharmacological supply primarily determine vitamin D levels. Vitamin D is then stored within the adipose tissue which influences its systemic availability. Pleiotropic vitamin D effects on the cardiovascular system are mediated by intracellular vitamin D receptor or other transmembrane receptors and include beneficial modulation of senescence, inflamm-ageing, and adiposopathy. Clinical translation of such findings still requires more complete preclinical evidence, especially on newly defined mechanisms such as epigenetic senescence, and also better standardization among clinical studies (e.g. definition of deficiency, distinctions between fortified food and pharmacologic supplementation).
Introduction
The role of vitamin D in cardiovascular (CV) health remains an elusive aspect of contemporary cardiology. A plethora of publications over the last decade have prompted a critical reappraisal of the evidence regarding the health benefits of vitamin D supplementation. Epidemiological observations have consistently linked low circulating levels of vitamin D to CV risk, but randomized clinical trials and meta-analyses have not shown a benefit of supplementation.1 Such a dichotomy raises many questions about potential confounders (e.g. limited outdoor physical activity, exogenous vitamin D supplementation, and calcium supplementation regimens).2 Furthermore, enlarging the gamut of CV risk factors beyond the traditional, e.g. including inflammation, might shed light on the role of this vitamin and help to understand its therapeutic potential. Obesity has long been considered a target for vitamin D supplementation,3,4 and qualitative fat abnormalities (i.e. adiposopathy) with different distributions of obesity have gained increasing recognition.5,6 This evolution has prompted a focus on the tissue microenvironment, a concept that may extend from visceral adipose tissue to ectopic fat depots, to the atherosclerotic plaque, the myocardium, and the brain.7 Beyond the recognition of its ‘non-classical’ effects, we now know that circulating concentrations of vitamins may not reflect their activity in tissues. Also, locally produced and stored vitamin D may have systemic functional consequences. This state-of-the-art review summarizes the evolving scientific knowledge regarding local and systemic effects of vitamin D in primary and secondary CV prevention. Furthermore, we propose that categorizing patients according to vitamin D levels might aid risk stratification and allocation of supplementation. Lastly, we provide a critical assessment of CV conditions proposed to derive benefit from vitamin D supplementation.
Vitamin D axis: systemic and local contribution to cardiovascular risk
Vitamin D biology and interaction with other cardiovascular relevant pathways
The substantial controversy surrounding vitamin D results from its complex biology (Figure 1). Sunlight exposure accounts for ∼80% of vitamin D synthesis (i.e. cholecalciferol). Exogenous dietary supply (i.e. cholecalciferol or ergocalciferol) has rather limited relevance under physiological conditions. The degree of endogenous production and exogenous intake depends on several factors. Indeed, the conversion from 7-dehydrocholesterol—the endogenous cholecalciferol precursor—to the active 1,25(OH)2D (i.e. calcitriol) requires multiple steps. The rate-limiting enzyme of this pathway, CYP27B1, is tightly regulated by feedback control mechanisms involving parathyroid hormone, calcitonin, the vitamin D catabolic enzyme CYP24A1, and activated vitamin D itself. Ageing and sequestration within adipose tissue add further layers to the complex vitamin D balance (Table 1). In particular, such sequestration critically determines vitamin D availability which relates to adipose tissue function/dysfunction rather than to its quantity.8 Ergocalciferol and cholecalciferol then require a two-step hydroxylation to become biologically active and reach target organs. Within the bloodstream, vitamin D mainly circulates as 25-hydroxyvitamin D combined with its carrier protein, vitamin D binding protein (VDBP). Under physiological conditions, circulating serum 25-hydroxyvitamin D level ranges between 20 and 50 ng/mL.9
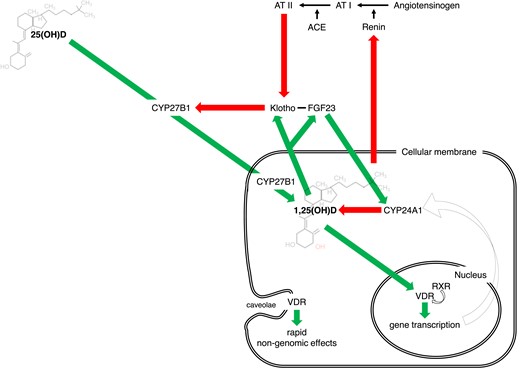
Vitamin D axis is a complex system regulated at several levels and connected with many signals of cardiovascular relevance. Beyond the renal control (under parathyroid hormone), local vitamin D axes involve intracrine/autocrine/paracrine feedforward and feedback circuits. Furthermore, vitamin D system intertwines closely with other pathways of cardiovascular relevance: renin–angiotensin–aldosterone system and Klotho-FGF23.
Determinants of circulating vitamin D levels
 |
|
Determinants of circulating vitamin D levels
|
|
No longer considered a mere vitamin but rather a lipophilic ‘steroid-like’ hormone, vitamin D exerts its biological response through the binding with the vitamin D receptor (VDR). The heterodimeric form of VDR—composed of the active vitamin D (1,25(OH)2D) and the retinoid × receptor (RXR α, β, or γ)—acts as transcription factor targeting specific vitamin D response elements (VDRE) located in the promoter region of target genes. VDR was initially detected in tissues involved in calcium/phosphate balance including bone, kidney, intestine, and parathyroid glands. The evidence of additional vitamin D effects prompted the recognition of VDR expression in cellular components of different organs such as the skin, immune, and CV systems. Within the CV system, VDR is expressed by endothelial cells (ECs), vascular smooth muscle cells (VSMCs), immune cells, and cardiomyocytes.10 Genomic actions of activated VDR then depend on allosteric modulation, VDRE location, and epigenetic modifications in different cell types.11 After the completion of the human genome, RNA sequencing and then microarrays have become the assays of choice for describing the vitamin D-transcriptome. The VDR affects the transcription of up to 5% of the whole human genome: some 20 000 target genes. However, primary target genes should be discriminated from delayed-reacting ‘secondary’ targets for which vitamin D encodes transcription factors, co-factors, and/or chromatin modifiers.12 Thus, the number of VDR target genes in a single cell type might be restricted to a few hundreds. Common target genes include those involved in calcium/phosphate homoeostasis (e.g. osteocalcin) and inflammation (e.g. cathelicidin antimicrobial peptide). Rapid non-genomic VDR effects depend on membrane receptors such as PDIA3 (protein disulphide isomerase family A member 3). PDIA3 is an endoplasmic reticulum chaperone located in cell membrane, cytoplasm, mitochondria, and nucleus that requires the interaction with caveolin 1 to trigger the rapid response to vitamin D. PDIA3 but not VDR is essential for the activation of protein kinase C-phospholipase A2 cascade and MAPK and WNT/NOTCH pathways. Disrupting the PDIA3 gene results in bone abnormalities and loss of protection against UV-induced DNA damage.13
Far from operating in isolation, vitamin D intertwines closely with other pathways pivotal in the CV system. Several experimental studies confirmed the inhibitory role of vitamin D on the renin–angiotensin–aldosterone system (RAAS) with effects on blood pressure and cardiac hypertrophy in mice.14,15 In turn, RAAS activation suppresses the renal expression of Klotho, essential for effective FGF23 signalling. These interactions comprise a complex feedback loop in which high FGF23 levels suppress vitamin D signal—by targeting both CYP27B1 and CYP24A1—eventually leading to RAAS activation16 (Figure 1).
Role of vitamin D in cellular senescence and ageing
New intriguing hypotheses link vitamin D deficiency—in conjunction with autophagy and Klotho-Nrf2 signalling—with cellular senescence, ageing, and age-related diseases17 (Figure 2). Cellular senescence develops as a cumulative effect of cell stressors eventually leading to a permanent cell growth arrest and the development of the senescence-associated secretory pattern (SASP). This programme contributes importantly to ‘inflamm-ageing’ the chronic low-grade inflammatory status that develops with ageing and participates in the pathophysiology of most CV diseases.18,19 Vitamin D can preserve mitochondrial function and limit oxidative stress by facilitating mitochondrial biogenesis, the electron transport chain, and autophagy.20,21 Specifically, vitamin D activates pro-survival autophagy as a protective mechanism to inhibit oxidative stress and apoptosis with critical effects on bone and CV health. Through autophagy, vitamin D mediates osteoclastogenesis and thus the risk of osteoporosis.21 Within the vessel wall, vitamin D restores oxidized low-density lipoprotein-impaired autophagy22 and prevents endothelial dysfunction.23 Furthermore, vitamin D-mediated autophagy also exerts beneficial effects on failing/ageing heart.24,25 With ageing, autophagy and vitamin D share the same trajectory, being characterized by inactivation and deficiency, respectively. Vitamin D deficiency may also accelerate ageing and ageing-related disease. Vitamin D may influence the ageing determinants NAD+-dependent protein acetylases sirtuins, calcium signalling homoeostasis, mitochondrial function, and oxidative stress regulation.17 By augmenting Klotho-Nrf2 signalling, vitamin D can limit oxidative stress by preventing the rise in intracellular Ca++. At the nuclear level, vitamin D reduces the rate of telomere shortening26 and controls the epigenetic landscape of its promoters by stimulating key DNA demethylases that alter epigenetic aspects of ageing.27 This effect likely involves the hypermethylation of Klotho’s promoter region causing a reduced level of this mediator and the progression of age-related phenomena.28 In addition to those mechanisms, vitamin D directly modulates inflammatory cell activation again blunting inflamm-ageing.29
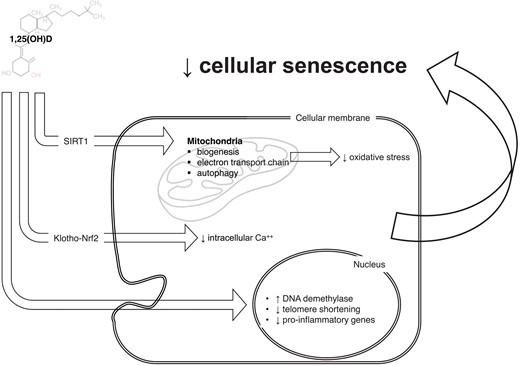
Vitamin D system might participate in senescence-ageing processes. Through a wide range of cellular and nuclear mechanisms, vitamin D can modulate many aspects of cellular senescence: mitochondrial survival and activity, intracellular calcium concentrations, and direct/epigenetic control on gene transcription. Those mechanisms may further explain the tight pathophysiological link between vitamin D axis and age-related diseases, including cardiovascular diseases.
The wide anti-inflammatory effects of vitamin D arise locally through intracrine, autocrine, and paracrine feedforward and feedback circuits.30 The stimulation of toll-like receptors (TLRs) amplifies CYP27B1 and VDR expression in macrophages in an autocrine manner, whereas interferon-γ produced by the Th1 lymphocyte subset determines the same effect through a paracrine pathway. In turn, when macrophages release 1,25(OH)2D, it drives VDR-dependent inhibitory feedback on lymphocyte activation.31 Such findings underscore the importance of 25(OH)D in macrophages, as it exerts direct effects on both innate and adaptive immunity. The co-existent expression of CYP27B1 and VDR characterizes many cell types and indicates the intracrine metabolism of vitamin D as a potential candidate to modulate their effects in different diseases.32 However, this plethora of experimental evidence about senolytic effect of vitamin D requires clinical translation in randomized trials.33
Vitamin D insufficiency/deficiency: definition and dispute
Defining reference ranges for vitamin D has long presented challenges. The preferential use of blood 25(OH)D levels reflects its relative abundance (1000-fold higher than 1,25(OH)2D) and stability (half-life: 15 days vs. 15 h for 1,25(OH)2D). 25(OH)D circulates both in its free form and bound to carrier proteins, mainly VDBP. Specifically, VDBP affinity is greater for the inactivated [24,25(OH)2D] and the depot [25(OH)D] forms of vitamin D rather than the active 1,25(OH)2D. VDBP deletion in mice does not lead to skeletal abnormalities or calcium imbalance so that a direct pathophysiological role for VDBP is unlikely.32 On the other hand, the measurement of the free, unbound form of 25(OH)D has been proved challenging, because of its low levels and the need for radioactive tracers. Yet, enzyme-linked immunosorbent assays for detecting free 25(OH)D are currently under development. Beyond analytical variability, 25(OH)D concentrations show seasonal variations due to their mechanisms of synthesis with higher levels during sunny seasons. Given such complexity, proposed vitamin D range calculators take into account different confounders, although their use still needs further validation.34
As a result, the thresholds defining vitamin D deficiency significantly differ across health organizations ranging from serum levels of 30–10 ng/mL. However, there is general agreement with those proposed by the former US Institute of Medicine now known as the Academy of Medicine9 (Table 2). The US population has a substantial prevalence of vitamin D insufficiency (41.4%) or deficiency (28.9%).38 These estimates increase substantially with the application of more stringent thresholds.39,40 Eastern European countries report lower levels of vitamin D than northern ones.37 Vitamin D status is also suboptimal in Western Europe, and especially in the UK. Finally, non-Western immigrants in European countries showed a very poor vitamin D status, with a mean 25(OH)D levels lower than locally born and people living in their countries of origin.37
Proposed thresholds for vitamin D deficiency definition
| 25(OH)D | Institute of Medicine | Endocrine Society | EFSA, 201635 | SACN, 201636 | ECTS, 201937 | US prevalencea | European prevalenceb |
|---|---|---|---|---|---|---|---|
| Normal values | ≥20 ng/mL | ≥30 ng/mL | ≥20 ng/mL | NA | ≥ 20 ng/mL | ||
| Insufficiency | 12–20 ng/mL | 20–29 ng/mL | – | – | 20–10 ng/mL | 41.4% |
|
| Deficiency | <12 ng/mL | <20 ng/mL | <20 ng/mL | <10 ng/mL | <10 ng/mL | 28.9% |
| 25(OH)D | Institute of Medicine | Endocrine Society | EFSA, 201635 | SACN, 201636 | ECTS, 201937 | US prevalencea | European prevalenceb |
|---|---|---|---|---|---|---|---|
| Normal values | ≥20 ng/mL | ≥30 ng/mL | ≥20 ng/mL | NA | ≥ 20 ng/mL | ||
| Insufficiency | 12–20 ng/mL | 20–29 ng/mL | – | – | 20–10 ng/mL | 41.4% |
|
| Deficiency | <12 ng/mL | <20 ng/mL | <20 ng/mL | <10 ng/mL | <10 ng/mL | 28.9% |
EFSA, European Food Safety Authority; SACN, Scientific Advisory Committee on Nutrition; ECTS, European Calcified Tissue Society.
Prevalence refers to the thresholds of the Institute of Medicine.
Prevalence of vitamin D deficiency refers to the threshold of the Endocrine Society.
Proposed thresholds for vitamin D deficiency definition
| 25(OH)D | Institute of Medicine | Endocrine Society | EFSA, 201635 | SACN, 201636 | ECTS, 201937 | US prevalencea | European prevalenceb |
|---|---|---|---|---|---|---|---|
| Normal values | ≥20 ng/mL | ≥30 ng/mL | ≥20 ng/mL | NA | ≥ 20 ng/mL | ||
| Insufficiency | 12–20 ng/mL | 20–29 ng/mL | – | – | 20–10 ng/mL | 41.4% |
|
| Deficiency | <12 ng/mL | <20 ng/mL | <20 ng/mL | <10 ng/mL | <10 ng/mL | 28.9% |
| 25(OH)D | Institute of Medicine | Endocrine Society | EFSA, 201635 | SACN, 201636 | ECTS, 201937 | US prevalencea | European prevalenceb |
|---|---|---|---|---|---|---|---|
| Normal values | ≥20 ng/mL | ≥30 ng/mL | ≥20 ng/mL | NA | ≥ 20 ng/mL | ||
| Insufficiency | 12–20 ng/mL | 20–29 ng/mL | – | – | 20–10 ng/mL | 41.4% |
|
| Deficiency | <12 ng/mL | <20 ng/mL | <20 ng/mL | <10 ng/mL | <10 ng/mL | 28.9% |
EFSA, European Food Safety Authority; SACN, Scientific Advisory Committee on Nutrition; ECTS, European Calcified Tissue Society.
Prevalence refers to the thresholds of the Institute of Medicine.
Prevalence of vitamin D deficiency refers to the threshold of the Endocrine Society.
Vitamin D and cardio-metabolic risk
Vitamin D activity on adipose tissue microenvironment
Visceral adipose tissue stores about three-quarters of the total cholecalciferol and one-third of 25(OH)D. But vitamin D depots within the adipose tissue depend little on circulating levels. Indeed, adipocytes express all key enzymes required for vitamin D hydroxylation. Within the adipose tissue, vitamin D signals in paracrine and autocrine manners. In its active form, 1,25(OH)2D improves glut4 expression and translocation, suppresses fatty acid synthesis from glucose, augments lipolytic enzymes, and inhibits adipokines and inflammatory signalling41 (Figure 3). Nevertheless, VDR knockout mice are lean and resist diet-induced obesity. Even adipose-specific VDR deletion did not resolve this apparent paradox.42 Less is known about the role of vitamin D in adipocyte hypertrophy, the hallmark of adiposopathy and associated dysfunction. Hypertrophic adipocytes show necrotic-like abnormalities as the vascular tree becomes deficient and hypoxia ensues. Lipolysis and insulin resistance characterize this microenvironment with effects on other tissues (e.g. liver and skeletal muscle) through free fatty acids and pro-inflammatory mediators released in the portal vein.43 More recent studies have further characterized the hypertrophic adipocyte as a senescent cell showing the typical pro-inflammatory SASP44 (Figure 3). As mentioned above, the vitamin D axis can influence several pathways involved in adipocyte senescence: mitochondrial respiration,45 autophagy,46 intracellular Ca++ concentration,47 and epigenetic clocks (i.e. methylation/acetylation of both DNA and histone and the expression of micro-RNAs).48,49 Vitamin D generally exerts anti-inflammatory effects on adipocytes. 1,25(OH)2D indeed prevents and even reverses monocyte chemotaxis.50 Nevertheless, dysfunctional hypertrophic adipocytes have blunting of the entire vitamin D axis and an associated feedback transcriptional response characterized by augmentation of CYP27A1 and VDR, and suppression of CYP27B1.51,52 Microarray approaches are deciphering the target cytokines and the role of miRNAs as components of adipocyte SASP.53
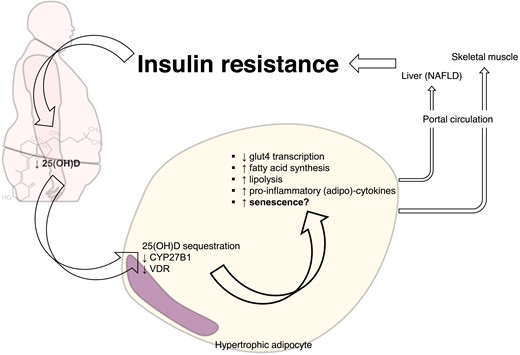
Dysfunctional hypertrophic adipocytes show blunted vitamin D signalling. Vitamin D receptor and the hydroxylase CYP27B1 are blunted in dysfunctional hypertrophic adipocyte. Alongside the sequestration of 25(OH)D within visceral adipose tissue, suppression of vitamin D signalling sustains metabolic disturbances not only in adipose tissue but also systemically.
Vitamin D deficiency and adiposopathy: clinical evidence
Visceral adiposity links consistently with vitamin D deficiency/insufficiency, as originally reported by the Framingham Heart Study.54 Furthermore, recent studies also showed an association with the unhealthy obesity pattern.55 A tight interplay exists between vitamin D axis and visceral adiposity. Not only do concentrations of 25(OH)D critically determine the VDR gene expression in adipocytes, but sun exposure and skin reflectance also associate with visceral adiposity.56,57 25(OH)D levels increase linearly as a function of visceral adiposity reduction during lifestyle modification programmes, with dynamic local changes in vitamin D metabolism.58–60 This effect is even more evident with massive weight loss achieved through a very low-calorie ketogenic diet61 or bariatric surgery.62 The expression of CYP27B1 indeed rises after weight loss and decreases during overweight along with vitamin D sequestration.63 As a result, obese subjects have a muted response to vitamin D supplementation.64 Further supporting a causative role in adiposopathy, vitamin D deficiency is associated with ectopic fat deposition. Few studies have so far focused on the association between vitamin D and epicardial adipose tissue (EAT), reporting an association with both thickness and dysfunction while resisting regression after replacement therapy.65,66 Similarly, preliminary data indicate an association between vitamin D deficiency, fatty degeneration of skeletal muscle,67 and the osteosarcopenic obese pattern.68 Although limited to association studies such evidence deserves further investigation. Recent evidence suggests a role for EAT in inflammation by hosting tertiary lymphoid organs (TLOs). In consideration of the role of TLOs as the regulator of B cell response with relevant effect on atherogenesis and recovery after myocardial infarction69–71 and in keeping with the immunomodulatory function of vitamin D, whether CV effects of such mediators may depend in part on local immune responses within ectopic fat is an intriguing question for future investigations.
Vitamin D deficiency and cardio-metabolic profile: clinical evidence
Extensive literature supports the association between vitamin D deficiency and non-alcoholic fatty liver disease (NAFLD). In this context, vitamin D may blunt the activation of hepatic stellate cells and have a broad immunomodulatory role for resident immune cells.72 Of importance, vitamin D deficiency might promote NAFLD progression independently of other risk factors (e.g. hypertriglyceridaemia and insulin resistance). In patients, analyses of polymorphisms in genes from the vitamin D axis further strengthened this association, whereas recent studies are deepening the effects on liver senescence mechanisms.73–75 Low serum 25(OH)D levels indeed correlate with the severity of steatosis and necro-inflammatory injury. This histopathological pattern is consistent with the suppression of TLRs by vitamin D and the increase of VDR expression in non-parenchymal liver cells including hepatic stellate cells, Kupffer cells, natural killer T cells, and sinusoidal ECs. Furthermore, vitamin D may protect against a high-fat diet by inducing autophagy. Yet, the causality of vitamin D deficiency in NAFLD is hard to prove. Indeed, the lack of vitamin D can be a consequence of its lower synthesis by the dysfunctional liver. This may explain why clinical trials of vitamin D supplementation generally reported a metabolic improvement without a significant effect on liver injury.76–81 Similarly, the notion that insulin resistance itself may account for vitamin D deficiency—rather than being its consequence—is gaining attention. Insulin resistance can suppress the expression of CYP2R1 in the liver and CYP27B1 in the kidney.82 Although vitamin D deficiency and insulin resistance can both be treated, beneficial effects derive only from increased insulin sensitivity. Whether improving insulin sensitivity may increase vitamin D levels and/or the effectiveness of supplementation strategies in CV prevention remains unresolved. Epidemiological data indicate that chronic deficit of vitamin D parallels with the clinical manifestations of insulin resistance. Vitamin D deficiency very frequently accompanies diabetes and associates with a higher risk of developing diabetes.83 The most recent meta-analysis and Mendelian randomization studies have evaluated more deeply the role of vitamin D status in the development and progression of diabetes. A significant, yet non-causal, association links the risk of developing diabetes with vitamin D deficiency in ∼300 000 participants.84 Yet, the same meta-analysis indicates adiposopathy and unhealthy lifestyles as risk factors for diabetes, confounding conclusions regarding the causality of vitamin D levels. Furthermore, clinical studies and meta-analyses agree on the close link of vitamin D deficiency with hypertension and dyslipidaemia as reviewed elsewhere.85,86 The pooled relative risk of incident hypertension in more than 300 000 participants has been estimated at 0.88 (0.81–0.97) per 10 ng/mL increment in 25(OH)D levels,87 whereas in 25 000 children and adolescents vitamin D associated with a favourable lipid profile.88 Yet, the effects of vitamin D supplementation on blood pressure and serum lipids remain unproven.89,90
The ‘intraplaque’ vitamin D system
Vitamin D activity in the atherosclerotic plaque microenvironment
Intimal inflammation drives many aspects of atherogenesis.91 The atherosclerotic plaque microenvironment represents the prototypical milieu in which the vitamin D axis locally exerts its ‘non-classical’ effects via paracrine and autocrine processes.92 The first recognition of VDR expression within the plaque dates to 2012.93 Our research group first localized VDR in human samples and observed how its expression was proportional to the infiltrate of pro-inflammatory M1 macrophages and the likelihood of future adverse CV events.94 As exposure to 1,25(OH)2D limited the expression of TLR-4 on M1 macrophages in vitro, we hypothesized that VDR expression in pro-inflammatory macrophages constituted a negative feedback loop. Intraplaque macrophages participate critically in the local intraplaque vitamin D system: they express all the enzymes for its synthesis as well as VDR thereby potentially regulating the axis in an intracrine, autocrine, and paracrine manner. The genome/epigenome of human monocytes shows regulation by vitamin D at multiple levels.95 The result is an extensive control on macrophage function, not limited to phenotype polarization and anti-inflammatory activity.30,96
Vitamin D prevents the formation of foam cells by intraplaque macrophages by inhibiting endoplasmic reticulum stress, the expression of scavenger receptors,97 and autophagy, triggered via the PTPN6/SHP-1 pathway.22 As with monocytes, the vitamin D axis may exert autocrine feedback on neutrophils as these granulocytes themselves express CYP27B1 and VDR. This point still lacks experimental demonstration.
Dendritic cells (DCs) provide another source of 1,25(OH)2D. Transplantation of apoptosis-resistant DCs into LDLR−/− recipient mice indeed slows atherosclerotic progression.98 This finding likely arises from an autocrine loop that results in DC regression to a less mature stage characterized by impaired chemotaxis and suppression of pro-inflammatory cytokines and costimulatory molecules (i.e. CD40, CD80, and CD86).99 T lymphocytes may have a further role, especially the regulatory (Treg) subtype, as the VDR direct controls their signature transcription factor FOXP3 under.100 Beyond the immune system, the pleiotropic activity of vitamin D involves different cell types of relevance in atherosclerosis. 1,25(OH)2D influences VSMCs directly via VDR. 1,25(OH)2D stimulates the expression of vascular endothelial growth factor by VSMC and regulates their proliferation, migration, calcification, and tissue factor expression. Furthermore, vitamin D signalling could prevent senescence in VSMCs, likely through the inhibition of local angiotensin-II signalling.101
Vitamin D and atherosclerotic burden: clinical evidence
Experiments with LDLR- and/or VDR-deficient mice demonstrated beneficial effects of vitamin D signalling on atherosclerosis progression. Likewise, clinical studies generally agree on the relationship between 25(OH)D and subclinical atherosclerosis. A recent pooled analysis of 21 studies matching ∼2000 vitamin D deficiency/insufficiency patients with controls reported a significant difference in carotid intima-media thickness and prevalence of carotid plaques.102 Two subsequent meta-analyses confirmed these findings and support the beneficial effects of vitamin D supplementation on this biomarker.103,104 The association between vitamin D levels and coronary atherosclerotic burden or composition remains less clear. A single systematic review dating 2014 did not find consistent evidence for the association with coronary artery calcification (CAC) and highlighted the relevant differences across studies.105 The heterogeneity in the definition of vitamin D deficiency/insufficiency and CAC criteria precluded a quantitative meta-analysis. Later studies did not solve this issue even when considering other biomarkers of plaque vulnerability106–113 (Table 3). Polymorphisms in the VDR gene were suggested as determinants of such a variability.114–116 A recent Mendelian randomization analysis supported a causal role for vitamin D deficiency as well as the beneficial effects of its supplementation on CV health. Of interest, this analysis found a non-linear L-shaped relationship, a feature formerly overlooked in randomized clinical trials and Mendelian randomization studies.117
Summary of the main studies addressing the association between vitamin D deficiency and coronary artery calcium score and biomarkers of atherosclerotic plaque vulnerability
| Authors | Year | Study design (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Martín-Reyes et al.106 | 2016 | Cross-sectional (270) | Categorized CAC |  |
| Sung et al.107 | 2016 | Cross-sectional (180,918) | CAC | |
| Lee et al.108 | 2016 | Cross-sectional (195) | ||
| Diederichsen et al.109 | 2017 | Prospective (1227) | Incident CAC CAC progression | |
| Sajjadieh et al.111 | 2020 | Cross-sectional (67) | CAC | There was no correlation between CAC and vitamin D levels (r = −0.03) |
| Anis et al.112 | 2020 | RCT (48w FU) (44) | CAC | CAC progression did not differ between treatment arms (calcitriol vs. paricalcitol; P = 0.76) |
| Rodrigues et al.113 | 2021 | Cross-sectional (140) | CAC | Excess visceral fat (OR 4.0 [1.4–11.7]) but not vitamin D is associated with subclinical atherosclerosis |
| Authors | Year | Study design (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Martín-Reyes et al.106 | 2016 | Cross-sectional (270) | Categorized CAC | |
| Sung et al.107 | 2016 | Cross-sectional (180,918) | CAC | |
| Lee et al.108 | 2016 | Cross-sectional (195) | ||
| Diederichsen et al.109 | 2017 | Prospective (1227) | Incident CAC CAC progression | |
| Sajjadieh et al.111 | 2020 | Cross-sectional (67) | CAC | There was no correlation between CAC and vitamin D levels (r = −0.03) |
| Anis et al.112 | 2020 | RCT (48w FU) (44) | CAC | CAC progression did not differ between treatment arms (calcitriol vs. paricalcitol; P = 0.76) |
| Rodrigues et al.113 | 2021 | Cross-sectional (140) | CAC | Excess visceral fat (OR 4.0 [1.4–11.7]) but not vitamin D is associated with subclinical atherosclerosis |
CAC, coronary artery calcification; OR, odds ratio; CI, confidence Interval; FU, follow-up.
Summary of the main studies addressing the association between vitamin D deficiency and coronary artery calcium score and biomarkers of atherosclerotic plaque vulnerability
| Authors | Year | Study design (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Martín-Reyes et al.106 | 2016 | Cross-sectional (270) | Categorized CAC | |
| Sung et al.107 | 2016 | Cross-sectional (180,918) | CAC | |
| Lee et al.108 | 2016 | Cross-sectional (195) | ||
| Diederichsen et al.109 | 2017 | Prospective (1227) | Incident CAC CAC progression | |
| Sajjadieh et al.111 | 2020 | Cross-sectional (67) | CAC | There was no correlation between CAC and vitamin D levels (r = −0.03) |
| Anis et al.112 | 2020 | RCT (48w FU) (44) | CAC | CAC progression did not differ between treatment arms (calcitriol vs. paricalcitol; P = 0.76) |
| Rodrigues et al.113 | 2021 | Cross-sectional (140) | CAC | Excess visceral fat (OR 4.0 [1.4–11.7]) but not vitamin D is associated with subclinical atherosclerosis |
| Authors | Year | Study design (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Martín-Reyes et al.106 | 2016 | Cross-sectional (270) | Categorized CAC | |
| Sung et al.107 | 2016 | Cross-sectional (180,918) | CAC | |
| Lee et al.108 | 2016 | Cross-sectional (195) | ||
| Diederichsen et al.109 | 2017 | Prospective (1227) | Incident CAC CAC progression | |
| Sajjadieh et al.111 | 2020 | Cross-sectional (67) | CAC | There was no correlation between CAC and vitamin D levels (r = −0.03) |
| Anis et al.112 | 2020 | RCT (48w FU) (44) | CAC | CAC progression did not differ between treatment arms (calcitriol vs. paricalcitol; P = 0.76) |
| Rodrigues et al.113 | 2021 | Cross-sectional (140) | CAC | Excess visceral fat (OR 4.0 [1.4–11.7]) but not vitamin D is associated with subclinical atherosclerosis |
CAC, coronary artery calcification; OR, odds ratio; CI, confidence Interval; FU, follow-up.
From vulnerable plaque to vulnerable patients: the role of vitamin D on blood and myocardium/brain vulnerability
Vitamin D deficiency and thrombogenicity
The evolution of an atherosclerotic plaque to produce a clinical thrombotic event requires a ‘perfect storm’ in which a pro-thrombotic milieu in the fluid phase of blood meets the ‘solid state’ of a culprit plaque91,118,119 (Figure 3). Endothelial functions contribute to the crosstalk between the atheroma and circulation and influence critically the vulnerability of the plaque and that of the patient. ECs express VDR constitutively, whereas in stress conditions (e.g. disturbed flow, pro-oxidant environment, hyperglycaemia) CYP27B1 may induce this receptor. Impaired vitamin D signalling in ECs enhances the interaction of leucocytes with the endothelium (i.e. rolling, adhesion, and migration).120 Furthermore, VDR silencing/vitamin D supplementation revealed the multifaceted actions of the vitamin D axis in ECs that involve: apoptosis, oxidative stress and nitric oxide generation, lipid metabolism, and interaction with platelets.121,122 The effect on platelets depends in part on the reduction of adhesion molecules on the EC surface and EC-derived microparticles and with integrin activation on the platelet surface. These broad effects of vitamin D on endothelial function can influence arterial stiffness (assessed by brachial artery flow-mediated dilation or by pulse wave velocity), as is common in patients with vitamin D deficiency. Conversely, the effects of vitamin D supplementation on markers of vascular function remain controversial, often showing only slight or insignificant alterations.123,124
Along with the aforementioned factors (i.e. chronic low-grade inflammatory status, chemokines/adhesion molecules, and EC dysfunction), the pro-thrombotic milieu depends on tissue factor expression, coagulation factors, and platelet activation. As these features do not yet clearly associate with vitamin D biology, this topic has become an intriguing research problem.125 The anti-thrombogenic effect of vitamin D was first documented in venous thromboembolism, whereas studies on VDR-deleted mice found increased platelet aggregation and imbalance between antithrombin/thrombomodulin and tissue factor expression.126–128 Accordingly, preliminary clinical studies reported an inverse association between vitamin D deficiency and mean platelet volume, a biomarker of platelet reactivity.129,130 A recent randomized clinical trial testing a weekly dose of 60 000 IU of 25(OH)D in diabetic patients reported an anti-inflammatory inhibitory effect on platelet activation and immune cell aggregate in the intervention arm.131
Vitamin D and its effects on myocardium and brain vulnerability and remodelling
Reperfusion strategies now comprise the cornerstone of the management of many acute ischaemic complications of atherosclerosis (i.e. ST-segment elevation myocardial infarction and many ischaemic strokes). Yet, the benefit of reperfusion comes at the cost of massive inflammatory and oxidative responses, potentially exacerbating damage to the hypoxic tissues, as well as bleeding risks following thrombolytic therapy or anti-platelet treatment in those undergoing percutaneous intervention. Optimal healing requires a timely orchestration of multiple processes.132,133 Cytokines and growth factors dynamically modulate resolution and repair, as well as local angiogenic responses. Also, the no-reflow phenomenon can impair the microcirculation in ischaemically injured tissues. Vitamin D deficiency may associate indirectly with impaired microvascular function as highlighted by the association of low coronary flow reserve.134,135
Moreover, given its immunomodulatory properties and the widespread expression of VDR in myocardial tissue, vitamin D may influence post-infarction cardiac remodelling. VDR deletion causes myocardial hypertrophy independently of RAAS activation and calcium abnormalities, while vitamin D supplementation may reverse cardiac steatosis and interstitial fibrosis.15,136,137 Although few experimental studies specifically focused on post-ischaemic injury, some suggest a beneficial effect of vitamin D on left ventricular remodelling.137–139 Animal experiments have supported population-based studies140,141 and the EVITA trial seemed to exclude a link between vitamin D and RAAS.142,143 The VINDICATE trial suggested a beneficial effect of vitamin D supplementation on the left ventricular structure and function144 but meta-analyses generally failed to show significant benefits145–149 (Table 4). Mechanistic studies on vitamin D in myocardial remodelling are lacking. It is, therefore, difficult to establish whether any clinical association reflects causality.152
Summary of randomized clinical trials investigating the effects of vitamin D supplementation on myocardial infarction
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Zittermann et al. EVITA trial150 | 2017 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
|  |
| Zittermann et al. EVITA trial143 | 2020 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| |
| Zittermann et al. EVITA trial142 | 2018 | Cholecalciferol 4000 IU/die i.m for 3 years (83 HF patients and control group) |
| Vitamin D did not affect RAAS system: renin (r = 0.157) or aldosteronee (r= 0.048) |
| Zittermann et al. EVITA trial151 | 2019 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| Vitamin D did not improve myocardial function |
| Witte et al. VINDICATE study144 | 2016 | Cholecalciferol 4000 IU/die i.m. (n = 80 HF patients and control group) |
|  |
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Zittermann et al. EVITA trial150 | 2017 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| |
| Zittermann et al. EVITA trial143 | 2020 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| |
| Zittermann et al. EVITA trial142 | 2018 | Cholecalciferol 4000 IU/die i.m for 3 years (83 HF patients and control group) |
| Vitamin D did not affect RAAS system: renin (r = 0.157) or aldosteronee (r= 0.048) |
| Zittermann et al. EVITA trial151 | 2019 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| Vitamin D did not improve myocardial function |
| Witte et al. VINDICATE study144 | 2016 | Cholecalciferol 4000 IU/die i.m. (n = 80 HF patients and control group) |
| |
CI, confidence interval; OR, odds ratio; HF, heart failure; MCS, mechanical circulatory support; RAAS, renin–angiotensin–aldosterone system; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; LVEF, left ventricular ejection fraction; 6MWT, 6-min walking test.
Summary of randomized clinical trials investigating the effects of vitamin D supplementation on myocardial infarction
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Zittermann et al. EVITA trial150 | 2017 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| |
| Zittermann et al. EVITA trial143 | 2020 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| |
| Zittermann et al. EVITA trial142 | 2018 | Cholecalciferol 4000 IU/die i.m for 3 years (83 HF patients and control group) |
| Vitamin D did not affect RAAS system: renin (r = 0.157) or aldosteronee (r= 0.048) |
| Zittermann et al. EVITA trial151 | 2019 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| Vitamin D did not improve myocardial function |
| Witte et al. VINDICATE study144 | 2016 | Cholecalciferol 4000 IU/die i.m. (n = 80 HF patients and control group) |
| |
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Zittermann et al. EVITA trial150 | 2017 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| |
| Zittermann et al. EVITA trial143 | 2020 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| |
| Zittermann et al. EVITA trial142 | 2018 | Cholecalciferol 4000 IU/die i.m for 3 years (83 HF patients and control group) |
| Vitamin D did not affect RAAS system: renin (r = 0.157) or aldosteronee (r= 0.048) |
| Zittermann et al. EVITA trial151 | 2019 | Cholecalciferol 4000 IU/die i.m for 3 years (200 HF patients and control group) |
| Vitamin D did not improve myocardial function |
| Witte et al. VINDICATE study144 | 2016 | Cholecalciferol 4000 IU/die i.m. (n = 80 HF patients and control group) |
| |
CI, confidence interval; OR, odds ratio; HF, heart failure; MCS, mechanical circulatory support; RAAS, renin–angiotensin–aldosterone system; LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; LVEF, left ventricular ejection fraction; 6MWT, 6-min walking test.
In distinction with the myocardium, the blood–brain barrier (BBB) tightly regulates the accessibility of immune cells to the parenchyma. Protection of BBB integrity153,154 and antioxidant activity155,156 are key effects of vitamin D in the early phases of ischaemic brain injury. Vitamin D might also contribute to late neuroprotection by preventing neuronal apoptosis157 and promoting angiogenesis.158 Experimental evidence agrees with clinical studies in which vitamin D levels correlate with the radiological estimation of ischaemic volume159–161 and with functional outcome.162 Yet, few studies have so far investigated the effects of vitamin D supplementation on stroke outcomes. Such studies have yielded inconsistent results, although they differed greatly in design and outcomes assessed163–168 (Table 5).
Summary of randomized clinical trials investigating the effects of vitamin D supplementation on stroke outcome
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Narasimhan et al.163 | 2017 | Cholecalciferol 6 lac IU i.m. (n = 30 per group) |
| Stroke outcome significantly improved in vitamin D deficient—but not insufficient—patients once supplemented (mean 6.39 ± 4.56 vs. 2.50 ± 2.20; P < 0.001) |
| Sari et al.164 | 2018 | Cholecalciferol 300 000 IU i.m. (n = 36 per group) |
| 3.09 ± 0.99 vs. 3.34 ± 5.28; P = 0.794 2.34 ± 1.31 vs. 1.60 ± 1.16; P = 0.018 59.18 ± 22.15 vs. 41.87 ± 20.42; P = 0.002 38.72 ± 14.14 vs. 24.75 ± 14.76; P < 0.001 |
| Momosaki et al.165 | 2019 | Cholecalciferol 2 000 IU/die i.m. (n = 49/48) |
| 19 ± 15 vs. 20 ± 13; P = 0.480 27%–20% vs. 25%–18%; P = 0.770;0.740 19%–20% vs. 23%–20%; P = 0.770;0.600 |
| Rist et al.166from the VITAL study | 2021 | Cholecalciferol 2 000 IU/die i.m. in primary prevention (n = 104/93 per group) | Nagi scale Rosow-Breslau scale Katz AD scale | OR 1.01 [95% CI 0.52–1.97] OR 0.92 [95% CI 0.59–1.67] OR 1.03 [95% CI 0.31–3.42] |
| Torrisi et al.167 | 2021 | Cholecalciferol 2 000 IU/die i.m. (n = 15/14) per group) | MADRS FIM GSE (all at 12 weeks) | Both groups improved mood and functional recovery independently of vitamin D supplementation |
| Wang et al.168 | 2021 | Cholecalciferol 600 IU/die i.m. (n = 72/51 per group) | mRS FSS (both at 3 months) | FSS and mRS significanlty lowered in the supplementation group (P < 0.01 for both) |
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Narasimhan et al.163 | 2017 | Cholecalciferol 6 lac IU i.m. (n = 30 per group) |
| Stroke outcome significantly improved in vitamin D deficient—but not insufficient—patients once supplemented (mean 6.39 ± 4.56 vs. 2.50 ± 2.20; P < 0.001) |
| Sari et al.164 | 2018 | Cholecalciferol 300 000 IU i.m. (n = 36 per group) |
| 3.09 ± 0.99 vs. 3.34 ± 5.28; P = 0.794 2.34 ± 1.31 vs. 1.60 ± 1.16; P = 0.018 59.18 ± 22.15 vs. 41.87 ± 20.42; P = 0.002 38.72 ± 14.14 vs. 24.75 ± 14.76; P < 0.001 |
| Momosaki et al.165 | 2019 | Cholecalciferol 2 000 IU/die i.m. (n = 49/48) |
| 19 ± 15 vs. 20 ± 13; P = 0.480 27%–20% vs. 25%–18%; P = 0.770;0.740 19%–20% vs. 23%–20%; P = 0.770;0.600 |
| Rist et al.166from the VITAL study | 2021 | Cholecalciferol 2 000 IU/die i.m. in primary prevention (n = 104/93 per group) | Nagi scale Rosow-Breslau scale Katz AD scale | OR 1.01 [95% CI 0.52–1.97] OR 0.92 [95% CI 0.59–1.67] OR 1.03 [95% CI 0.31–3.42] |
| Torrisi et al.167 | 2021 | Cholecalciferol 2 000 IU/die i.m. (n = 15/14) per group) | MADRS FIM GSE (all at 12 weeks) | Both groups improved mood and functional recovery independently of vitamin D supplementation |
| Wang et al.168 | 2021 | Cholecalciferol 600 IU/die i.m. (n = 72/51 per group) | mRS FSS (both at 3 months) | FSS and mRS significanlty lowered in the supplementation group (P < 0.01 for both) |
CI, confidence interval; OR, odds ratio; SSS, Scandinavian Stroke Scale; BRS, Brunnstrom recovery staging; FAS, functional ambulation scale; MBI, modified Barthel index; BBS, Berg balance scale; MADRS, Montgomery Aasberg depression rating scale; FIM, functional independent measures; GSE, general self-efficacy; mRS, modified Rankin score; FSS, fatigue severity scale.
Summary of randomized clinical trials investigating the effects of vitamin D supplementation on stroke outcome
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Narasimhan et al.163 | 2017 | Cholecalciferol 6 lac IU i.m. (n = 30 per group) |
| Stroke outcome significantly improved in vitamin D deficient—but not insufficient—patients once supplemented (mean 6.39 ± 4.56 vs. 2.50 ± 2.20; P < 0.001) |
| Sari et al.164 | 2018 | Cholecalciferol 300 000 IU i.m. (n = 36 per group) |
| 3.09 ± 0.99 vs. 3.34 ± 5.28; P = 0.794 2.34 ± 1.31 vs. 1.60 ± 1.16; P = 0.018 59.18 ± 22.15 vs. 41.87 ± 20.42; P = 0.002 38.72 ± 14.14 vs. 24.75 ± 14.76; P < 0.001 |
| Momosaki et al.165 | 2019 | Cholecalciferol 2 000 IU/die i.m. (n = 49/48) |
| 19 ± 15 vs. 20 ± 13; P = 0.480 27%–20% vs. 25%–18%; P = 0.770;0.740 19%–20% vs. 23%–20%; P = 0.770;0.600 |
| Rist et al.166from the VITAL study | 2021 | Cholecalciferol 2 000 IU/die i.m. in primary prevention (n = 104/93 per group) | Nagi scale Rosow-Breslau scale Katz AD scale | OR 1.01 [95% CI 0.52–1.97] OR 0.92 [95% CI 0.59–1.67] OR 1.03 [95% CI 0.31–3.42] |
| Torrisi et al.167 | 2021 | Cholecalciferol 2 000 IU/die i.m. (n = 15/14) per group) | MADRS FIM GSE (all at 12 weeks) | Both groups improved mood and functional recovery independently of vitamin D supplementation |
| Wang et al.168 | 2021 | Cholecalciferol 600 IU/die i.m. (n = 72/51 per group) | mRS FSS (both at 3 months) | FSS and mRS significanlty lowered in the supplementation group (P < 0.01 for both) |
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Narasimhan et al.163 | 2017 | Cholecalciferol 6 lac IU i.m. (n = 30 per group) |
| Stroke outcome significantly improved in vitamin D deficient—but not insufficient—patients once supplemented (mean 6.39 ± 4.56 vs. 2.50 ± 2.20; P < 0.001) |
| Sari et al.164 | 2018 | Cholecalciferol 300 000 IU i.m. (n = 36 per group) |
| 3.09 ± 0.99 vs. 3.34 ± 5.28; P = 0.794 2.34 ± 1.31 vs. 1.60 ± 1.16; P = 0.018 59.18 ± 22.15 vs. 41.87 ± 20.42; P = 0.002 38.72 ± 14.14 vs. 24.75 ± 14.76; P < 0.001 |
| Momosaki et al.165 | 2019 | Cholecalciferol 2 000 IU/die i.m. (n = 49/48) |
| 19 ± 15 vs. 20 ± 13; P = 0.480 27%–20% vs. 25%–18%; P = 0.770;0.740 19%–20% vs. 23%–20%; P = 0.770;0.600 |
| Rist et al.166from the VITAL study | 2021 | Cholecalciferol 2 000 IU/die i.m. in primary prevention (n = 104/93 per group) | Nagi scale Rosow-Breslau scale Katz AD scale | OR 1.01 [95% CI 0.52–1.97] OR 0.92 [95% CI 0.59–1.67] OR 1.03 [95% CI 0.31–3.42] |
| Torrisi et al.167 | 2021 | Cholecalciferol 2 000 IU/die i.m. (n = 15/14) per group) | MADRS FIM GSE (all at 12 weeks) | Both groups improved mood and functional recovery independently of vitamin D supplementation |
| Wang et al.168 | 2021 | Cholecalciferol 600 IU/die i.m. (n = 72/51 per group) | mRS FSS (both at 3 months) | FSS and mRS significanlty lowered in the supplementation group (P < 0.01 for both) |
CI, confidence interval; OR, odds ratio; SSS, Scandinavian Stroke Scale; BRS, Brunnstrom recovery staging; FAS, functional ambulation scale; MBI, modified Barthel index; BBS, Berg balance scale; MADRS, Montgomery Aasberg depression rating scale; FIM, functional independent measures; GSE, general self-efficacy; mRS, modified Rankin score; FSS, fatigue severity scale.
Global cardiovascular risk related to vitamin D deficiency and supplementation: clinical evidence
Considerable observational evidence links vitamin D deficiency with poor CV prognosis. In the ARIC study, the risk of incident heart failure during 21 years of follow-up was two-fold higher in the lowest quintile (≤17 ng/mL) of 25(OH)D.169 A large body of observational studies has further characterized the CV risk associated with vitamin D deficiency. The data consistently associate vitamin D deficiency with increased incidence and prevalence of subclinical and clinical atherosclerotic disease, myocardial infarction, ischaemic stroke, and peripheral artery disease.2 However, the poor prognosis associated with vitamin D deficiency is not CV-specific, as NHANES III reported a 26% higher overall mortality in those with lower levels of vitamin D.170 Indeed, a critical reappraisal of observational studies raises issues of reverse causation. Vitamin sufficiency may reflect good health in subjects engaged in physical activity and healthy diet. Whereas the Cooper Center Longitudinal Study recently confirmed a benefit of physical activity of vitamin D sufficiency,171 the negative effect on %fat would further contribute to the rise of circulating 25(OH)D in the EpiFloripa Aging Cohort Study.172 Similarly, unhealthy dietary patterns associate not only directly with vitamin D deficiency but also promote other aspects of dysmetabolism further reducing 25(OH)D levels.173
We thus possess a plethora of observational and experimental studies relating to the health effects of vitamin D. Yet, from a practical clinical perspective, what matters most is whether screening for vitamin D status, and supplementation in those found to be vitamin D deficient, can improve patient outcomes. In line with this ‘D-lemma’—as repeatedly quoted by Holick—substantial evidence now shows that vitamin D supplementation does not confer CV benefit.
The origins of tailored randomized clinical trials date back to 2008, when the first conflicting data emerged from interventional studies.174–176 Vitamin D Assessment (ViDA) study and VITamin D and OmegA-3 TriaL (VITAL) were registered almost simultaneously. They both reassuringly reported similar results despite different vitamin D dosing schedules, monthly for ViDA and daily for VITAL. Despite a significant rise in serum 25(OH)D concentrations, the interventions had no effect on the cumulative incidence of CV events (11.8% vs. 11.5% and 6.5% vs. 6.4% for ViDA and VITAL, respectively). Accumulating evidence that vitamin D supplementation benefits primarily deficient people initially rose questions regarding the ViDA and VITAL trials, due to the broad range of serum 25(OH)D levels in the enrolled cohorts, although it now seems established that their findings do not depend on the baseline 25(OH)D levels or CV history.177 Furthermore, evidence from Mendelian randomization studies converges in supporting vitamin D’s inefficacy in reducing CV risk.178,179 An extensive meta-analysis of 21 randomized clinical trials enrolling 83 291 participants reported no benefit of vitamin D supplementation on major adverse CV events, myocardial infarction, stroke, CV, or overall mortality.1 Two other contemporary meta-analyses have provided similar results.180,181 Similarly, recent randomized clinical trials failed to prove a benefit of vitamin D supplementation on diabetes risk (D2d study)182 and associated renal impairment (ancillary study to the VITAL),183 hypertension,184 or CV mortality (D-Health and Finnish vitamin D trials)185,186 (Table 6). On the contrary, most recent trials showed beneficial effects of vitamin D supplementation on body composition and cardio-metabolic/inflammatory biomarkers.187–189 Such results were not unexpected as vitamin D supplementation recently showed favourable effects on atherosclerotic CV disease score without reducing major adverse CV event incidence.190
Summary of randomized clinical trials investigating the effects of vitamin D supplementation on CV risk
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Pittas et al. D2d study182 | 2019 | Cholecalciferol 4000 IU/die i.m (1211patients with pre-diabetes) |
|  |
| de Boer et al. VITAL trial183 | 2019 | Cholecalciferol 2000 IU/die i.m (333 patients with diabetes) |
| |
| Neale et al. D-Health trial185 | 2022 | Cholecalciferol 60 000 IU/month i.m (10662 older people and control group) |
| |
| Theiler-Schwetz et al. Styrian Vitamin D Hypertension Trial184 | 2022 | Cholecalciferol 2800 IU/die i.m (100 hypertensive patients and control group) |
|  |
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Pittas et al. D2d study182 | 2019 | Cholecalciferol 4000 IU/die i.m (1211patients with pre-diabetes) |
| |
| de Boer et al. VITAL trial183 | 2019 | Cholecalciferol 2000 IU/die i.m (333 patients with diabetes) |
| |
| Neale et al. D-Health trial185 | 2022 | Cholecalciferol 60 000 IU/month i.m (10662 older people and control group) |
| |
| Theiler-Schwetz et al. Styrian Vitamin D Hypertension Trial184 | 2022 | Cholecalciferol 2800 IU/die i.m (100 hypertensive patients and control group) |
| |
CI, confidence interval; OR, odds ratio; HR, hazard ratio; eGFR, estimated glomerular filtration rate; ACR, albumin–creatinine ratio; BP, blood pressure; PWV, pulse wave velocity.
Summary of randomized clinical trials investigating the effects of vitamin D supplementation on CV risk
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Pittas et al. D2d study182 | 2019 | Cholecalciferol 4000 IU/die i.m (1211patients with pre-diabetes) |
| |
| de Boer et al. VITAL trial183 | 2019 | Cholecalciferol 2000 IU/die i.m (333 patients with diabetes) |
| |
| Neale et al. D-Health trial185 | 2022 | Cholecalciferol 60 000 IU/month i.m (10662 older people and control group) |
| |
| Theiler-Schwetz et al. Styrian Vitamin D Hypertension Trial184 | 2022 | Cholecalciferol 2800 IU/die i.m (100 hypertensive patients and control group) |
| |
| Authors | Year | Intervention (patient number) | Outcome(s) | Results |
|---|---|---|---|---|
| Pittas et al. D2d study182 | 2019 | Cholecalciferol 4000 IU/die i.m (1211patients with pre-diabetes) |
| |
| de Boer et al. VITAL trial183 | 2019 | Cholecalciferol 2000 IU/die i.m (333 patients with diabetes) |
| |
| Neale et al. D-Health trial185 | 2022 | Cholecalciferol 60 000 IU/month i.m (10662 older people and control group) |
| |
| Theiler-Schwetz et al. Styrian Vitamin D Hypertension Trial184 | 2022 | Cholecalciferol 2800 IU/die i.m (100 hypertensive patients and control group) |
| |
CI, confidence interval; OR, odds ratio; HR, hazard ratio; eGFR, estimated glomerular filtration rate; ACR, albumin–creatinine ratio; BP, blood pressure; PWV, pulse wave velocity.
Shortcomings and outlook
Vitamin D deficiency affects populations worldwide. It appears prevalent in Western countries which show a strong relationship of vitamin D deficiency with CV disease risk and prevalence. Yet, such associations do not establish a causal relationship. During the past decade, experimental evidence on the vitamin D axis and CV health generated important advances, but a wide gap still exists between the research realm and practical clinical measures. Many mechanisms may link vitamin D deficiency with cardio-metabolic risk, atherosclerosis, and its complications (Graphical Abstract). Experimental studies require more rigour. For example, VDR deletion does not address the rapid actions of vitamin D that do not require altered gene transcription. Molecular and epigenetic senescence are newly appreciated mechanisms likely regulated by the vitamin D axis worthy of further investigation. Very recent studies indeed support a potential senolytic activity of vitamin D supplementation, mainly targeting epigenetic ageing and telomerase activity.27,191 The epigenetic clocks might then furnish the intermediate link between vitamin D and CV outcome. Such a change of view—interpreting vitamin D as senolytic—may then help to design future experimental clinical studies.
Ultimately, the field needs to address the inconsistencies in clinical translation. Determinants of vitamin D synthesis, intake, and body distribution require standardization in the design of future clinical trials. Distinctions between fortified food and pharmacologic supplementation is another critical point, compounded by the widespread use of calcium supplements.2 The available data do not justify the screening for vitamin D deficiency in asymptomatic adults.172,192 Vitamin D supplementation has no proven CV benefit with the exception of end-stage renal disease, and even the current thresholds for vitamin D deficiency are still bone-driven. Framing ‘non-classical’—often local—effects of the vitamin D axis with traditional categories will not allow to clarify the extent to which vitamin D status influences cardio-metabolic risk or serves as an independent biomarker of good health. The putative link between vitamin D and cardio-metabolic risk is likely complex and future studies should take reverse causation into account rigorously.
Acknowledgements
None declared.
Data availability
No new data were generated or analysed in support of this research.
Funding
L.L. receives funding support from the Swiss Heart Foundation. P.L. receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892, 1R01HL163099-01, and 1R01HL163099-01), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund. This work was supported by grants from Italian Ministry of Health—5 × 1000 funds 2020 to F.M.
References
Author notes
Conflict of interest L.L. and P.L. are coinventors on the International Patent WO/2020/226993 filed in April 2020. L.L. received speaker fees outside of this work from Daichi-Sankyo. P.L. is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Novartis, Pfizer, and Sanofi-Regeneron. P.L. is a member of the scientific advisory board for Amgen, Caristo Diagnostics, Cartesian Therapeutics, CSL Behring, DalCor Pharmaceuticals, Dewpoint Therapeutics, Kancera, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, PlaqueTec, TenSixteen Bio, Soley Therapeutics, and XBiotech, Inc. P.L.’s laboratory has received research funding in the last 2 years from Novartis. P.L. is on the Board of Directors of XBiotech, Inc. P.L. has a financial interest in Xbiotech, a company developing therapeutic human antibodies. P.L.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. P.L. has a financial interest in TenSixteen Bio, a company targeting somatic mosaicism and clonal haematopoiesis of indeterminate potential (CHIP) to discover and develop novel therapeutics to treat age-related diseases. P.L. has a financial interest in Soley Therapeutics, a biotechnology company that is combining artificial intelligence with molecular and cellular response detection for discovering and developing new drugs, currently focusing on cancer therapeutics. P.L.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Mass General Brigham in accordance with their conflict-of-interest policies. F.M. and F.C. declared no conflict of interest.


{kind=link}
{kind=link}
{kind=link}
{kind=link}