





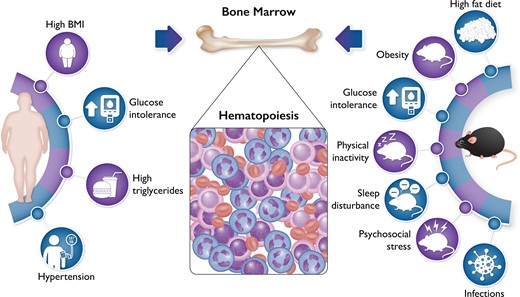
Bone marrow haematopoiesis increases in response to diverse drivers of cardiovascular disease. Numerous experiments in mice have demonstrated that several traditional risk factors tested, including hypertension, hypercholesterolaemia, and hyperglycaemia, can augment haematopoiesis (listed on the right). Other stimuli to bone marrow activation relevant to cardiovascular disease include sleep fragmentation and psychosocial stress. The validation in humans of these observations in mice has lagged behind. The new findings from PESA help to close this gap by showing that the components of the metabolic syndrome listed on the left can increase glucose analogue FDG uptake in bone marrow as determined by positron emission tomographic imaging. These observations on chronic risk factors, in an apparently well population, complement observations in humans undergoing acute coronary syndrome that show increased FDG uptake in both the bone marrow and the spleen. The experimental and human observations together provide strong support for the concept that bone marrow activation and increased haematopoiesis can contribute to the development of atherosclerosis.
This editorial refers to ‘Bone marrow activation in response to metabolic syndrome and early atherosclerosis’, by A. Devesa et al., https://doi.org/10.1093/eurheartj/ehac102.
The bone marrow, once the exclusive province of haematologists, has emerged from the wings to occupy centre stage as a protagonist in cardiovascular pathophysiology. The role of bone marrow-derived leucocytes and of inflammation in vascular and cardiac disease no longer engenders controversy.1,2 A rich experimental literature and manifold human biomarker studies have implicated inflammatory processes in atherosclerosis, arterial and venous thrombosis, and many forms of myocardial disease. The Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) demonstrated the causality of inflammation in humans with atherosclerotic cardiovascular disease. Follow-on studies with colchicine have confirmed inflammation as a viable therapeutic target in secondary prevention of atherosclerosis.3
Despite the increasingly detailed experimental exploration of the mechanisms that link inflammation to atherosclerosis, the translation from the laboratory to humans has remained primarily indirect and speculative. Devesa and colleagues from the Centro Nacional de Investigaciones Cardiovasculares (CNIC) in Madrid now present, in this issue of the European Heart Journal, data from the ‘Progression of Early Subclinical Atherosclerosis’ (PESA) study that helps to close this gap between the laboratory and the clinic, and provides insight into the causality and directionality of the relationship of inflammation to human atherosclerosis.4 PESA, masterminded by Professor Valentin Fuster, enrolled >4000 apparently well, predominantly male, bank employees in Madrid. These participants underwent multimodality imaging including fluorodeoxyglucose positron emission tomography (FDG PET). The investigators used uptake of this radio-labelled glucose analogue in lumbar vertebrae as an index of bone marrow activation.5 They found a remarkable and quite consistent association of components of the metabolic syndrome including central obesity, elevated blood triglycerides, hypertension, indices of insulin resistance, and enhanced FDG uptake in bone marrow (Graphical Abstract). Dichotomous analyses disclosed these associations, and they generally followed rank order in quintile groupings.
Bone marrow FDG uptake was also associated with a variety of circulating inflammatory markers including C-reactive protein measured with a highly sensitive assay (hsCRP). Yet, some of the participants had evidence for bone marrow activation even in the absence of elevation of this integrative biomarker of inflammation. This dissociation indicates that locally increased metabolic activity, which is associated with higher blood stem cell activity and thus leucocyte production in the bone marrow, can occur independently of—and perhaps before—discernible systemic inflammation. Moreover, many participants had evidence of bone marrow activation without having atherosclerotic lesions in several arterial beds, as gauged by imaging with magnetic resonance, computed tomography, and ultrasound. These observations occurred in the absence of other instigators of haematopoiesis such as infection, and therefore imply that mischief in the bone marrow can precede systemic inflammation or accumulation of atherosclerotic plaque, consistent with a causal precursor relationship. While experimental evidence shows that atherosclerosis can stimulate haematopoiesis, these findings from PESA in humans suggest that boosted haematopoiesis can occur before lesion formation, informing the directionality of this relationship.6,7 The experimental and human observations together suggest the operation of a positive feedback loop: haematopoiesis boosts atherogenesis, and atheroma can promote haematopoiesis (Figure 1).
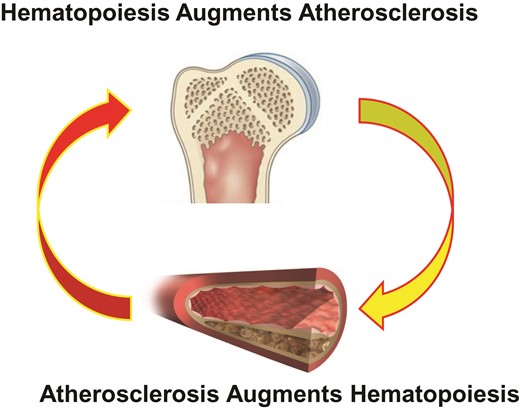
A combination of experimental results and the new human observations of Devesa et al. indicate that haematopoiesis can drive atherosclerosis. Laboratory evidence shows that hypercholesterolaemia in mice can boost haematopoiesis. The bi-directional positive feedback loop depicted here could contribute to progression of atherosclerosis and ongoing risk in individuals with atherosclerosis. Taming bone marrow activation may furnish a new biomarker for therapeutic effects of interventions and a new target for treatment.
Exposures beyond those studied here can also modulate haematopoiesis, including physical activity, sleep disturbance, and mental and social stress.8,9 These emerging links of mental and social stress to haematopoiesis provide novel insight into the link between mind and body. The association between mental stress, adversity, and cardiovascular disease has a venerable and anecdotal history. We are now beginning to piece together the fundamental mechanisms which link the brain to the cardiovascular system. Activation of the bone marrow has emerged as a pivotal piece of this puzzle.10
The pioneering study of Devesa et al. raises a few issues that merit particular comment. The authors do not mention FDG uptake by the spleen. Work in mice shows that the spleen can serve as a source of leucocytes mobilized during stress.1 Patients with acute coronary syndromes show increased splenic as well as heightened bone marrow FDG avidity.11 These human observations suggest that chronic low-grade inflammation in the apparently well cohort of bank functionaries stimulates leucocyte production in the bone marrow, whereas splenic extramedullary haematopoiesis or mobilization of leucocytes from the spleen contribute to leucocytosis, the response to an acute stress such as myocardial infarction.
The notion that FDG uptake correlates directly with inflammation has gained acceptance. This association makes sense as macrophages exhibit augmented dependence on glycolysis, as do tumour cells (the Warburg phenomenon). Thus, the increased glucose uptake may merely reflect accumulation of macrophages. Yet, smooth muscle cells can also contribute to arterial FDG uptake. Moreover, moderate hypoxia, a condition that prevails in plaques, robustly increases glucose uptake by cultured human mononuclear phagocytes to a greater extent than inflammatory activation, increasing reliance of these cells on glycolytic energy production.12 In addition, an enhanced microvasculature within arterial plaque and perhaps bone marrow can selectively increase delivery of the isotopically labelled glucose analogue, augmenting the radiotracer signal.7,13 In support of this concept, hypertension, a risk factor that drives atherosclerosis, leads to mild angiogenesis, endothelial dysfunction, and increased haematopoiesis in the bone marrow of mice.7 Indeed, in the bone marrow haematopoietic niche, experimental atherosclerosis and coronary artery ligation in mice drive leucocyte production.7 Such processes, if replicated in humans, could contribute to the increased vertebral body FDG signal observed by Devesa et al. With respect to clinical translation, the proinflammatory cytokine interleukin-6 (IL-6) contributes to enhanced myelopoiesis in the bone marrow in mice with atherosclerosis or acute myocardial infarction. Inhibition of IL-6 signalling has become a practical target in cardiovascular conditions, and effects at the level of the bone marrow haematopoietic niche might contribute to clinical benefit.3
This investigation in the PESA population showed a particular rise in granulocytes among the leucocyte populations in those with evidence of bone marrow activation. This observation has clinical significance, as the neutrophil–lymphocyte ratio (NLR) furnishes a simple index of inflammation readily calculable from a routine haemogram.14 The study of Devesa et al. provides some mechanistic insight into the genesis of this convenient biomarker of immediate clinical utility.
Overall, this important study provides key translational support for the burgeoning findings in mice that implicate altered bone marrow function and enhanced haematopoiesis as a previously unrecognized driver of atherogenesis in humans (Graphical Abstract). This new work from PESA validates concepts that have emerged from recent laboratory experiments. The production of inflammatory leucocytes in haematopoietic organs has emerged as a newly recognized mechanism promoting cardiovascular diseases. This ‘primary prevention’ study in PESA provides us with novel information gleaned from assessment of a marker of bone marrow activation in humans. We have a new therapeutic target on the horizon in cardiovascular prevention: mitigating mischief in the bone marrow.
Funding
P.L. receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), the RRM Charitable Fund, and the Simard Fund. M.N. receives support from the National Heart, Lung, and Blood Institute (HL142494 and HL139598). F.K.S. reports funding from the National Heart, Lung, and Blood Institute (HL135752).
Conflict of interest: P.L. is an unpaid consultant to, or involved in clinical trials for, Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Novartis, Pfizer, and Sanofi-Regeneron. He is a member of the scientific advisory board for Amgen, Caristo Diagnostics, Cartesian Therapeutics, CSL Behring, DalCor Pharmaceuticals, Dewpoint Therapeutics, Kancera, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, PlaqueTec, TenSixteen Bio, and XBiotech, Inc. His laboratory has received research funding in the last 2 years from Novartis. He is on the Board of Directors of XBiotech, Inc., and has a financial interest in Xbiotech, a company developing therapeutic human antibodies. His interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies. M.N. has received funds or material research support from Alnylam, Biotronik, CSL Behring, GlycoMimetics, GSK, Medtronic, Novartis, and Pfizer, as well as consulting fees from Lilly, Biogen, Gimv, IFM Therapeutics, Molecular Imaging, Sigilon, and Verseau Therapeutics. F.K.S. has received funds or material research support from Verseau Therapeutics, Pfizer, Novartis, GSK, Partner Therapeutics, and Astra Zeneca.
References
Author notes
The opinions expressed in this article are not necessarily those of the Editors of the European Heart Journal or of the European Society of Cardiology.


{kind=link}
{kind=link}