





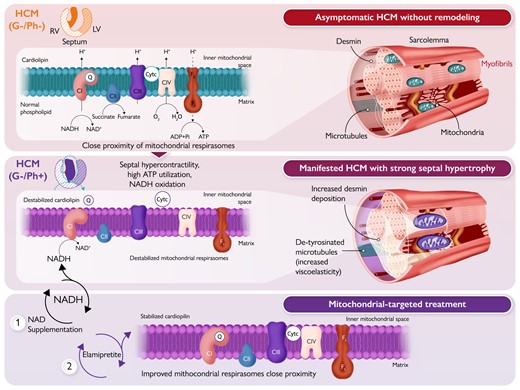
Mitochondrial-targeted therapies in hypertrophic cardiomyopathy (HCM). The top panel illustrates a healthy cardiomyocyte–mitochondrial organization in asymptomatic genotype-negative (G–/Ph–) HCM individuals. The 2D cross-section heart illustration shows the right ventricle (RV), septum, and left ventricle (LV). The cardiomyocyte–mitochondrial organization in G–/Ph– HCM is characterized by near normal production of viscoelastic proteins, such as desmin and microtubules, and the close proximity of mitochondrial supercomplexes or respirasomes in the presence of cardiolipin. The inner mitochondrial membrane (IMM) is traversed by NADH dehydrogenase (complex I, CI), cytochrome b–c1 complex (complex III, CIII), cytochrome c oxidase (complex IV, CIV), and F0F1-ATP synthase (complex V). Reduced forms of NADH and FADH2 (derived from succinate) donate electrons to the electron transport system via CI or CII (not membrane spanning), which are sequentially transferred to electron carriers, such as coenzyme Q (CoQ or Q), CIII, and cytochrome c (Cytc). CIV accepts electrons from the electron transport system and transforms oxygen into water. As electrons move along the electron transfer system, protons (H+) are pumped from the mitochondrial matrix into the mitochondrial IMM (at CI, CIII, and CIV; CII lacks a proton pumping mechanism), establishing an electrochemical proton gradient across the IMM. This gradient is used to regenerate ATP from ADP at complex V. The middle panel illustrates the potential consequences of septal hypercontractility, high ATP utilization, and increased NADH oxidation, to induce septal enlargement in G–/Ph+ HCM individuals. This progression is characterized by increased production of viscoelastic elements, such as microtubule densification, desmin deposition, and hypertrophied myofibrils. Consequently, this results in poor myofibril and mitochondrial architecture, and disorganized mitochondrial respirasomes. The lower panel highlights the potential benefits of improving the provision of reducing equivalents in the Krebs cycle with NAD supplementation or improving respiratory chain function with elamipretide to stabilize cardiolipin in the IMM. These therapies may improve the proximity of mitochondrial respirasomes, reduce electron leakage and oxidative stress, and enhance ATP regeneration.
This editorial refers to ‘Mitochondrial dysfunction in human hypertrophic cardiomyopathy is linked to cardiomyocyte architecture disruption and corrected by improving NADH-driven mitochondrial respiration’, by E.E. Nollet et al., https://doi.org/10.1093/eurheartj/ehad028.
Hypertrophic cardiomyopathy (HCM) is the most prevalent inherited form of heart failure (1:200 prevalence) and is associated with >1500 gene mutations. Over 95% of these mutations are found in >11 genes encoding sarcomeric proteins, i.e. the heart’s contractile building blocks.1,2 Consequently, mutations of sarcomeric genes are a primary cause of HCM. The clinical definition of HCM is a wall thickness of 15 mm or more in one or more left ventricular (LV) segments that cannot be explained by loading conditions alone.2 A poor clinical prognosis is associated with marked diastolic dysfunction progressing to heart failure with preserved ejection fraction and ventricular arrhythmias, with HCM being a common cause of sudden cardiac death, particularly in young people with sarcomeric gene mutations.3
HCM is characterized by extensive hypertrophy of the left ventricle, most notably affecting the interventricular septum. Although there is a moderate correlation between the prognosis of HCM and the degree of cardiac hypertrophy and fibrosis in the septum, the relationship between genotype and phenotype is rather weak. In patients with HCM, onset and presentation of disease can be quite diverse, ranging from asymptomatic mutation carriers (genotype-positive, phenotype-negative; G+/Ph–) to symptomatic patients (G+/Ph+) with severe cardiac remodelling, including hypertrophy, cellular disarray, fibrosis, and vascular dysfunction.3 Nearly two-thirds of HCM cases can be linked to pathogenic mutations, while the cause of the remaining third often remains unknown (genotype-negative, phenotype-positive; G–/Ph+).1,2 About 70% of patients have classical asymmetric septal hypertrophy with LV outflow tract obstruction [hypertrophic obstructive cardiomyopathy (HOCM)] at rest or during exercise.3 A small percentage of patients (5%) develop heart failure with a reduced ejection fraction (<50%) with cavity dilation and regression of hypertrophy, i.e. the dilated-hypokinetic (end-stage) evolution of HCM.
In hereditary forms of HCM (G+), the majority of affected individuals have an autosomal dominant genetic disorder, or are heterozygous for the disease (they carry one mutant and one wild-type allele). Since the same pathogenic mutation that causes HCM in one individual (G+/Ph+) can be harmless in another (HCM carriers, G+/Ph–),2 secondary disease-modifying factors may be required to initiate and/or modulate the progression of HCM. In fact, even mildly increased body weight during late adolescence can contribute to the penetrance of HCM during adulthood,4 indicating that, in particular, metabolic disorders may play an important role as disease modifiers.
In most hereditary forms of HCM, the sarcomeric mutations increase the Ca2+ affinity of the myofilaments, which on the one hand causes hypercontractility during systole, and on the other hand impairs relaxation during diastole.5 Since both systolic and diastolic force development consume substantial amounts of ATP (and oxygen), the energetic burden in the myocardium of patients with HCM is substantial and contributes to energetic depletion in patients with HCM even before the onset of relevant symptoms, as indicated by reduced levels of phosphocreatine (PCr) in relation to ATP.6
In cardiomyocytes, mitochondria are the main suppliers of cellular ATP by oxidative phosphorylation (OXPHOS).7 Primarily fatty acids, but also glucose (and other substrates), fuel the Krebs cycle, which produces the reduced form of NAD (NADH). By donating electrons to the electron transport system (ETS), NADH becomes oxidized to NAD+, while the electron flux along the ETS creates a proton gradient across the inner mitochondrial membrane which is harnessed for the production of ATP (Graphical Abstract, top panel).7 While the increased energetic demand imposed by hypercontractility may readily explain energetic depletion in G+/Ph+ HCM patients, it is unclear whether in patients without sarcomeric mutations (G–/Ph+), hypercontractility (caused by other mechanisms) or, alternatively, (primary) mitochondrial dysfunction may also contribute to the energetic deficit. Furthermore, it is unclear whether the chronic energetic burden can eventually also cause mitochondrial dysfunction in patients with genetic HCM.
In this issue of the European Heart Journal, Nollet et al. took a deep dive to address these open issues.8 In a careful, yet extensive, experimental approach, using mostly fresh biopsies from the myocardium of patients with HOCM who are either positive (G+/Ph+) or negative (G–/Ph+) for sarcomeric mutations, the authors made three key observations: (i) in patients with HOCM, lower mitochondrial respiration correlated with increased septum mass in G–/Ph+ but not G+/Ph+ HCM patients; (ii) improper spatial organization of mitochondria (which are usually located in a regular pattern close to the ATP-demanding myofibrils) was closely associated with dysfunctional mitochondrial respiration; and (iii) ex vivo treatment with NAD+, supposedly increasing mitochondrial NADH availability, or elamipretide, a tetrapeptide that stabilizes ETS supercomplexes,9 improved mitochondrial respiration.
Considering that at least a third of symptomatic HCM cases are negative for sarcomeric mutations (G–/Ph+) and, thereby, have an unknown aetiology, the study by Nollet and co-workers is the first to show an association between mitochondrial dysfunction, potentially related to disrupted cardiomyocyte mitochondrial localization, and septal hypertrophy in genotype-negative HCM (Graphical Abstract, middle panel). From a therapeutic perspective, it is interesting to notice improved mitochondrial respiration following NAD+ or elamipretide supplementation in HCM samples. Despite significant myocardial remodelling, mitochondria were receptive to ex vivo treatments designed to restore mitochondrial respiration. These results generate the hypothesis that mitochondria could be pharmacological targets to prevent and mitigate HCM severity in patients (Graphical Abstract, lower panel).
Previous studies from the same group10 found abnormalities in NAD+ homeostasis in cardiac biopsies of human HCM, suggesting that mitochondrial NAD+ (‘backbone’) levels may be reduced compared with non-failing controls, hence reducing the availability of NADH. In fact, in rodent models of ageing, metabolic syndrome, and hypertension, restoration of cellular NAD+ levels in vivo enhanced mitochondrial bioenergetics and improved diastolic dysfunction, which was to some extent mediated by improvement of cardiac myocyte stiffness and Ca2+-dependent active relaxation.11 Furthermore, supplementation with nicotinamide riboside prevented heart failure development in a mouse model of dilated cardiomyopathy.12 These findings are consistent with additional studies demonstrating benefits of several ‘NAD+ boosters’ in various heart failure models.13 Therefore, these findings and the current results by Nollet et al. provide a rationale to test the impact of NAD+ supplementation also in animal models of and patients with HCM (Graphical Abstract, lower panel).
Furthermore, the study of Nollet et al. also showed that ex vivo incubation of myocardial tissue from HCM patients with the tetrapeptide elamipretide (also known as SS-319) improved mitochondrial NADH-coupled respiration, which was associated with stabilization of complex I within the ETS supercomplexes in human HCM. Cardiolipin is a unique type of phospholipid (four-tail lipid) that is exclusively found at the inner (up to 22%) and outer (3%) mitochondrial membrane and improves electron transfer and ATP regeneration, while simultaneously reducing electron leakage and formation of reactive oxygen species.9 Cardiolipin is susceptible to lipid peroxidation and the formation of 4-hydroxynonenal (4-HNE), which is elevated in the myocardium of HCM patients.14 In principle, disruption of mitochondrial supercomplexes (by, for example, peroxidation) may hamper mitochondrial electron flow, decreasing ATP production and increasing aberrant formation of superoxide (Graphical Abstract, middle panel). Elamipretide accumulates in mitochondria and stabilizes cardiolipin, which is thought to tighten proximity gaps between the ETS supercomplexes to minimize electron leakage during electron transport.9 In fact, Nollet et al. observed that integration of complex I into the ETS supercomplex increased after incubation of the myocardium with elamipretide, which was associated with higher complex I-linked respiration (Graphical Abstract, lower panel). These data are in principal agreement with the observation that compared with mutation carriers without a cardiac phenotype (G+/Ph–), patients with HOCM have decreased O2 consumption per myocardial mass.15 While the underlying reasons for the complex I defect in G–/Ph+ patients in the current study remain unresolved, they suggest, however, that approaches to improve mitochondrial respiration may be a promising therapeutic strategy.
The authors need to be congratulated for addressing the pathophysiological basis of energy starvation in patients with HOCM in a large and clinically well-characterized cohort of patients with HOCM. However, several issues remain. First, for the functional measurements, a control group of normal myocardium is lacking (since such tissue is obviously difficult to obtain) and, therefore, it is not fully clear whether, on average, mitochondrial function (i.e. respiration) is really compromised at all in patients with HCM compared with healthy hearts. Furthermore, the results are somewhat at odds with the clinical observation that energy depletion (indexed by PCr/ATP) is more severe in HCM patients with, compared with those without, pathogenic mutations.6 Potentially, the fact that LV outflow tract obstruction was more severe in genotype-negative compared with genotype-positive patients may have contributed to more severe respiratory defects in the mitochondria, although no correlation was observed between the LV outflow tract gradients and mitochondrial function. Finally, in the acute treatment experiments, 100 µM elamipretide was used; it is currently unclear whether such high concentrations can be achieved by (clinical) treatment with elamipretide in vivo.
Nevertheless, the current study provides convincing evidence that respiratory defects of cardiac mitochondria may contribute to energy depletion and the pathophysiology of H(O)CM, in particular when LV hypertrophy progresses. Improving the provision of reducing equivalents in the Krebs cycle with the delivery of NAD+ (precursors13) or modifying cardiac substrate metabolism (as has been performed previously in HCM with perhexilline16), or improving respiratory chain function with elamipretide9 could be valuable treatment options that may further improve the course of the disease beyond the currently available approaches.
Acknowledgements
We thank Dr Hubert Seggewiß for valuable input to the manuscript.
Funding
C.M. is funded by the Deutsche Forschungsgemeinschaft (DFG; SFB-1525/project nos 453989101 and Ma 2528/8-1). V.S. is supported by a research fund from Bristol Myers Squibb. A.B. is funded by the DFG Clinician Scientist program UNION-CVD (project no. 413657723).
References
Author notes
The opinions expressed in this article are not necessarily those of the Editors of the European Heart Journal or of the European Society of Cardiology.
Conflict of interest: C.M. is an advisor to Amgen, Boehringer Ingelheim, Bristol Myers Squibb, NovoNordisk, and Servier, and received speaker honoraria from AstraZeneca, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, Berlin Chemie, Novartis, and NovoNordisk. The other authors have no conflicts to declare.


{kind=link}