





Abstract
The advent of single-cell biology opens a new chapter for understanding human biological processes and for diagnosing, monitoring, and treating disease. This revolution now reaches the field of cardiovascular disease (CVD). New technologies to interrogate CVD samples at single-cell resolution are allowing the identification of novel cell communities that are important in shaping disease development and direct towards new therapeutic strategies. These approaches have begun to revolutionize atherosclerosis pathology and redraw our understanding of disease development. This review discusses the state-of-the-art of single-cell analysis of atherosclerotic plaques, with a particular focus on human lesions, and presents the current resolution of cellular subpopulations and their heterogeneity and plasticity in relation to clinically relevant features. Opportunities and pitfalls of current technologies as well as the clinical impact of single-cell technologies in CVD patient care are highlighted, advocating for multidisciplinary and international collaborative efforts to join the cellular dots of CVD.
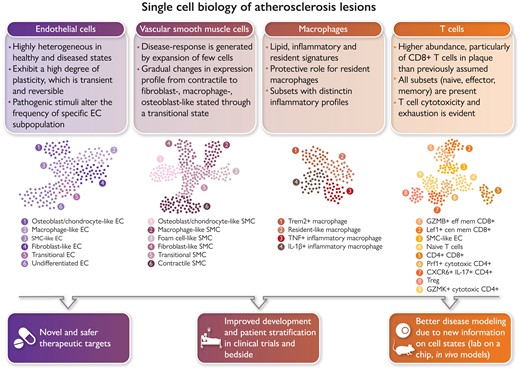
Single-cell biology is bringing new clinical meaning to patient heterogeneity in many disciplines. Atlases of the cellular building blocks of the human atherosclerotic plaque have so far shown that: (i) cellular identity is overall preserved, albeit overlapping transcriptional programmes are activated; (ii) changes in cellular cluster abundance appear between healthy and diseased vascular states; (iii) renewed evidence emerged for a role of T cells in human cardiovascular disease (CVD); and (iv) macrophage heterogeneity supports targeting inflammation and lipids while sparing protective subsets. Vascular single-cell biology has clear translational implications for CVD in terms of identification of the molecular pathways of disease resistance vs. disease propensity and genetic risk, guidance in designing new therapies, vaccines and repurposing drugs for CVD, the study of the therapy-induced adaptation of plaque and circulating cells in clinical trials, improved modelling of human CVD through the availability of metagenomic data sets, and advances in patient selection and stratification. GMZB, Granzyme B; Lef1, lymphoid enhancer binding factor 1; Prf1 Perforin 1; Trem2, triggering receptor expressed on myeloid cells 2.
All figures were drawn by the authors either on PowerPoint or on BioRender. No additional permissions are required.
Introduction
Cardiovascular disease (CVD) is the major cause of death worldwide, despite risk factor management and interventional procedures.1 Atherosclerosis is the key disease process underlying CVD mortality, and it causes the accumulation of lipids, inflammatory cells, smooth muscle cells (SMCs), and extracellular matrix (ECM) components within the intima of large and muscular arteries.2 Although atherosclerotic plaques can develop ubiquitously and silently over the lifetime of an individual, a minority of plaques evolves into a variety of culprit lesions causing the bulk of cardiovascular events. Identifying vulnerable plaques and patients in advance is still the biggest challenge of modern cardiology.
The cellular landscape of human atherosclerosis and its relation to disease development and clinical complications remains enigmatic. Conventional approaches like histochemistry and immunohistochemistry led to the definition of stages of the disease and the broad cellular composition of culprit lesions.3–5 Individual tissue markers for subpopulations of cells were linked to patient characteristics or plaque features.2,6,7 However, vascular tissues exhibit significant heterogeneity due to haemodynamic factors, cell ontogeny, cell recruitment, and cell transdifferentiation such as endothelial-to-mesenchymal cell transition (EndoMT; Figure 1). Traditional methodologies cannot keep up with the degree of complexity of cellular architecture that atherosclerosis presents.
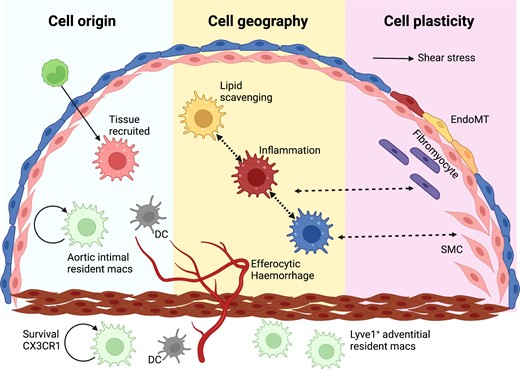
The basis of cellular heterogeneity in atherosclerosis There are three main determinants of cellular heterogeneity in arteries and atherosclerosis. (i) Cell origin. Arteries have different ontogeny. They originate from the neural crest (carotid and proximal aorta), the proepicardium (coronary arteries) or the mesoderma (rest of the body). Also, macrophages are either resident, or increasingly bone marrow-derived with age. (ii) Cell geography. Cell location imprints cells, either via exposure to different shear stress and other haemodynamic forces, as is the case with ECs, or through cell residence within distinct niches in health and disease. Macrophages have distinct phenotypes in the intima or adventitia or respond to intraplaque events such as haemorrhage or lipid accumulation. (iii) Cell plasticity. EndoMT is characterized by a downregulation of EC-specific gene expression and/or the full disappearance of EC fate marker genes along with the appearance of gene expression programmes associated with other cell types, including fibroblasts, SMCs and macrophages, among others.8–10 Often such ‘foreign’ gene expression is associated with activation of EC proliferation and migration and loss of the protective quiescent metabolic state as well as of the ability to exert normal EC function, such as a response to blood flow, regulation of permeability, and antioxidant capacity. These changes in EC gene expression show a remarkable degree of plasticity and can be temporary activated shortly after myocardial infarction,11 or turn into a permanent EndoMT.12 A number of pathologic factors including disturbed flow, oxidative stress, hypoxia, and inflammation (e.g. activation of endothelial TGFβ or IL-beta signalling) can initiate EndMT.13–16 Pro-atherogenic cues trigger SMC phenotypic modulation and atherosclerotic plaque investment by oligoclonal expansion. Plasticity between different SMC-derived states suggests that SMC phenotypic states might be niche-dependent, transitory, and/or interconvertible. A macrophage-to-mesenchymal transition has also been described.
The Human Cell Atlas is an ambitious initiative to build a comprehensive reference map of all human cells and is bringing about a monumental shift in human physiology and pathology. These efforts have led to the continuous discovery of new cell types. For example, single-cell RNA sequencing (scRNA-seq) has identified the aerocyte as a capillary endothelial cell (EC) responsible for gas exchange in the lungs17 and is changing the face of cancer biology.18 Groundbreaking studies published in CVD so far have shown us what is possible, but they are only the beginning. In this review, we aim to present a timely and comprehensive insight into single-cell biology-driven knowledge on cell communities in atherosclerosis. We discuss the limitations, challenges, and opportunities afforded by single-cell biology for CVD and its impact on basic discovery, translational science, reverse translation, and clinical insights.
The cellular landscape of atherosclerosis
Here we provide a concise overview of insights obtained through single-cell biology into the currently defined vascular cell populations (Figure 2). Boxes 1 and 2 contain an overview of the technical aspects of the most common single-cell biology platforms and the characteristics of major cell populations in atherosclerosis, respectively.
Recent technological breakthroughs allow detailed definition of the cellular composition of atherosclerotic lesions (reviewed in Hartman et al.22) Mass cytometry (CyTOF Helios) is enabling the analysis of 45 (the theoretical limit is 100, depending on the availability of pure rare metal) parameters on cells simultaneously using metal-labelled antibodies on single-cell suspensions from tissues, allowing identification of a plethora of subsets and activation states. Using metal-labelled antibodies avoids tissue autofluorescence, which is a serious limitation of vascular and lipid-rich tissues. Spectral analysers such as the CyTek Aurora use fluorescence to capture 28 (theoretical limit is 50 depending on the spectral resolution) parameters.
Single-cell RNA sequencing (scRNA-seq) is defining the full transcriptional repertoire of individual cells. RNA-seq is a technique to quantify and analyse RNA molecules in a sample by next generation sequencing. It allows detailed characterization of gene expression programmes in individual cells. It builds on the RNA-seq analysis of cells in isolated cell populations, collected from blood or tissues from patients. By performing single-cell RNA-seq, we can profile cellular populations present in tissues, identify characteristics of these cellular populations and define associations between these populations and disease or clinically relevant parameters. It will lead to a better understanding of cellular interactions in plaques, the signalling pathways that control detrimental populations and interventions (i.e. drugs) to modulate cell-relevant processes such as inflammation in atherosclerosis.
Factors that drive choices and costs of scRNA-seq include desired cell numbers, cells of interest, isolation strategy, choice of tissue, and sequencing depth. Different scRNA-seq technologies are available and offer pros and cons with respect to cell numbers captured, sequencing depth and accompanying detection of less abundant genes and populations and costs. Detailed summaries of experimental guidelines and technical platforms have previously been published23–29 and will not be discussed here.
Fate mapping is a technique borrowed from developmental biology whereby a cell is marked genetically, usually by encoding a fluorescent tracer, and is used to study its progeny after induction of disease. Fate mapping is now used in combination with scRNA-seq to trace the origin of cells. This methodology is unfeasible in man and it is an area where murine models are required.
ScRNA-seq is now sometimes integrated with CITE-seq, whereby antibodies detecting specific cellular proteins are integrated in the pipeline, allowing both the detection of RNA molecules and selected proteins. CITE-seq platforms have achieved validation of >100 Ab oligo-labelled cocktails. Additionally, scATACseq assesses genome-wide chromatin accessibility, giving insights into active chromatin differences and transcription factor used by motif analysis of identified open chromatin. Aptamer technology combined with scRNA-seq (Apt-Seq)30 allows the discrimination between cells based on aptamer binding and differences in gene expression. Most of its applications are at present in cancer. This technology could be instrumental in developing personalized atherosclerosis drug treatments.
Spatial omics is an emerging technology that provides geolocation as well as cell heterogeneity information. At present, first-generation transcriptomics platforms such as 10× Visium lack single-cell depth analysis, but still provide robust information on geolocation, which is missing from scRNA-seq. Spatial data will be very valuable to define, for instance, the luminal ECs around the plaque, which exhibit a different biology from the ones that participate in the intraplaque neovessel formation. The same applies for other plaque cell subtypes, rendering spatial omics highly complementary to dissociative methodologies such as scRNA-seq and CyTOF Helios. Proteomics alternatives such as CyTOF-Hyperion and methoxyisobutyl isonitrile approximate single-cell resolution with detection of metal-labelled antibodies bypassing the famed vascular autofluorescence. Combination CITE-seq-CyTOF-Hyperion Imaging mass spectrometry will reveal geography of cells and cell-specific pathways of interest coming from Cite/sc/RNA-seq next generation spatial methods (e.g. hybridization-based in situ sequencing such as Vizgen MERSCOPE or spatial molecular imaging platform NanoString CosMx31) approximate single-cell resolution and also allow for the analysis of larger sample sizes (10× platforms are limited to 6 × 6 mm). Practically, they are not genome-wide currently, but allow ∼1000 genes and ∼100 proteins to be simultaneously detected at 50 nm resolution with good sensitivity. Hence, in combination with scRNA-seq clustering—marker signatures for a large number of cell states can be analysed in each sample. Overall, scRNA-seq provides superb opportunities to define pathological mechanisms occurring in tissues that control disease development and provide ways to investigate novel therapeutic approaches.
Endothelial cells: The endothelium plays a pivotal role in arterial health and disease by regulating the transport of cells, lipoproteins, and other molecules from the circulation to the arterial wall and vice versa and by regulating key processes including inflammation, vascular smooth muscle cell proliferation and migration, vascular tone, and thrombosis. Endothelial cells (ECs) also govern vascular responses to mechanical forces generated by flowing blood that are important in atherosclerosis initiation and disease progression.15,32 Disturbed flow is known to induce multiple pathophysiological changes in EC, including endothelial-to-mesenchymal transition (EndoMT), vascular inflammation and apoptosis, which collectively increase arterial permeability to cholesterol-rich lipoproteins.33–36 By contrast, laminar flow is protective.
Smooth muscle cells: Smooth muscle cells (SMCs) are important for vascular integrity and tone, yet they retain remarkable adaptive capacities and dynamically modulate their phenotype in response to environmental changes. Pro-atherogenic cues trigger SMC phenotypic modulation and atherosclerotic plaque investment by oligoclonal expansion.37 Fate mapping studies in mouse revealed that SMC account for a predominant fraction of plaque cells. Over 80% of lesion SMC do not express conventional SMC marker genes (e.g. ACTA2),38 challenging previous paradigms restricting SMC function to protective fibrous cap generation and ECM synthesis. Instead, functional studies provided evidence that SMC played active roles in pro-inflammatory cytokine production, lipid accumulation, apoptotic cell efferocytosis, and calcification. SMC phenotypic states might be niche-dependent, transitory, and/or interconvertible. By combining single-cell RNA sequencing (scRNA-seq) with genetic tracing of SMC-derived cells and immunohistological validation, relevant transcript signatures for identification of proposed atheroprotective or atheropromoting SMC transitions have been suggested. However, information about how atheroprotective and atheropromoting phenotypes are controlled is currently lacking.
Macrophages: Macrophages, as key inflammatory cells, play a central role in the pathogenesis of atherosclerosis in all its stages. In health the arterial wall contains vascular resident macrophages in the adventitia (Lyve 1+ macrophages), which perform homeostatic functions.39 The accumulation of lipid-laden mononuclear phagocytes within the intimal layer of the arterial wall is the hallmark of atherosclerosis and is critical for its development.40,41 The variety of macrophage subsets described in atherosclerotic plaques is testament to the plasticity of this cell type, with cell reprogramming driven by lipid accumulation, inflammation, and haemorrhage (reviewed in Willemsen and de Winther42 and Chinetti-Gbaguidi et al.43). However, little is known about the functional significance of emerging specific subsets in atherosclerosis.
T cells: CD4+ and CD8+ T cells play subset-dependent roles in atherosclerosis (reviewed in Schafer and Zernecke,44 Saigusa et al.,45 and Ali et al.46). Increasing evidence argues for the presence of CD4+ T cells in mice and humans responding to atherosclerosis-relevant antigens.47–49 Autoimmune T-cell responses in atherosclerosis have been mostly characterized as pro-inflammatory Th1 cell responses, e.g. secretion of pro-atherosclerotic cytokines such as interferon-gamma. Previous knowledge was based on low-parametric flow cytometry for T-cell lineage-defining transcription factors, cytokines, or chemokine receptors. Fate mapping in mouse models and scRNA sequencing now suggest that many of these pro-inflammatory atherosclerosis antigen-specific CD4+ T cells seem to have derived from previous regulatory CD4+ T cells, which have known atheroprotective functions.
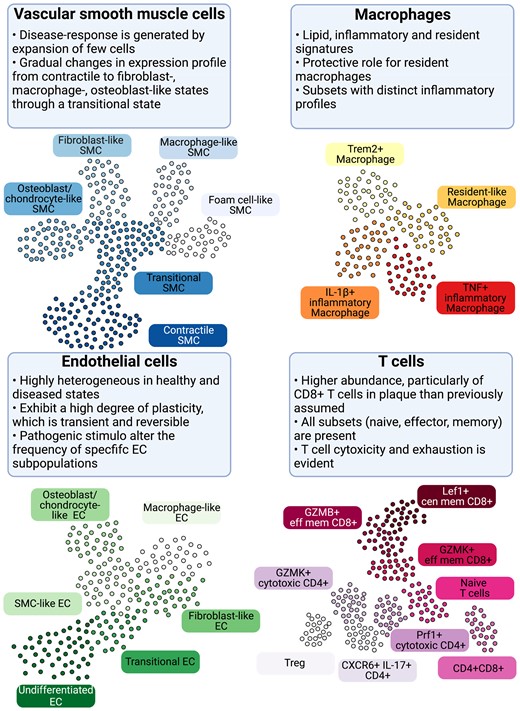
Cellular populations and plasticity in atherosclerosis. Cellular diversity uncovered by scRNA-seq analysis in human and murine atherosclerosis with focus on consensus populations found in both species. Macrophages are key inflammatory immune cells in atherosclerotic plaques. Four macrophage populations have been identified with different functional features, including: Trem2hi macrophages, resident-like macrophages, and inflammatory macrophages. Aortic macrophages identified by scRNA-seq can have different origins and derive from infiltrating monocytes, proliferation of embryonically derived macrophages, and lastly from SMCs and fibroblasts obtaining macrophage features. The greatest cellular diversity is found among T cells. Multiple CD4 and CD8 effector and central memory (eff mem) and cytotoxic subsets have been identified that likely contribute to atheroprogression. Regulatory T cells (Tregs) have known anti-inflammatory and atherosclerotic properties. CXCR6+ CD4+ T cells have a multi-lineage signature and can likely differentiate into other subtypes. Contrasting to T-cell diversity, only one B-cell population has been identified in human atherosclerotic plaques with yet unknown function. EC and SMC undergo various phenotypic transitions likely stage- and micro-environment dependent within the plaque. They can display either atheroprotective or atheropromoting phenotypes. SMCs (top left, adapted from Worssam and Jorgensen19) in atherosclerotic lesions undergo progressive cell transitions with loss of the expression profile of contractile cells found in healthy arteries. Instead, SMC-derived cells adopt a range of cell states, displaying some aspects of fibroblasts, mesenchymal cells, and chondrocytes. Evidence for a transient, intermediate cell state with stem-cell, endothelial, and monocyte features (termed SEM)20,21 and transition of SMC towards a macrophage-like state has been provided by lineage tracing in mouse models. GMZB, Granzyme B; Lef1, Lymphoid Enhancer Binding Factor 1; Prf1, Perforin 1; Trem2, Triggering Receptor Expressed On Myeloid Cells 2.
Endothelial cells have multiple faces in vascular health and disease
ScRNA-seq has been essential for the identification of a spectrum of EC subsets in both health and disease.50–52 scRNA-seq studies have demonstrated that vascular cells such as ECs adopt an even larger spectrum of phenotypes than anticipated in both healthy and diseased vessels, some of which are primed for disease, and that they change in a gradual manner rather than in discrete states, probably through reversible transitional states.11 Data from a recent cell atlas of cardiac arteries from patients undergoing heart transplantation identified four subtypes of ECs at steady state: three larger vascular EC subsets and a smaller lymphatic subpopulation of ECs.52 The first subset highly expressed inflammatory genes (i.e. ICAM, VCAM1, CRP) and were found in greater abundance in atherosclerotic coronary arteries than in non-diseased controls. The second exhibited enrichment in genes involved in endothelium development and regulation of vasculature development. The gene expression signature of the third EC population was linked to the ECM organization. This subpopulation decreased in atherosclerosis compared with non-diseased conditions, suggesting protective functions. Finally, the fourth population expressed the lymphatic EC marker LYVE1 and was mainly implicated in neutrophil-mediated immune responses, such as neutrophil activation and degranulation. Interestingly, the expression of atherosclerosis-associated genes appeared to be constitutive also in non-atherosclerotic ECs reflecting donor selection and/or disease predisposition.52,53
Recent scRNA-seq analysis of human carotid atherosclerotic plaques identified very similar populations as seen in the heart.13 However, an additional subset not found in the coronaries presented signs of EndoMT [i.e. expression of typical SMC markers, such as α-smooth muscle actin (ACTA2), NOTCH3, and smooth muscle myosin heavy chain 11 (MYH11)]. Expression of both ACTA2, which is a marker of SMC identity, and CD34 was found in cells lining the intraplaque vasculature on sequential histological slides in different patients,13 further supporting the concept that transdifferentiation of ECs is likely to occur in progression of advanced atherosclerosis lesions. In line with this notion, Chen et al.54 demonstrated using scRNA-seq that EndoMT subsets are induced by high fat feeding of apoe−/− mice. Similarly, Newman et al.14 recently revealed that also the fibrous cap in murine and human lesions contains 20–40% of ACTA2+ cells, from a non-SMC origin including EndoMT33 or macrophage-to-mesenchymal transition. These findings suggest that the presence of transdifferentiated cells needs to be factored in to understand the mechanisms of fibrous cap stability. However, EndoMT is rarely a full, permanent transition to a mesenchymal cell state in atherogenesis but rather represents activation of the mesenchymal gene programme and the acquisition of novel functions. As we extend our reach towards the full transcriptome analysis at the single-cell level, we expand our capability to derive exact therapeutic targets and our potential for cell fate manipulations.
Smooth muscle cell transition towards pathogenic subsets in atherosclerosis
ScRNA-seq studies have demonstrated that SMCs adopt a spectrum of phenotypes rather than just a few discrete states of synthetic and contractile cells. The application of scRNA-seq to SMCs, in combination with fate mapping (Box 1), showed that small, pre-existing SMC populations take over in response to pathogenic stimuli during disease development and progression. Single-cell profiling of SMC-lineage-traced cells in mouse-detected cell states characterized by signatures overlapping with that of fibromyocytes, osteochondrocytes, ECs, foam cells, and mesenchymal stem cells (Table 1).20,55–58 Moreover, the range of SMC-derived cell states observed in mouse models changes over time, indicating that the developing plaque environment induces further state perturbations as the disease progresses.20,57 Computational data integration has suggested the existence of equivalent cell states in human data sets,20,57–59 but the annotation of cell types is not trivial because of the pronounced plasticity and heterogeneity.13 Further hampering the definition of what constitutes an SMC plaque state is the fact that lineage labelling demonstrates that other cells can also transition towards an SMC state within lesions14,54,60 and vice versa.38,61,62
Vascular subsets in human and mouse atherosclerotic tissue
| Functional prediction | Markers (mouse/human) | |
|---|---|---|
| Endothelial cells | ||
| ȃEndoMT (human) | ECM production | ACTA2, VIM, COL1A2, NOTCH3, MYH11 |
| ȃEndoMT (human) | Pro-inflammatory | CCL2, CCL7CXCL1, CXCL2, VCAM1, ICAM1, ITGA5, FGF18, HEG1 |
| ȃEndoMT (mouse) | Pro-inflammatory and ECM production | Vcam1, Sele, Selp, Cxcl15, Fbln5, Col8A1, Vim, Fn, Mmp2, Mmp14 |
| ȃMSC like (mouse) | Ly6a, Cd44, Cd34 | |
| ȃNormal and Western diet-fed mice | Lymphatic EC | Lyve1 |
| Smooth muscle cells | ||
| ȃFibrous cap SMCs | ECM production | Acta2, Phactr1, ACTA2, PHACTR1, PDGFR |
| ȃFibromyocyte/myofibroblast | ECM production | Lum, Tnfrsf11b, TNFRSF11B |
| ȃOsteogenic-like/fibrochondrocytes | Mineralization, calcification | lbsp, Dcn, Sox9, SOX9 |
| ȃ‘MSC like’/SEM | Cell expansion, transition to other states | Ly6a, Vcam1, Lgals3, Cd73, Cd90, Cd105, C3 |
| ȃFoam cells/phagocytes/macrophage like | Lipid uptake, pro-inflammatory, ingestion of debris/apoptotic cells | Cd68, Lgals3, Ly6c1, CD68, LGALS3 |
| Functional prediction | Markers (mouse/human) | |
|---|---|---|
| Endothelial cells | ||
| ȃEndoMT (human) | ECM production | ACTA2, VIM, COL1A2, NOTCH3, MYH11 |
| ȃEndoMT (human) | Pro-inflammatory | CCL2, CCL7CXCL1, CXCL2, VCAM1, ICAM1, ITGA5, FGF18, HEG1 |
| ȃEndoMT (mouse) | Pro-inflammatory and ECM production | Vcam1, Sele, Selp, Cxcl15, Fbln5, Col8A1, Vim, Fn, Mmp2, Mmp14 |
| ȃMSC like (mouse) | Ly6a, Cd44, Cd34 | |
| ȃNormal and Western diet-fed mice | Lymphatic EC | Lyve1 |
| Smooth muscle cells | ||
| ȃFibrous cap SMCs | ECM production | Acta2, Phactr1, ACTA2, PHACTR1, PDGFR |
| ȃFibromyocyte/myofibroblast | ECM production | Lum, Tnfrsf11b, TNFRSF11B |
| ȃOsteogenic-like/fibrochondrocytes | Mineralization, calcification | lbsp, Dcn, Sox9, SOX9 |
| ȃ‘MSC like’/SEM | Cell expansion, transition to other states | Ly6a, Vcam1, Lgals3, Cd73, Cd90, Cd105, C3 |
| ȃFoam cells/phagocytes/macrophage like | Lipid uptake, pro-inflammatory, ingestion of debris/apoptotic cells | Cd68, Lgals3, Ly6c1, CD68, LGALS3 |
Vascular subsets in human and mouse atherosclerotic tissue
| Functional prediction | Markers (mouse/human) | |
|---|---|---|
| Endothelial cells | ||
| ȃEndoMT (human) | ECM production | ACTA2, VIM, COL1A2, NOTCH3, MYH11 |
| ȃEndoMT (human) | Pro-inflammatory | CCL2, CCL7CXCL1, CXCL2, VCAM1, ICAM1, ITGA5, FGF18, HEG1 |
| ȃEndoMT (mouse) | Pro-inflammatory and ECM production | Vcam1, Sele, Selp, Cxcl15, Fbln5, Col8A1, Vim, Fn, Mmp2, Mmp14 |
| ȃMSC like (mouse) | Ly6a, Cd44, Cd34 | |
| ȃNormal and Western diet-fed mice | Lymphatic EC | Lyve1 |
| Smooth muscle cells | ||
| ȃFibrous cap SMCs | ECM production | Acta2, Phactr1, ACTA2, PHACTR1, PDGFR |
| ȃFibromyocyte/myofibroblast | ECM production | Lum, Tnfrsf11b, TNFRSF11B |
| ȃOsteogenic-like/fibrochondrocytes | Mineralization, calcification | lbsp, Dcn, Sox9, SOX9 |
| ȃ‘MSC like’/SEM | Cell expansion, transition to other states | Ly6a, Vcam1, Lgals3, Cd73, Cd90, Cd105, C3 |
| ȃFoam cells/phagocytes/macrophage like | Lipid uptake, pro-inflammatory, ingestion of debris/apoptotic cells | Cd68, Lgals3, Ly6c1, CD68, LGALS3 |
| Functional prediction | Markers (mouse/human) | |
|---|---|---|
| Endothelial cells | ||
| ȃEndoMT (human) | ECM production | ACTA2, VIM, COL1A2, NOTCH3, MYH11 |
| ȃEndoMT (human) | Pro-inflammatory | CCL2, CCL7CXCL1, CXCL2, VCAM1, ICAM1, ITGA5, FGF18, HEG1 |
| ȃEndoMT (mouse) | Pro-inflammatory and ECM production | Vcam1, Sele, Selp, Cxcl15, Fbln5, Col8A1, Vim, Fn, Mmp2, Mmp14 |
| ȃMSC like (mouse) | Ly6a, Cd44, Cd34 | |
| ȃNormal and Western diet-fed mice | Lymphatic EC | Lyve1 |
| Smooth muscle cells | ||
| ȃFibrous cap SMCs | ECM production | Acta2, Phactr1, ACTA2, PHACTR1, PDGFR |
| ȃFibromyocyte/myofibroblast | ECM production | Lum, Tnfrsf11b, TNFRSF11B |
| ȃOsteogenic-like/fibrochondrocytes | Mineralization, calcification | lbsp, Dcn, Sox9, SOX9 |
| ȃ‘MSC like’/SEM | Cell expansion, transition to other states | Ly6a, Vcam1, Lgals3, Cd73, Cd90, Cd105, C3 |
| ȃFoam cells/phagocytes/macrophage like | Lipid uptake, pro-inflammatory, ingestion of debris/apoptotic cells | Cd68, Lgals3, Ly6c1, CD68, LGALS3 |
Despite this extensive plasticity, specific signatures have been linked to clinical traits. For example, ACTA2 and PHACTR1 are markers of atheroprotective SMCs located in the fibrous cap with high expression in stable plaques. Conversely, the expression of Sox9, TRPV4, and S100b (in an osteochondrogenic SMC cluster) marks unstable plaques.63 Inclusion of such signatures in the characterization of both mouse and human plaques will help in improving plaque vulnerability assessment.57 Pathway analysis has identified novel regulators and known factors that control SMC state (e.g. TCF21,58KLF4,57PDGFRB,14 retinoic acid signalling,20 and metabolism14) and could be targeted to promote a stabilizing phenotype. The transcriptomic characterization of a hyperproliferative state may also facilitate manipulation and restriction of cell expansion.56
ScRNA-seq data indicate that cellular transitions are often gradual over time and probably happen through transitional states as opposed to punctate events. This discovery emphasizes that the expression of a few proteins in the context of immuno-histochemistry is not sufficient to draw conclusions about cellular identity. For instance, the detection of macrophage or chondrocyte markers in SMCs does not necessarily imply that a full transdifferentiation has been achieved. The transition of SMCs to macrophages or macrophage-to-mesenchymal cells needs further investigation in the context of CVD.
Macrophages include inflammatory, resident, and lipid-laden cell communities
Single-cell studies from human and murine atherosclerosis identified four main macrophage populations.13,64,65 One inflammatory macrophage population includes macrophages expressing the therapeutically relevant cytokine interleukin 1 beta (IL1B)66 and components required for IL1B production (IL1B, CASP1, CASP4, and NLRP3), as well as other major inflammatory mediators of relevance to atherogenesis such as the calgranulins (S100A8/S1009/S10012), the inflammation-inducing toll-like receptors and receptor for advanced glycation endproducts ligands. Another inflammatory subset expresses tumour necrosis factor α, indicating a certain degree of heterogeneity in the inflammatory responses adopted by the plaque.
A resident macrophage cluster expresses classical markers of resident macrophages such as LYVE1, FOLR2, VSIG4, and F13A1, complements of the C1Q family (C1QA, C1QB, and C1QC), and genes of the major histocompatibility complex (HLA-DRA, HLA-DRB1, and CD74). Predicted functions of these macrophages include antigen processing and presentation as well as complement activation and phagocytosis, suggesting a role in activating additional and accessory immune cells in the atherosclerotic plaque as well as a homeostatic role in vascular housekeeping. Murine studies show significant heterogeneity in the vascular resident macrophage compartment. In mouse, LYVE1+ vascular resident macrophages are engaged in crosstalk with medial SMCs, regulating production of ECM and arterial diameter67 and Clec4a2+ vascular resident macrophages were shown to have an anti-atherogenic function.68
The fourth subset comprises TREM2hi macrophages, which express genes involved in lipid metabolism as well as genes regulating cholesterol transport and efflux. Their pronounced lipid and cholesterol signature identified them as the candidate foam cells. The TREM2hi macrophages have been identified in human plaques as well as murine aortas,13,69,70 adipose tissue, and liver,71–73 suggesting important similarities across metabolic diseases and tissues. The lack of inflammatory gene expression challenges the concept of lipid-driven inflammation. Additionally, TREM2hi macrophages have been linked to pro-fibrotic characteristics,72 making them potentially relevant for plaque stability (Table 2).
Immune subsets in human and mouse atherosclerotic tissue
| Functional prediction | Markers (mouse/human) | |
|---|---|---|
| Macrophages | ||
| ȃInflammatory macrophages | Lesional inflammation | S100A8/S100A9, IL1B, CASP1, CASP4, NLRP3 |
| ȃResident-like macrophages | Efferocytosis, endocytosis antigen processing and presentation | LYVE1, CX3CR1, FOLR2, C1QA/C1QB/C1QC, CD74, HLA-DRA/HLA-DRB1 |
| ȃTREM2 ‘Foamy’ macrophages | Lipid processing, antioxidant | TREM2, CD9, FABP5, APOE, APOC1 |
| ȃIFNIC macrophagesa | Induced in disease progression | Isg15, Irf7, Ifit3, Ifit1 |
| ȃProliferating macrophagesa | Macrophage expansion or renewal | Stmn1, Top2a, Mki67, Tuba1b, Tubb5 |
| T cells | ||
| ȃMouse cytotoxic CD8+ T cells | Cytotoxicity | Cd8a/b, Nkg7, Ms4ab4, Ccl5, Gzmk, Eomes, Cd3d |
| ȃMouse CXCR6+ CD4+ T cells | Immune response to lipids | Cxcr6, Rora, Tmem176, Ramp3, Il7a, Gata3, Il1rl1 |
| ȃMouse thymocyte-like CD4+CD8+ cells | Unknown | Cd8b, Cd4, Tcf7, Ccr9, Rag1, Dntt, Lck |
| ȃMouse naïve T cells | Steady state | Cd28, Cr7, Lef1, Dapl1, Tcf7, Cd2, Sell/Il7r |
| ȃHuman GZMK+ effector/memory CD8+ T cells (CD8.0) | Effector memory | GZMK, ITM2C, CCL4, CD74, MYADM |
| ȃHuman GZMB+ cytotoxic CD8+ T cells (CD8.1) | Cytotoxicity | FGFBP2, ADGRG1, GZMB, CX3CR1, GNLY, FCGR3A |
| ȃHuman LEF1+ central memory CD8+ T cells (CD8.3) | Central memory | LEF1, LTB, SELL, IL7R, PDE3B, EEF1A1 |
| ȃPRF1+ cytotoxic CD4+ T cells (CD4.0) | Cytotoxicity | NKG7, CCL5, PRF1, GNLY, GZMA |
| ȃGZMK+ cytotoxic CD4+ T cells | Cytotoxicity | GZMK, GZMA |
| ȃIL7R+ naïve CD4+ T cells | Steady state | ZNF480, IL7R, CTSB |
| ȃFOXP3+ regulatory CD4+ T cells | Immunomodulation | FOXP3, IL2RA, ICOS, TIGIT |
| ȃLef1+ central memory CD4+ Tc ells | Central memory | LEF1, LTB, CCR7, TCF7 |
| Functional prediction | Markers (mouse/human) | |
|---|---|---|
| Macrophages | ||
| ȃInflammatory macrophages | Lesional inflammation | S100A8/S100A9, IL1B, CASP1, CASP4, NLRP3 |
| ȃResident-like macrophages | Efferocytosis, endocytosis antigen processing and presentation | LYVE1, CX3CR1, FOLR2, C1QA/C1QB/C1QC, CD74, HLA-DRA/HLA-DRB1 |
| ȃTREM2 ‘Foamy’ macrophages | Lipid processing, antioxidant | TREM2, CD9, FABP5, APOE, APOC1 |
| ȃIFNIC macrophagesa | Induced in disease progression | Isg15, Irf7, Ifit3, Ifit1 |
| ȃProliferating macrophagesa | Macrophage expansion or renewal | Stmn1, Top2a, Mki67, Tuba1b, Tubb5 |
| T cells | ||
| ȃMouse cytotoxic CD8+ T cells | Cytotoxicity | Cd8a/b, Nkg7, Ms4ab4, Ccl5, Gzmk, Eomes, Cd3d |
| ȃMouse CXCR6+ CD4+ T cells | Immune response to lipids | Cxcr6, Rora, Tmem176, Ramp3, Il7a, Gata3, Il1rl1 |
| ȃMouse thymocyte-like CD4+CD8+ cells | Unknown | Cd8b, Cd4, Tcf7, Ccr9, Rag1, Dntt, Lck |
| ȃMouse naïve T cells | Steady state | Cd28, Cr7, Lef1, Dapl1, Tcf7, Cd2, Sell/Il7r |
| ȃHuman GZMK+ effector/memory CD8+ T cells (CD8.0) | Effector memory | GZMK, ITM2C, CCL4, CD74, MYADM |
| ȃHuman GZMB+ cytotoxic CD8+ T cells (CD8.1) | Cytotoxicity | FGFBP2, ADGRG1, GZMB, CX3CR1, GNLY, FCGR3A |
| ȃHuman LEF1+ central memory CD8+ T cells (CD8.3) | Central memory | LEF1, LTB, SELL, IL7R, PDE3B, EEF1A1 |
| ȃPRF1+ cytotoxic CD4+ T cells (CD4.0) | Cytotoxicity | NKG7, CCL5, PRF1, GNLY, GZMA |
| ȃGZMK+ cytotoxic CD4+ T cells | Cytotoxicity | GZMK, GZMA |
| ȃIL7R+ naïve CD4+ T cells | Steady state | ZNF480, IL7R, CTSB |
| ȃFOXP3+ regulatory CD4+ T cells | Immunomodulation | FOXP3, IL2RA, ICOS, TIGIT |
| ȃLef1+ central memory CD4+ Tc ells | Central memory | LEF1, LTB, CCR7, TCF7 |
Immune subsets in human and mouse atherosclerotic tissue
| Functional prediction | Markers (mouse/human) | |
|---|---|---|
| Macrophages | ||
| ȃInflammatory macrophages | Lesional inflammation | S100A8/S100A9, IL1B, CASP1, CASP4, NLRP3 |
| ȃResident-like macrophages | Efferocytosis, endocytosis antigen processing and presentation | LYVE1, CX3CR1, FOLR2, C1QA/C1QB/C1QC, CD74, HLA-DRA/HLA-DRB1 |
| ȃTREM2 ‘Foamy’ macrophages | Lipid processing, antioxidant | TREM2, CD9, FABP5, APOE, APOC1 |
| ȃIFNIC macrophagesa | Induced in disease progression | Isg15, Irf7, Ifit3, Ifit1 |
| ȃProliferating macrophagesa | Macrophage expansion or renewal | Stmn1, Top2a, Mki67, Tuba1b, Tubb5 |
| T cells | ||
| ȃMouse cytotoxic CD8+ T cells | Cytotoxicity | Cd8a/b, Nkg7, Ms4ab4, Ccl5, Gzmk, Eomes, Cd3d |
| ȃMouse CXCR6+ CD4+ T cells | Immune response to lipids | Cxcr6, Rora, Tmem176, Ramp3, Il7a, Gata3, Il1rl1 |
| ȃMouse thymocyte-like CD4+CD8+ cells | Unknown | Cd8b, Cd4, Tcf7, Ccr9, Rag1, Dntt, Lck |
| ȃMouse naïve T cells | Steady state | Cd28, Cr7, Lef1, Dapl1, Tcf7, Cd2, Sell/Il7r |
| ȃHuman GZMK+ effector/memory CD8+ T cells (CD8.0) | Effector memory | GZMK, ITM2C, CCL4, CD74, MYADM |
| ȃHuman GZMB+ cytotoxic CD8+ T cells (CD8.1) | Cytotoxicity | FGFBP2, ADGRG1, GZMB, CX3CR1, GNLY, FCGR3A |
| ȃHuman LEF1+ central memory CD8+ T cells (CD8.3) | Central memory | LEF1, LTB, SELL, IL7R, PDE3B, EEF1A1 |
| ȃPRF1+ cytotoxic CD4+ T cells (CD4.0) | Cytotoxicity | NKG7, CCL5, PRF1, GNLY, GZMA |
| ȃGZMK+ cytotoxic CD4+ T cells | Cytotoxicity | GZMK, GZMA |
| ȃIL7R+ naïve CD4+ T cells | Steady state | ZNF480, IL7R, CTSB |
| ȃFOXP3+ regulatory CD4+ T cells | Immunomodulation | FOXP3, IL2RA, ICOS, TIGIT |
| ȃLef1+ central memory CD4+ Tc ells | Central memory | LEF1, LTB, CCR7, TCF7 |
| Functional prediction | Markers (mouse/human) | |
|---|---|---|
| Macrophages | ||
| ȃInflammatory macrophages | Lesional inflammation | S100A8/S100A9, IL1B, CASP1, CASP4, NLRP3 |
| ȃResident-like macrophages | Efferocytosis, endocytosis antigen processing and presentation | LYVE1, CX3CR1, FOLR2, C1QA/C1QB/C1QC, CD74, HLA-DRA/HLA-DRB1 |
| ȃTREM2 ‘Foamy’ macrophages | Lipid processing, antioxidant | TREM2, CD9, FABP5, APOE, APOC1 |
| ȃIFNIC macrophagesa | Induced in disease progression | Isg15, Irf7, Ifit3, Ifit1 |
| ȃProliferating macrophagesa | Macrophage expansion or renewal | Stmn1, Top2a, Mki67, Tuba1b, Tubb5 |
| T cells | ||
| ȃMouse cytotoxic CD8+ T cells | Cytotoxicity | Cd8a/b, Nkg7, Ms4ab4, Ccl5, Gzmk, Eomes, Cd3d |
| ȃMouse CXCR6+ CD4+ T cells | Immune response to lipids | Cxcr6, Rora, Tmem176, Ramp3, Il7a, Gata3, Il1rl1 |
| ȃMouse thymocyte-like CD4+CD8+ cells | Unknown | Cd8b, Cd4, Tcf7, Ccr9, Rag1, Dntt, Lck |
| ȃMouse naïve T cells | Steady state | Cd28, Cr7, Lef1, Dapl1, Tcf7, Cd2, Sell/Il7r |
| ȃHuman GZMK+ effector/memory CD8+ T cells (CD8.0) | Effector memory | GZMK, ITM2C, CCL4, CD74, MYADM |
| ȃHuman GZMB+ cytotoxic CD8+ T cells (CD8.1) | Cytotoxicity | FGFBP2, ADGRG1, GZMB, CX3CR1, GNLY, FCGR3A |
| ȃHuman LEF1+ central memory CD8+ T cells (CD8.3) | Central memory | LEF1, LTB, SELL, IL7R, PDE3B, EEF1A1 |
| ȃPRF1+ cytotoxic CD4+ T cells (CD4.0) | Cytotoxicity | NKG7, CCL5, PRF1, GNLY, GZMA |
| ȃGZMK+ cytotoxic CD4+ T cells | Cytotoxicity | GZMK, GZMA |
| ȃIL7R+ naïve CD4+ T cells | Steady state | ZNF480, IL7R, CTSB |
| ȃFOXP3+ regulatory CD4+ T cells | Immunomodulation | FOXP3, IL2RA, ICOS, TIGIT |
| ȃLef1+ central memory CD4+ Tc ells | Central memory | LEF1, LTB, CCR7, TCF7 |
The above-mentioned single-cell studies revealed that human populations share striking similarities with those identified in the murine models,13 highlighting the relevance of mouse models to study atherosclerotic disease. Nevertheless, several differences between human and murine myeloid cell populations in atherosclerosis were identified. Whereas macrophages make up the bulk of immune cells in the murine aorta (around 50% of CD45+ cells), macrophage content decreases to 16%–20% of total CD45+ live cells in human carotid endarterectomies. Fate mapping during murine atherogenesis and regression has shown that both resident and bone marrow-derived contribute to each macrophage subset.74 Finally, single-cell techniques identified a proliferating and an interferon (IFN)-responsive macrophage cluster in mouse models, whereas a human counterpart of these subsets was not yet observed in human.65 These discrepancies might be due to the nature of the analysed tissue itself: in humans, cells are collected only from the intimal layer, whereas in most mouse studies, whole aortas encompassing the intima, media, and adventitial layers were included. In addition, physiological differences as well as differences in disease progression and timeline of disease progression (i.e. weeks vs. years) between the two species might contribute to the species differences. Further alignment focusing on similarities and differences between human and murine subsets is needed.
Lymphocytes outnumber macrophages in human atherosclerotic lesions
An integrative analysis of multiple single-cell transcriptome data sets of atherosclerotic mouse aortas23,65,69 found four major aortic T-cell populations: cytotoxic CD8+ T cells, a multi-lineage CXCR6+ CD4+ population, thymocyte-like CD4+CD8+, and naïve T cells. Two scRNA-seq studies of human atherosclerotic plaques uncovered an even larger diversity of T-cell populations and found three13 to eight64 cytotoxic CD8+ T-cell subpopulations and five13 to thirteen64 CD4+ T-cell populations. In human plaques, T cells outnumber macrophages by almost 2:1 within the live CD45+ immune cell pool. Histological studies have confirmed that tissue dissociation and asymmetrical cell loss are not the only explanation for the T-cell bias in the CD45+ live cell populations in the human atherosclerotic intima.13 Plaque T cells display an effector memory phenotype with multi-omics features of activation compared with the blood. CD8 T cells are highly represented in human atherosclerotic lesions compared with the blood, and in symptomatic patients who suffered a cerebral ischaemic event within 6 months before carotid atherosclerotic plaque collection display features of exhaustion.64
Tissue-resident CD4+ T cells showed a migratory gene signature in symptomatic patients. Whereas a higher enrichment in pro-inflammatory cytokine pathways (tumour necrosis factor α, IFNγ, IL12, and IL6) was found in asymptomatic patients, challenging the view that T-cell activation is univocally pro-atherogenic. Moreover, an effector memory CD4+ T-cell population was enriched in atherosclerotic plaques from patients with previous cerebrovascular event,64 providing insights and differences of the immune response in different patient categories. The transcriptome of this population overlaid with the transcriptome of ApoB-reactive CD4+ T cells,47 which also expressed hallmark genes of multiple T-cell lineages. These cells particularly show great plasticity in the course of atherosclerosis as they gradually lose their regulatory phenotype and acquire pro-inflammatory traits. As expansion of ApoB-reactive CD4+ T cells with a regulatory phenotype could be the basis for a vaccine against atherosclerosis, a deeper understanding of T-cell phenotypes is necessary to avoid expansion of pro-atherosclerotic T-cell subsets.75 Further formal integration, combining atherosclerotic plaque leucocyte data sets across species, will help model T-cell functional diversity.
B cells also belong to the adaptive immune system. Upon activation, B cells differentiate into short-lived plasma cells, which possess great potential to produce antibodies and long-lived memory cells. Atherosclerotic individuals have high levels of naturally occurring—or germline encoded—antibodies with high affinity for oxidized epitopes found on lipoproteins and dying cells. Although these exert anti-atherosclerotic mechanisms, B-cell responses in atherosclerosis are complex and dependent on the subset, thus playing pro- or anti-atherosclerotic effects.76,77 ScRNA-seq experiments identified two B-cell clusters in the aortas of atherosclerotic mice65 and only one B-cell cluster in human atherosclerotic plaques,13,64 and it is not yet clear how they aligns with the murine populations. In both species, B cells constituted only a very minor fraction of leucocytes, in line with previous reports where the majority of B cells resided in the adventitia.78 Notably, B cells drive atherogenesis by their activation in lymphoid organs and cavities and have a minor presence in the plaque itself.
Challenges and limitations of vascular single-cell biology
The unprecedented hypothesis-generating potential provided by vascular single-cell biology in atherosclerotic lesions is accompanied by limitations and challenges, often related to tissue handling, data generation, and computation analysis. A typical multidisciplinary scRNA-seq workflow is summarized in Figure 3.
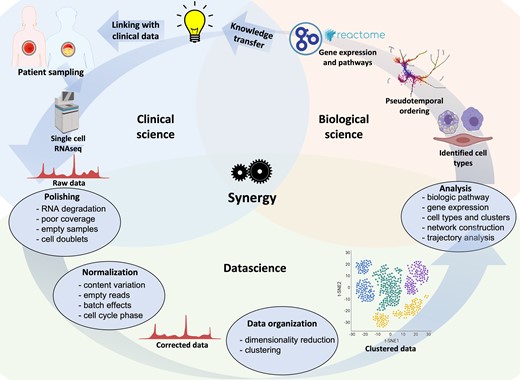
An iterative and multidisciplinary technical pipeline for scRNA-seq of human atherosclerosis. Samples from patients and healthy individuals can be processed, sequenced, and the resulting raw data filtered and normalized prior to organization into clusters to identify potential cell types involved in the disease. After obtaining raw sequencing data, subsequent bioinformatic steps involve quality controls (e.g. RNA degradation, unmapped reads or mitochondrial genes, or technical errors including cell doublets), normalization of data, removal of poorly sequenced cells, and possibly imputation of empty reads. Additional information, e.g. molecular pathways, differential gene expression, and networks, can be obtained. Novel findings can be transferred to scientific and clinical stakeholders, or used to optimize patient selection, specimen sampling and analysis.
Cell and tissue handling pitfalls
The analysis of cell populations in tissues by single-cell biology requires the generation of single-cell suspensions either by enzymatic digestion or by mechanical forces tailored to the type of tissue and disease state. Obtaining cell suspensions from vascular and atherosclerotic tissues, rich in calcifications and ECM is particularly challenging. Differences in digestion protocols can skew cell composition and different timing between sample collections may also affect the results, rendering the representation of real-life heterogeneity challenging. Despite divergence in the relative cellular composition,65 protocols have been established for whole mouse aorta digestion79 and after separation of the adventitia from the intima and media.80 In humans, specialized protocols for digestion have also been established.13,64
High viability of target cells is essential for robust scRNA-seq data. Small cell subsets can be enriched by cell sorting of the target cells of interest. Tissue dissociation can alter the gene expression profile. Processing-induced cell stress signatures81,82 can be counteracted by transcriptional inhibitors during tissue dissociation.13 Finally, sequencing of nuclei correlates with the total cell transcriptome83 and it offers the advantage of enabling analysis of pre-existing tissue libraries.
Computational analysis challenges
Access to single-cell data was until quite recently the preserve of specialized laboratories, but this is changing. With a growing number of analytical strategies, major steps have been taken to streamline and benchmark bioinformatic pipelines.24,84–89 Several web-based tutorials and user-friendly interfaces allow the visualization of data sets by non-bioinformaticians.90–92 However, it is recommended that a computational scientist be involved in the processing in a truly multidisciplinary collaboration (Figure 3). Quality control is a key feature of data analysis and perhaps the less transparent to the non-bioinformatic user. The occurrence of cell damage during dissociation or inefficient reverse transcription or polymerase chain reaction amplification can be inferred by the tell-tale signs of low total event counts, few expressed genes per cell, enrichment for nuclear RNAs, and low mitochondrial genome proportions. Batch effects arising from pre-analytical variations are common confounders that can be attenuated by increasing the number of replicates and/or using computational methods to test for batch variation.24,86
Single-cell data sets have a high level of dimensionality, which can be reduced using unsupervised hierarchical clustering algorithms, e.g. uniform manifold approximation and projection for dimension reduction , which allow the identification of clusters of cells with similar expression programmes. Although these do not reveal the identities of clusters, this can be inferred from the expression of specific markers that are known to hallmark a particular cell type or by referencing expression profiles obtained from isolated cells with known identity, such as leucocyte subtypes.93,94 This step represents one of the greatest hurdles in chronic human disease settings such as atherosclerosis, where tissue adaptations of unknown phenotype occur over time (Figure 1).
Low-quality scRNA-seq libraries can contribute to misleading results in downstream analyses. For instance, if the largest variances are driven by differences between low- and high-quality cells, the effectiveness of dimensionality reduction is decreased, leading to the inclusion of spurious cell clusters. The identification of rare cell subsets or transcriptional states relies on a significant number of cells and data points. Congruent data sets obtained across centres need to be merged to optimize information availability in the cardiovascular community. Data will need to be integrated across different types of single-cell measurements, organisms, anatomical location, and sites. These steps require a partnership between cardiovascular clinicians, surgeons, vascular biologists, and computational scientists (Figure 3). An embedded approach with a shared laboratory and office is the most conducive to true discovery.
Current limitations and future technological evolutions
Conventional scRNA-seq (e.g. illumina/10×) is biased towards 3′ transcripts and highly expressed transcripts. Thus, primary microRNAs, microRNAs, long noncoding RNAs, circular RNAs, alternatively spliced RNAs, and transcripts that are not highly expressed cannot be monitored by the current scRNA-seq technologies. The combination of scRNA-seq and bulk RNA sequencing of enriched target clusters is required to achieve in-depth information about the cell-specific whole transcriptome. Future efforts will focus on the development of higher depth RNA sequencing or single-cell direct RNA sequencing (also known as long-read RNA sequencing). Evolving smart-seq modalities offer the possibility of full-length sequencing, isoform detection, and detection of 5′-UTR mutations. The increase in resolution at the single-cell level results in a reduction of depth of sequencing and data stability compared with bulk RNA sequencing, and mechanistically relevant but non-abundant molecules might not be identified. This well-recognized pitfall can be mitigated either by scaling the number of cells up (which is desirable but possibly unfeasible with small human vascular tissue samples) or by combining bulk and single-cell data sets using deconvolution or other alignment computations, or by applying multi-parametric approaches such as mass cytometry against specific molecules of interest.
The cellular phenotypic heterogeneity and markers described so far are often based on gene expression profiling, which means they do not necessarily correlate with protein expression. Therefore, scRNA-seq should be complemented by proteomics methods, including single-cell proteomics or mass cytometry. Moreover, the transcriptional signature of a cell might diverge from the protein surface markers we usually rely on for their identification. CITE-seq is currently solving this divergence, but it requires a higher number of cells and more effort to evolve the antibody panels.
Cellular heterogeneity is often the result of niche heterogeneity and the need for tissue dissociation causes an inherent loss of geographic information. Tissue positional information of cells needs to be integrated to understand how cell–cell contacts affect cell state and function as well as disease progression. Multi-parametric tissue imaging and spatial transcriptomics will quickly fill this particular gap.95 Spatial transcriptomics is evolving to achieve true single-cell resolution. In the near future, integrated spatial approaches that give positional information on cellular communities and dissociative approaches that provide information on larger numbers of cells will go hand in hand.
Finally, recent technological advancements have allowed combining scRNA-seq and single-cell chromatin accessibility sequencing (e.g. scATACseq), thus enhancing the functional understanding of the role of transcription factors in cellular plasticity and intercellular communication at the site of disease.13,96 Future studies are needed to investigate the contribution of genomic (e.g. single-cell DNA seq97 and genome-wide association studies, disease-related genetic variants), transcriptomic (e.g. single-cell RNA editing,98 RNA modifications99), and proteomic posttranslational modifications (modifications including phosphorylation, glycosylation, ubiquitination, citrullination, nitrosylation, methylation, acetylation, and lipidation) in cellular subset phenotypes.
Clinical and translational insights emerging from vascular single-cell biology
Single-cell biology has opened novel opportunities for translational approaches in medicine and CVD will be next (Figure 4). While the generation of the first-cell atlases helped shift our perspectives on atherosclerosis, a clear gap exists in understanding how this technology can be exploited in the clinical setting for the direct benefit of patients.
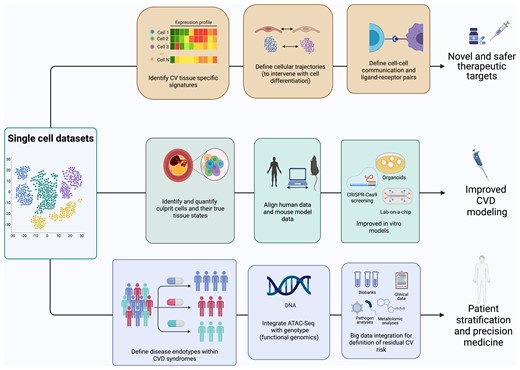
Translational opportunities for single-cell biology. Single-cell data sets allow a multitude of opportunities to better understand and define cardiovascular disease. Single-cell data flows will contribute to the translation from the laboratory to the cardiovascular disease clinics across several avenues. Firstly, they will inform the discovery of novel disease-associated therapeutic targets and drug repositioning. Secondly, they will transform our ability to model human disease in vivo and in vitro, by defining similarities and differences between human and experimental disease irrespective of species and at the single-cell level, and by defining cell phenotyping in situ for in vitro modelling. Finally, vascular single-cell biology will resolve heterogenous atherosclerotic syndromes in endotypes driven by specific cellular processes, identify cellular factors driving patient heterogeneity, and identify residual risk after optimal therapy.
Single-cell biology studies have recapitulated many of the features of key cell types in CVD, including their plasticity and acquisition of new functions, such as lipid uptake, phagocytosis, and calcification. The analysis of atherosclerotic lesions so far has shown that cellular identity is overall preserved, albeit overlapping transcriptional programmes are activated (Figure 2). scRNA-seq has the unique advantage over bulk RNA sequencing of capturing changes in cellular cluster abundance, giving us a better idea of the relative proportion of different subtypes between healthy and diseased states. The identification of transcriptional states with specific functions comes with clues to their transitions and adaptations over time, as well as the drivers associated with each state for selective targeting. Future mechanistic insights emerging also in association with scATACseq will lead to the identification of the specific molecular pathways that induce disease resistance vs. disease propensity and a deeper understanding of health to disease transition. This wealth of knowledge has great potential for precision medicine and improved therapeutic safety, as it would provide us with information on how to change pathogenic cellular fate decisions and promote transcriptional stages that drive health. Understanding cellular transitions will also shed light on genetic risk. For instance, genetic variants associated with lower expression of the transcription factor TCF21 have a high risk of cardiovascular events, and the loss of TCF21 in mice reduces the number of fibroblast-like SMCs, leading to a thinner fibrous cap.58
Single-cell biology has offered tremendous insight into the next steps of immunomodulation in CVD. The discovery that distinct subsets of T cells are present in clinically symptomatic carotid artery plaques associated with cerebrovascular events points towards the need for the development of cardiovascular immunotherapies that are tailored to T-cell activation, e.g. targeting immune checkpoints.64 Expansion of ApoB-reactive CD4+ T cells with a regulatory phenotype could represent the basis for a vaccine against atherosclerosis, and a deeper understanding of T-cell phenotypes is necessary to expand the correct cells and avoid expansion of pro-atherosclerotic T-cell subsets.75 The existence of distinct inflammatory and lipid-laden macrophage subsets provides a cellular basis for the evidence from clinical trials that targeting inflammation on top of lipid-lowering conveys outcome improvements in a non-redundant manner.100 Further dissection of macrophage heterogeneity may help us design athero-specific drugs that preserve the functions of atheroprotective resident macrophages,68 while targeting pro-atherogenic populations.
Single-cell analysis in human biosamples allows identification of relevant signalling pathways in disease and accelerates drug discovery and identification of biomarkers for disease prognosis53 and/or therapy response,101,102 further paving the way towards precision medicine in CVD. Integration of GWAS genetic variants with single-cell data and ATAC-seq will enhance our understanding of the mechanistic basis of heritable genetic risk.13,63 ScRNA-seq could provide the information needed to repurpose currently used drugs in cardiovascular treatments for specific patient groups, as well as enhance our understanding of the cellular and molecular effects of therapies currently used in CVD.
Although scRNA-seq analyses are widely used to characterize tissue specimens derived from humans and patients with atherosclerotic disease, future studies are needed to evaluate how cardiovascular therapy affects cellular responses at the single-cell level and to quantify the residual risk post-treatment. By combining scRNA-seq with machine learning approaches in human cohorts and available biobanking, researchers will be able to identify patient- and cell-specific mechanisms of disease and drug mechanisms of action. This approach has been successful in cancer,18 where the deep (scRNA-seq) analysis of tumour biopsies before and after a treatment delineated multiple spatially distinct tumours composed of genetically and functionally distinct neoplastic subpopulations as well as diverse non-neoplastic cell types. More intriguingly, biological features revealed by scRNA-seq enhanced the prediction of clinical outcomes in independent cohorts, thus highlighting how therapy-induced adaptation of the multi-cellular ecosystem of metastatic cancer shapes clinical outcomes.18 At this point in time, elevated costs, complex logistics (blood preservation, cell sorting needs, etc.), concentrated know-how, a lack of bioinformaticians or big data analysis specialists, a lack of standard protocols, and a lack of bedside technologies represent challenges to the application of these approaches to CVD. Examples are emerging. scRNA-seq of circulating blood is a sensitive method to evaluate treatment impact in clinical trials.102 In the future, we will see more clinical trial-driven sequencing as protocols and standardization evolve quickly, especially when driven by large-scale collaboration and knowledge sharing. Single-cell profiling of circulating cells before and during treatment could provide new tools for patient selection in clinical trials and showed promise in distinguishing responders from non-responders to the tested treatment.
Inflammation has been well established as a risk factor for atherosclerosis and its complications, including acute myocardial infarction.103 Thus, scRNA-seq of circulating leucocytes at various time points during the disease’s development or post therapy or medical treatment could be instrumental in identifying the leucocyte subtypes involved in the residual risk of patients with chronic or acute coronary syndromes, carotid or cerebral atherosclerotic vascular disease, and peripheral arterial disease. ScRNA-seq of circulating ECs in blood may enable the early identification of vascular dysfunction at a molecular level. ScRNA-seq may be used for therapy response monitoring, mining factors involved in sex-dependent differences in CVD, investigating specific anti-inflammatory treatments, towards future diversified preventative and therapeutic strategies. Identifying specific cells that associate with clinical events or poor disease outcomes in patients may aid in developing stratified therapies for the treatment of disease. A recent study identified a new subset of SLAN+ CXCR6+ monocytes that correlate with coronary artery disease.104 Further studies aligning plaque and blood cellular phenotypes will be needed to reveal whether functional changes in circulating cell subsets mirror changes occurring in the atherosclerotic plaque.
In the future additional pathophysiological insights may be obtained integrating sex-specific gene regulatory networks with scRNA-seq data of atherosclerotic plaques from humans and mice models to examine whether there will be sex differences involving phenotypic switching of atherosclerotic plaque EC subtypes as recently suggested for SMCs.22 Establishing congruent protocols and data sets and integrating large cohorts of patients where scRNA-seq data are linked to individual clinical patient data are essential to reaching the sample size and data quality needed for progress in this respect. Training of both scientists and clinicians in these novel technologies will be essential for progressing new, individualized cell-guided therapies.
Vascular single-cell biology will also help us improve our modelling of CVD in vitro and in vivo. Plaque-specific cell phenotypes will give us clues for organoid models and tissue signals. Mouse models allow to decipher basic mechanisms of cell transitions, including the assessment of the impact of lesion stage (early, late, regression), of the effect of manipulation (drugs, genetic), and of the cell of origin (lineage tracing). While gene regulatory networks are generally conserved between mouse and human cells, interspecies differences demand studies in humans. These are confounded and complicated by factors like vascular location (i.e. data from the aorta vs. carotid artery) and genetic variation in the human population. A public repository of scRNA-seq data with genotype metainformation would represent a major advance in the field, allowing stratification of states according to genomics and positional information.
Concluding remarks
Vascular single-cell biology is redrawing the cellular building blocks of the atherosclerotic plaque.105 By doing so, it has already challenged many of our assumptions. T cells outnumber macrophages in human atherosclerotic lesions.13 Macrophages can perform atheroprotective functions.68 The majority of human intimal myeloid cells expressing CD11c (a traditional dendritic cell marker) are in fact macrophages.106 SMCs and ECs are more plastic and dynamic than we ever imagined.58,107,108 This is only the beginning. The further detailing of the cellular landscape of the human plaque will accelerate drug discovery by identifying culprit cells rather than culprit lesions, discovering pathways of cellular activation and transdifferentiation, and finding new ligand–receptor interactions as a means of cellular crosstalk (Graphical Abstract).
The field of vascular single-cell biology is in its infancy and CVD is lagging behind other specialties in reaping its potential. A central atlas for vascular diseased tissues is still lacking. Due to the multifactorial nature of CVD, the integration of multiple human data sets and biobanks, as well as human and murine data sets, genetics, clinical characteristics, imaging profiles, and systemic markers, is paramount. A collaborative effort of unprecedented magnitude is required to collate data from multiple patients, disease stages, treatment regime and vascular beds to represent real-life heterogeneity with sufficient granularity to achieve translational usefulness.109 Growing improvements in medical treatment and increasing use of percutaneous endovascular procedures pose a time limit for the window of opportunity for vascular single-cell biology. No human vascular tissue sample should go to waste in the next decade, as it is becoming more and more precious. Biobanking needs updated protocols for cryopreservation of cells and tissues to match the vast range of new and emerging technologies. The CVD community and funders should redouble their efforts to enrich and establish human tissue biobanks at the national and international level. Collaborative and integrated progress is the only way forward to face the challenges and opportunities afforded by vascular single-cell biology and open new avenues for the treatment of CVD.
Funding
All authors declare no funding for this contribution.
Data availability
No new data were generated or analysed in support of this research.
References
Author notes
Nucleus member of the Working Group on Atherosclerosis and Vascular Biology.
Conflict of interest M.P.J.W. is funded by The Netherlands Heart Foundation (CVON 2017–20); The Netherlands Heart Foundation and Spark-Holding BV [2019B016]; Foundation Leducq (LEAN Transatlantic Network Grant); Amsterdam Cardiovascular Sciences; Amsterdam UMC; ZonMW (Open competition 09120011910025). P.E. is supported by the British Heart Foundation [RG/19/10/34506]. D.G. is funded by the National Institute of Health R01HL146465 and the American Heart Association 20IPA35310394 grants. I.G. is supported by the Swedish Research Council, the Swedish Heart and Lung Foundation, Skåne University Hospital funds, Lund University Diabetes Center (the Swedish Foundation for Strategic Research Dnr IRC15-006) and Region Skåne. H.F.J. is funded by the British Heart Foundation [PG/19/6/34153, RM/13/3/30159]. E.L. is supported by the European Research Council (ERC consolidator grant) and the Deutsche Forschungsgemeinschaft [CRC 1123]. G.D.N. is supported by: Telethon Foundation [GGP19146], Progetti di Rilevante Interesse Nazionale [PRIN 2017 K55HLC], Ricerca Finalizzata, Ministry of Health [RF-2019-12370896], and PNRR Missione 4 [Progetto CN3—National Center for Gene Therapy and Drugs based on RNA Technology]. E.O. is supported by the Swiss National Science Foundation (PRIMA: PR00P3_179861/1), the Swiss Life Foundation, the Alfred and Annemarie von Sick Grants for Translational and Clinical Research Cardiology and Oncology, the Heubergstiftung and the Swiss Heart Foundation. M.S. is funded by NIH grant HL139582. S.Y.H. is funded by the European Research Council Advanced Grant No 844382. H.W. is supported by the Neven-DuMont Foundation and the Deutsche Forschungsgemeinschaft: SFB TRR259 [397484323] and GRK2407 [360043781]. M.L.B.P. is supported by the Swiss National Science Foundation # 310030_185370/1. C.M. is funded by the British Heart Foundation [PG/18/1/33430 and PG/19/41/34426], the European Commission (TAXINOMISIS; grant agreement H2020-SC1-2016-2017, and 797788 STRIKING STREAKS), The Kennedy Trustees, Novo Nordisk (Oxford Novo Nordisk Fellowship), and the Novo Nordisk Foundation [NNF15CC0018346 and NNF0064142]. Figures were made with BioRender.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}